
Targeting Protein Kinases to Protect Beta-Cell Function and Survival in Diabetes

Abstract
1. Introduction
2. Targeting Protein Kinases to Protect Beta-Cell Function and Survival from Inflammation
2.1. Targeting the Serine/Threonine Kinase Mammalian Sterile 20-like Kinase 1 (MST1)
2.2. Targeting the Serine/Threonine Kinase Transforming Growth Factor-β Activated Kinase-1 (TAK1, or MAP3kinase 7)
2.3. Targeting the Tumor Progression Locus 2 Kinase (TPL2, or MAP3kinase 8)
2.4. Targeting the Cellular Abelson Tyrosine Kinase (c-Abl)
2.5. Targeting the Tyrosine Kinase 2 (TYK2)
2.6. Targeting the Mammalian Janus Kinase 1 (JAK1)
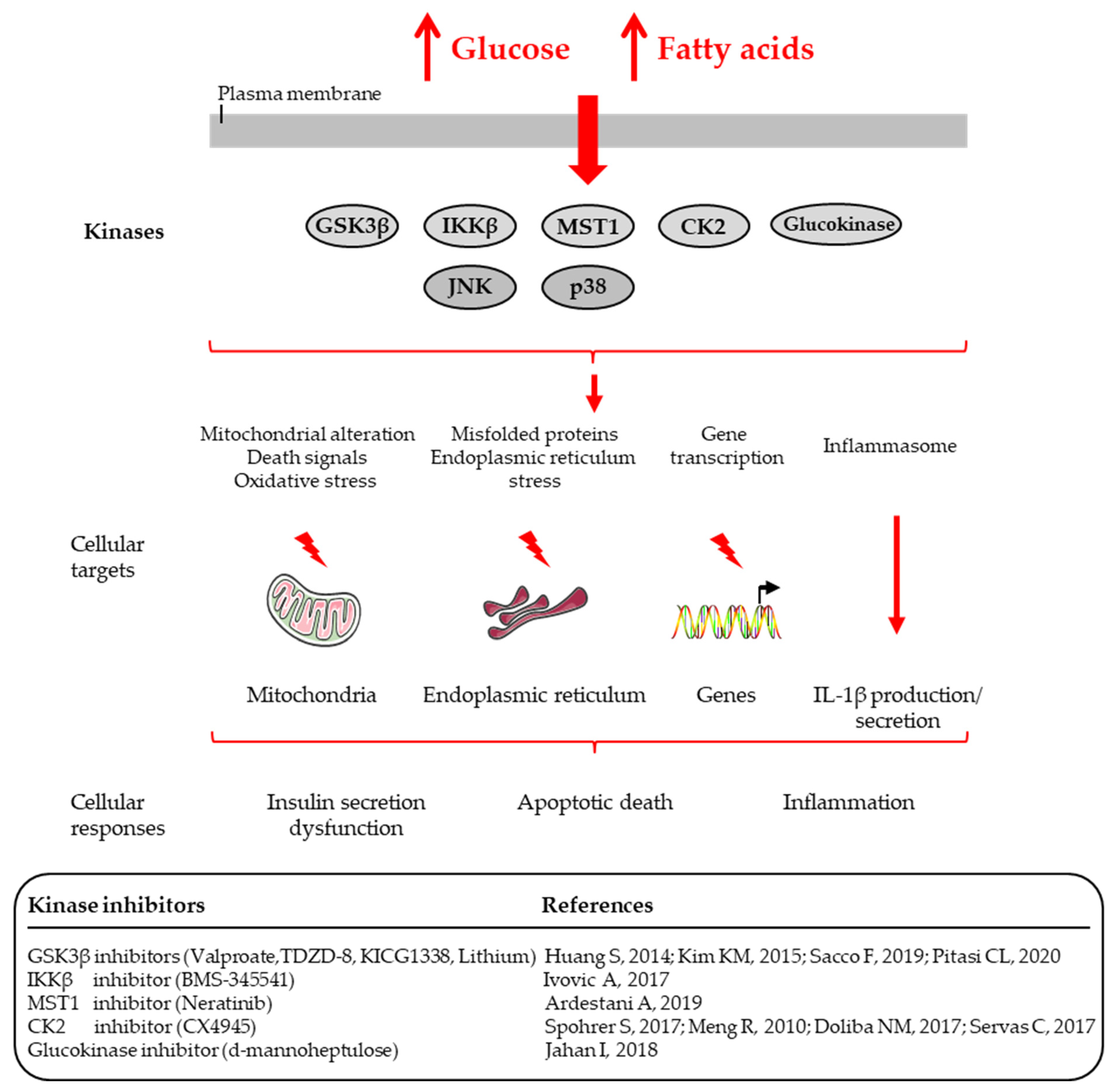
3. Targeting Protein Kinases to Protect Beta-Cell Function and Survival from Glucotoxicity and Glucolipotoxicity
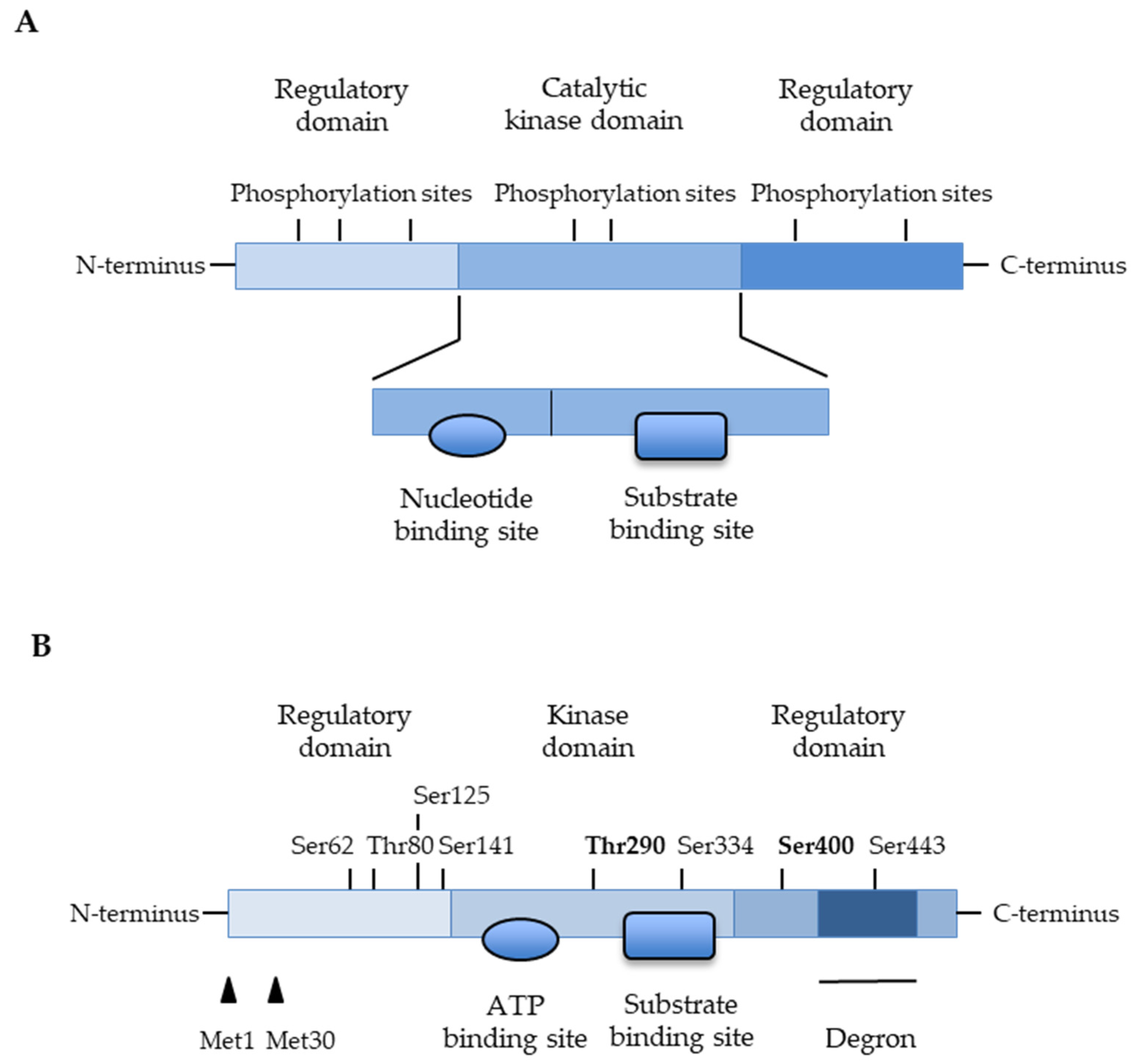
3.1. Targeting the Glycogen Synthase Kinase-3β (GSK-3β)
3.2. Targeting the Inhibitor of Nuclear Factor Kappa-B Kinase Subunit Beta (IKKβ)
3.3. Targeting the Serine/Threonine Kinase Mammalian Sterile 20-like Kinase 1 (MST1)
3.4. Targeting the Adenosine Monophosphate-Activated Protein Kinase (AMPK)
3.5. Targeting the Protein Kinase CK2 (Previously Called Casein Kinase 2 or CK-II)
3.6. Targeting the Glucokinase
3.7. Targeting the Protein Kinase RNA-like Endoplasmic Reticulum Kinase (PERK)
4. Targeting Protein Kinase to Induce Beta-Cell Proliferation and Regeneration
5. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107. [Google Scholar] [CrossRef] [PubMed]
- Mandrup-Poulsen, T. Interleukin-1 antagonists and other cytokine blockade strategies for type 1 diabetes. Rev. Diabet. Stud. 2012, 9, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.I.G.; DeVries, J.H.; Hess-Fischl, A.; Hirsch, I.B.; Kirkman, M.S.; Klupa, T.; Ludwig, B.; Nørgaard, K.; Pettus, J.; Renard, E.; et al. The management of type 1 diabetes in adults. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2021, 64, 2609–2652. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Zraika, S.; Utzschneider, K.M.; Hull, R.L. The beta cell lesion in type 2 diabetes: There has to be a primary functional abnormality. Diabetologia 2009, 52, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.R.; Clark, A.; Rorsman, P. β-cell secretory dysfunction: A key cause of type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Weir, G.C.; Gaglia, J.; Bonner-Weir, S. Inadequate β-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 249–256. [Google Scholar] [CrossRef]
- Bensellam, M.; Laybutt, D.R.; Jonas, J.C. The molecular mechanisms of pancreatic β-cell glucotoxicity: Recent findings and future research directions. Mol. Cell. Endocrinol. 2012, 364, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rohm, M.; Clark, A.; Brereton, M.F. Is type 2 diabetes a glycogen storage disease of pancreatic β-cells. Cell. Metab. 2017, 26, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C. Glucolipotoxicity, β-cells, and diabetes: The emperor has no clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Peyot, M.L.; Masiello, P.; Madiraju, S.R.M. Nutrient-induced metabolic stress, adaptation, detoxification, and toxicity in the pancreatic β-Cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Bertrand, G.; Ravier, M.A. Mechanisms of beta-cell apoptosis in type 2 diabetes-prone situations and potential protection by GLP-1-based therapies. Int. J. Mol. Sci. 2021, 22, 5303. [Google Scholar] [CrossRef] [PubMed]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Jörns, A.; Arndt, T.; Meyer zu Vilsendorf, A.; Klempnauer, J.; Wedekind, D.; Hedrich, H.J.; Marselli, L.; Marchetti, P.; Harada, N.; Nakaya, Y.; et al. Islet infiltration, cytokine expression and beta cell death in the NOD mouse, BB rat, Komeda rat, LEW.1AR1-iddm rat and humans with type 1 diabetes. Diabetologia 2014, 57, 512–521. [Google Scholar] [CrossRef]
- Barton, F.B.; Rickels, M.R.; Alejandro, R.; Hering, B.J.; Wease, S.; Naziruddin, B.; Oberholzer, J.; Odorico, J.S.; Garfinkel, M.R.; Levy, M.; et al. Improvement in outcomes of clinical islet transplantation: 1999–2010. Diabetes Care 2012, 35, 1436–1445. [Google Scholar] [CrossRef]
- Marfil-Garza, B.A.; Imes, S.; Verhoeff, K.; Hefler, J.; Lam, A.; Dajani, K.; Anderson, B.; O’Gorman, D.; Kin, T.; Bigam, D.; et al. Pancreatic islet transplantation in type 1 diabetes: 20-year experience from a single-centre cohort in Canada. Lancet Diabetes Endocrinol. 2022, 10, 519–532. [Google Scholar] [CrossRef]
- Lablanche, S.; Borot, S.; Wojtusciszyn, A.; Bayle, F.; Tétaz, R.; Badet, L.; Thivolet, C.; Morelon, E.; Frimat, L.; Penfornis, A.; et al. Five-year metabolic, functional, and safety results of patients with type 1 diabetes transplanted with allogenic islets within the swiss-french GRAGIL network. Diabetes Care 2015, 38, 1714–1722. [Google Scholar] [CrossRef]
- Barshes, N.R.; Wyllie, S.; Goss, J.A. Inflammation-mediated dysfunction and apoptosis in pancreatic islet transplantation: Implications for intrahepatic grafts. J. Leukoc. Biol. 2005, 77, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Urano, F. Endoplasmic reticulum stress in beta cells and autoimmune diabetes. Curr. Opin. Immunol. 2016, 43, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, A.K.; Heimberg, H.; Heremans, Y.; Leeman, R.; Kutlu, B.; Kruhøffer, M.; Ørntoft, T.; Eizirik, D.L. A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J. Biol. Chem. 2001, 276, 48879–48886. [Google Scholar] [CrossRef] [PubMed]
- Heimberg, H.; Heremans, Y.; Jobin, C.; Leemans, R.; Cardozo, A.K.; Darville, M.; Eizirik, D.L. Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes 2001, 50, 2219–2224. [Google Scholar] [CrossRef] [PubMed]
- Pirot, P.; Cardozo, A.K.; Eizirik, D.L. Mediators and mechanisms of pancreatic beta-cell death in type 1 diabetes. Arq. Bras. Endocrinol. Metabol. 2008, 52, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Meyerovich, K.; Ortis, F.; Cardozo, A.K. The non-canonical NF-kappaB pathway and its contribution to beta-cell failure in diabetes. J. Mol. Endocrinol. 2018, 61, F1–F6. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Buckner, J.H.; Herold, K.C. Immunotherapy: Building a bridge to a cure for type 1 diabetes. Science 2021, 373, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Li, S.; Annamalai, K.; Lupse, B.; Geravandi, S.; Dobrowolski, A.; Yu, S.; Zhu, S.; Baguley, T.D.; Surakattula, M.; et al. Neratinib protects pancreatic beta cells in diabetes. Nat. Commun. 2019, 10, 5015. [Google Scholar] [CrossRef]
- Cao, H.; Lu, J.; Du, J.; Xia, F.; Wei, S.; Liu, X.; Liu, T.; Liu, Y.; Xiang, M. TAK1 inhibition prevents the development of autoimmune diabetes in NOD mice. Sci. Rep. 2015, 13, 14593. [Google Scholar] [CrossRef]
- Varin, E.M.; Wojtusciszyn, A.; Broca, C.; Muller, D.; Ravier, M.A.; Ceppo, F.; Renard, E.; Tanti, J.F.; Dalle, S. Inhibition of the MAP3 kinase Tpl2 protects rodent and human β-cells from apoptosis and dysfunction induced by cytokines and enhances anti-inflammatory actions of exendin-4. Cell Death Dis. 2016, 7, e2065. [Google Scholar] [CrossRef] [PubMed]
- Hägerkvist, R.; Sandler, S.; Mokhtari, D.; Welsh, N. Amelioration of diabetes by imatinib mesylate (Gleevec): Role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J. 2007, 21, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Gitelman, S.E.; Bundy, B.N.; Ferrannini, E.; Lim, N.; Blanchfield, J.L.; DiMeglio, L.A.; Felner, E.I.; Gaglia, J.L.; Gottlieb, P.A.; Long, S.A.; et al. Imatinib therapy for patients with recent-onset type 1 diabetes: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2021, 9, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Coomans de Brachène, A.; Castela, A.; Op de Beeck, A.; Mirmira, R.G.; Marselli, L.; Marchetti, P.; Masse, C.; Miao, W.; Leit, S.; Evans-Molina, C.; et al. Preclinical evaluation of tyrosine kinase 2 inhibitors for human beta-cell protection in type 1 diabetes. Diabetes Obes. Metab. 2020, 22, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Jhala, G.; Fynch, S.; Akazawa, S.; Litwak, S.; Pappas, E.G.; Catterall, T.; Vakil, I.; Long, A.J.; Olson, L.M.; et al. The JAK1 selective inhibitor ABT 317 blocks signaling through interferon-γ and common γ chain cytokine receptors to reverse autoimmune diabetes in NOD mice. Front. Immunol. 2020, 11, 588543. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Phung, A.L.; Jhala, G.; Trivedi, P.; Principe, N.; De George, D.J.; Pappas, E.G.; Litwak, S.; Sanz-Villanueva, L.; Catterall, T.; et al. Diabetes induced by checkpoint inhibition in nonobese diabetic mice can be prevented or reversed by a JAK1/JAK2 inhibitor. Clin. Transl. Immunol. 2022, 11, e1425. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Vandewalle, B.; Tourrel-Cuzin, C.; Broca, C.; Linck, N.; Bertrand, G.; Kerr-Conte, J.; Portha, B.; Pattou, F.; Bockaert, J.; et al. Degradation of cAMP-responsive element-binding protein by the ubiquitin-proteasome pathway contributes to glucotoxicity in beta-cells and human pancreatic islets. Diabetes 2009, 58, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Gurlo, T.; Ravier, M.A.; Wojtusciszyn, A.; Mathieu, J.; Brown, M.R.; Broca, C.; Bertrand, G.; Butler, P.C.; Matveyenko, A.V.; et al. Proteasomal degradation of the histone acetyl transferase p300 contributes to beta-cell injury in a diabetes environment. Cell Death Dis. 2018, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in type 2 diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Lytrivi, M.; Ghaddar, K.; Lopes, M.; Rosengren, V.; Piron, A.; Yi, X.; Johansson, H.; Lehtiö, J.; Igoillo-Esteve, M.; Cunha, D.A.; et al. Combined transcriptome and proteome profiling of the pancreatic β-cell response to palmitate unveils key pathways of β-cell lipotoxicity. BMC Genom. 2020, 21, 590. [Google Scholar] [CrossRef]
- You, S.; Zheng, J.; Chen, Y.; Huang, H. Research progress on the mechanism of beta-cell apoptosis in type 2 diabetes mellitus. Front. Endocrinol. 2022, 13, 976465. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, M.; Wu, W.; Rashid, A.; Liang, Y.; Hou, L.; Ning, Q.; Luo, X. Valproate pretreatment protects pancreatic β-cells from palmitate-induced ER stress and apoptosis by inhibiting glycogen synthase kinase-3β. J. Biomed. Sci. 2014, 21, 38. [Google Scholar] [CrossRef]
- Kim, K.M.; Lee, K.S.; Lee, G.Y.; Jin, H.; Durrance, E.S.; Park, H.S.; Choi, S.H.; Park, K.S.; Kim, Y.B.; Jang, H.C.; et al. Anti-diabetic efficacy of KICG1338, a novel glycogen synthase kinase-3β inhibitor, and its molecular characterization in animal models of type 2 diabetes and insulin resistance. Mol. Cell. Endocrinol. 2015, 409, 1–10. [Google Scholar] [CrossRef]
- Sacco, F.; Seelig, A.; Humphrey, S.J.; Krahmer, N.; Volta, F.; Reggio, A.; Marchetti, P.; Gerdes, J.; Mann, M. Phosphoproteomics reveals the GSK3-PDX1 axis as a key pathogenic signaling node in diabetic Islets. Cell Metab. 2019, 29, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Pitasi, C.L.; Liu, J.; Gausserès, B.; Pommier, G.; Delangre, E.; Armanet, M.; Cattan, P.; Mégarbane, B.; Hanak, A.S.; Maouche, K.; et al. Implication of glycogen synthase kinase 3 in diabetes-associated islet inflammation. J. Endocrinol. 2020, 244, 133–148. [Google Scholar] [CrossRef]
- Ivovic, A.; Oprescu, A.I.; Koulajian, K.; Mori, Y.; Eversley, J.A.; Zhang, L.; Nino-Fong, R.; Lewis, G.F.; Donath, M.Y.; Karin, M.; et al. IKKβ inhibition prevents fat-induced beta cell dysfunction in vitro and in vivo in rodents. Diabetologia 2017, 60, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Spohrer, S.; Gross, R.; Nalbach, L.; Schwind, L.; Stumpf, H.; Menger, M.D.; Ampofo, E.; Montenarh, M.; Götz, C. Functional interplay between the transcription factors USF1 and PDX-1 and protein kinase CK2 in pancreatic β-cells. Sci. Rep. 2017, 7, 16367. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.; Al-Quobaili, F.; Müller, I.; Götz, C.; Thiel, G.; Montenarh, M. CK2 phosphorylation of Pdx-1 regulates its transcription factor activity. Cell Mol. Life Sci. 2010, 67, 2481–2489. [Google Scholar] [CrossRef]
- Servas, C.; Kiehlmeier, S.; Hach, J.; Gross, R.; Gotz, C.; Montenarh, M. The mammalian STE20-like kinase 1 (MST1) is a substrate for the apoptosis inhibiting protein kinase CK2. Cell Signal. 2017, 36, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Doliba, N.M.; Liu, Q.; Li, C.; Chen, P.; Liu, C.; Naji, A.; Matschinsky, F.M. Inhibition of cholinergic potentiation of insulin secretion from pancreatic islets by chronic elevation of glucose and fatty acids: Protection by casein kinase 2 inhibitor. Mol. Metab. 2017, 6, 1240–1253. [Google Scholar] [CrossRef]
- Jahan, I.; Corbin, K.L.; Bogart, A.M.; Whitticar, N.B.; Waters, C.D.; Schildmeyer, C.; Vann, N.W.; West, H.L.; Law, N.C.; Wiseman, J.S.; et al. Reducing glucokinase activity restores endogenous pulsatility and enhances insulin secretion in islets from db/db mice. Endocrinology 2018, 159, 3747–3760. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E.H.; Higgins, J.M.G. Pharmacological approaches to understanding protein kinase signaling networks. Front. Pharmacol. 2023, 14, 1310135. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.J.; Linley, A.; Hammond, D.E.; Hood, F.E.; Coulson, J.M.; MacEwan, D.J.; Ross, S.J.; Slupsky, J.R.; Smith, P.D.; Eyers, P.A.; et al. New perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res. 2018, 78, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Buljan, M.; Ciuffa, R.; van Drogen, A.; Vichalkovski, A.; Mehnert, M.; Rosenberger, G.; Lee, S.; Varjosalo, M.; Pernas, L.E.; Spegg, V.; et al. Kinase interaction network expands functional and disease roles of human kinases. Mol. Cell 2020, 79, 504–520. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Tumor immunotherapy directed at PD-1. N. Engl. J. Med. 2012, 366, 2517–2519. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Fallahi, P.; Elia, G.; Ragusa, F.; Ruffilli, I.; Patrizio, A.; Galdiero, M.R.; Baldini, E.; Ulisse, S.; Marone, G.; et al. Autoimmune endocrine dysfunctions associated with cancer immunotherapies. Int. J. Mol. Sci. 2019, 20, 2560. [Google Scholar] [CrossRef] [PubMed]
- Delangre, E.; Liu, J.; Tolu, S.; Maouche, K.; Armanet, M.; Cattan, P.; Pommier, G.; Bailbé, D.; Movassat, J. Underlying mechanisms of glucocorticoid-induced β-cell death and dysfunction: A new role for glycogen synthase kinase 3. Cell Death Dis. 2021, 12, 1136. [Google Scholar] [CrossRef]
- Rauch, J.; Volinsky, N.; Romano, D.; Kolch, W. The secret life of kinases: Functions beyond catalysis. Cell Commun. Signal. 2011, 9, 23. [Google Scholar] [CrossRef]
- Xu, D.; Matsumoto, M.L.; McKenzie, B.S.; Zarrin, A.A. TPL2 kinase action and control of inflammation. Pharmacol. Res. 2018, 129, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J.; Zhou, D.; Fitamant, J.; Bardeesy, N.; Mou, F.; Barrufet, L.R. Protein kinases of the Hippo pathway: Regulation and substrates. Semin. Cell. Dev. Biol. 2012, 23, 770–784. [Google Scholar] [CrossRef]
- Bi, W.; Xiao, L.; Jia, Y.; Wu, J.; Xie, Q.; Ren, J.; Ji, G.; Yuan, Z. c-Jun N-terminal kinase enhances MST1-mediated pro-apoptotic signaling through phosphorylation at serine 82. J. Biol. Chem. 2010, 285, 6259–6264. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Paroni, F.; Azizi, Z.; Kaur, S.; Khobragade, V.; Yuan, T.; Frogne, T.; Tao, W.; Oberholzer, J.; Pattou, F.; et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat. Med. 2014, 20, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Maedler, K. MST1: A promising therapeutic target to restore functional beta cell mass in diabetes. Diabetologia 2016, 59, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya-Tsuji, J.; Kishimoto, K.; Hiyama, A.; Inoue, J.; Cao, Z.; Matsumoto, K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 1999, 398, 252–256. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Chi, H.; Xie, M.; Schneider, M.D.; Flavell, R.A. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 2006, 7, 851–858. [Google Scholar] [CrossRef]
- Gantke, T.; Sriskantharajah, S.; Sadowski, M.; Ley, S.C. IkB kinase regulation of the TPL-2/ERK pathway. Immunol. Rev. 2012, 246, 168–182. [Google Scholar] [CrossRef]
- Gavrin, L.K.; Green, N.; Hu, Y.; Janz, K.; Kaila, N.; Li, H.Q.; Tam, S.Y.; Thomason, J.R.; Gopalsamy, A.; Ciszewski, G.; et al. Inhibition of Tpl2 kinase and TNF-alpha production with 1,7-naphthyridine-3-carbonitriles: Synthesis and structure-activity relationships. Bioorg. Med. Chem. Lett. 2005, 15, 5288–5292. [Google Scholar] [CrossRef]
- Hu, Y.; Green, N.; Gavrin, L.K.; Janz, K.; Kaila, N.; Li, H.Q.; Thomason, J.R.; Cuozzo, J.W.; Hall, J.P.; Hsu, S.; et al. Inhibition of Tpl2 kinase and TNFalpha production with quinoline-3-carbonitriles for the treatment of rheumatoid arthritis. Bioorg. Med. Chem. Lett. 2006, 16, 6067–6072. [Google Scholar] [CrossRef]
- Buchdunger, E.; Zimmermann, J.; Mett, H.; Meyer, T.; Müller, M.; Druker, B.J.; Lydon, N.B. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996, 56, 100–104. [Google Scholar]
- Manley, P.W.; Cowan-Jacob, S.W.; Buchdunger, E.; Fabbro, D.; Fendrich, G.; Furet, P.; Meyer, T.; Zimmermann, J. Imatinib: A selective tyrosine kinase inhibitor. Eur. J. Cancer 2002, 38 (Suppl. 5), S19–S27. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Coomans de Brachène, A.; Alvelos, M.I.; Szymczak, F.; Zimath, P.L.; Castela, A.; Marmontel de Souza, B.; Roca Rivada, A.; Marín-Cañas, S.; Yi, X.; Op de Beeck, A.; et al. Interferons are key cytokines acting on pancreatic islets in type 1 diabetes. Diabetologia 2024, 67, 908–927. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Farquharson, M.A.; Meager, A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet 1987, 2, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.C.; Guo, H.; Coulson, R.M.; Smyth, D.J.; Pekalski, M.L.; Burren, O.S.; Cutler, A.J.; Doecke, J.D.; Flint, S.; McKinney, E.F.; et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 2014, 63, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Dos Santos, R.S.; Op de Beeck, A.; Coomans de Brachène, A.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Haegeman, G.; Elewaut, D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharm. 2010, 10, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Gulliford, M.C.; Charlton, J.; Latinovic, R. Risk of diabetes associated with prescribed glucocorticoids in a large population. Diabetes Care 2006, 29, 2728–2729. [Google Scholar] [CrossRef]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-κB-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef]
- Szkudelski, T.; Szkudelska, K. The relevance of AMP-activated protein kinase in insulin secreting β cells: A potential target for improving β cell function? J. Physiol. Biochem. 2019, 75, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK activation: A therapeutic target for type 2 diabetes? Diabetes Metab. Syndr. Obes. 2014, 7, 241–253. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a therapeutic target for treating metabolic diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Weikel, K.A.; Ruderman, N.B.; Cacicedo, J.M. Unraveling the actions of AMP-activated protein kinase in metabolic diseases: Systemic to molecular insights. Metabolism 2016, 65, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, I.; Woltersdorf, W.W.; da Silva Xavier, G.; Rowe, R.L.; Cross, S.E.; Korbutt, G.S.; Rajotte, R.V.; Smith, R.; Rutter, G.A. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: Impact on glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1023–E1031. [Google Scholar] [CrossRef] [PubMed]
- da Silva Xavier, G.; Leclerc, I.; Varadi, A.; Tsuboi, T.; Moule, S.K.; Rutter, G.A. Role for AMP-activated protein kinase in glucose stimulated insulin secretion and preproinsulin gene expression. Biochem. J. 2003, 371, 761–774. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, S.Y.; Baek, W.K.; Lim, B.; Park, J.H.; Sung, H.Y.; Kim, Y.K.; Bae, K.C.; Bae, J.H.; Song, D.K. Regulation of glucose-dependent insulin secretion by insulin: Possible role of AMP-activated protein kinase. Life Sci. 2009, 85, 178–183. [Google Scholar] [CrossRef]
- Rourke, J.L.; Hu, Q.; Screaton, R.A. AMPK and friends: Central regulators of β cell biology. Trends Endocrinol. Metab. 2018, 29, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Liu, Y.; Zhou, F.; Zhang, Y.; Zhu, Q.; Zhang, L.; Zhang, Q.; Wang, S.; Zhu, K.; Wang, X.; et al. Berberine inhibits glucose oxidation and insulin secretion in rat islets. Endocr. J. 2018, 65, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; You, Y.H.; Ham, D.S.; Yang, H.K.; Yoon, K.H. The paradoxical effects of AMPK on insulin gene expression and glucose-induced insulin secretion. J. Cell. Biochem. 2016, 117, 239–246. [Google Scholar] [CrossRef]
- Wang, C.Z.; Wang, Y.; Di, A.; Magnuson, M.A.; Ye, H.; Roe, M.W.; Nelson, D.J.; Bell, G.I.; Philipson, L.H. 5-amino-imidazole carboxamide riboside acutely potentiates glucose-stimulated insulin secretion from mouse pancreatic islets by KATP channel-dependent and -independent pathways. Biochem. Biophys. Res. Commun. 2005, 330, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Sarno, S.; Cesaro, L.; Nakamura, H.; Pinna, L.A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim. Biophys. Acta 2009, 1793, 847–859. [Google Scholar] [CrossRef]
- Nunez de Villavicencio-Diaz, T.; Rabalski, A.J.; Litchfield, D.W. Protein kinase CK2: Intricate Relationships within regulatory cellular networks. Pharmaceuticals 2017, 10, 27. [Google Scholar] [CrossRef]
- Ampofo, E.; Nalbach, L.; Götz, C. Protein kinase CK2—A putative target for the therapy of diabetes mellitus? Int. J. Mol. Sci. 2019, 20, 4398. [Google Scholar] [CrossRef]
- Boldyreff, B.; Meggio, F.; Pinna, L.A.; Issinger, O.G. Protein kinase CK2 structure-function relationship: Effects of the beta subunit on reconstitution and activity. Cell. Mol. Biol. Res. 1994, 40, 391–399. [Google Scholar]
- Wirkner, U.; Voss, H.; Lichter, P.; Ansorge, W.; Pyerin, W. The human gene (CSNK2A1) coding for the casein kinase II subunit alpha is located on chromosome 20 and contains tandemly arranged Alu repeats. Genomics 1994, 19, 257–265. [Google Scholar] [CrossRef]
- Ackermann, K.; Neidhart, T.; Gerber, J.; Waxmann, A.; Pyerin, W. The catalytic subunit alpha’ gene of human protein kinase CK2 (CSNK2A2): Genomic organization, promoter identification and determination of Ets1 as a key regulator. Mol. Cell. Biochem. 2005, 274, 91–101. [Google Scholar] [CrossRef]
- Raaf, J.; Brunstein, E.; Issinger, O.G.; Niefind, K. The interaction of CK2alpha and CK2beta, the subunits of protein kinase CK2, requires CK2beta in a preformed conformation and is enthalpically driven. Protein Sci. 2008, 17, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Boldyreff, B.; Marin, O.; Pinna, L.A.; Issinger, O.G. CK2: Role of the beta subunit on the stability and specificity of the recombinant reconstituted holoenzyme. Eur. J. Biochem. 1992, 204, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Boldyreff, B.; Meggio, F.; Pinna, L.A.; Issinger, O.G. Casein kinase-2 structure-function relationship: Creation of a set of mutants of the beta subunit that variably surrogate the wildtype beta subunit function. Biochem. Biophys. Res. Commun. 1992, 188, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.A.; Contreras, C.; Bolanos-Garcia, V.; Allende, J.E. Protein kinase CK2 as an ectokinase: The role of the regulatory CK2beta subunit. Proc. Natl. Acad. Sci. USA 2008, 105, 5693–5698. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.; Götz, C.; Montenarh, M. The role of protein kinase CK2 in the regulation of the insulin production of pancreatic islets. Biochem. Biophys. Res. Commun. 2010, 401, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Matschinsky, F.M.; Magnuson, M.A.; Zelent, D.; Jetton, T.L.; Doliba, N.; Han, Y.; Taub, R.; Grimsby, J. The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes 2006, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Matschinsky, F.M.; Wilson, D.F. The central role of glucokinase in glucose homeostasis: A perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Front. Physiol. 2019, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Grupe, A.; Hultgren, B.; Ryan, A.; Ma, Y.H.; Bauer, M.; Stewart, T.A. Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell 1995, 83, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Terauchi, Y.; Sakura, H.; Yasuda, K.; Iwamoto, K.; Takahashi, N.; Ito, K.; Kasai, H.; Suzuki, H.; Ueda, O.; Kamada, N.; et al. Pancreatic beta-cell-specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J. Biol. Chem. 1995, 270, 30253–30256. [Google Scholar] [CrossRef]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef]
- Hussain, K. Mutations in pancreatic β-cell glucokinase as a cause of hyperinsulinaemic hypoglycaemia and neonatal diabetes mellitus. Rev. Endocr. Metab. Disord. 2010, 11, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Chakera, A.J.; Steele, A.M.; Gloyn, A.L.; Shepherd, M.H.; Shields, B.; Ellard, S.; Hattersley, A.T. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 2015, 38, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Terauchi, Y. Present status of clinical deployment of glucokinase activators. J. Diabetes Investig. 2015, 6, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Omori, K.; Terauchi, Y. Glucokinase activation or inactivation: Which will lead to the treatment of type 2 diabetes? Diabetes Obes. Metab. 2021, 23, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Thilagavathi, R.; Hosseini-Zare, M.S.; Malini, M.; Selvam, C. A comprehensive review on glucokinase activators: Promising agents for the treatment of type 2 diabetes. Chem. Biol. Drug Des. 2022, 99, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Toulis, K.A.; Nirantharakumar, K.; Pourzitaki, C.; Barnett, A.H.; Tahrani, A.A. Glucokinase activators for type 2 diabetes: Challenges and future developments. Drugs 2020, 80, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Matschinsky, F.M. GKAs for diabetes therapy: Why no clinically useful drug after two decades of trying? Trends Pharmacol. Sci. 2013, 34, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Wilding, J.P.; Leonsson-Zachrisson, M.; Wessman, C.; Johnsson, E. Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes. Metab. 2013, 15, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Whitticar, N.B.; Nunemaker, C.S. Reducing glucokinase activity to enhance insulin secretion: A counterintuitive theory to preserve cellular function and glucose homeostasis. Front. Endocrinol. 2020, 11, 378. [Google Scholar] [CrossRef]
- Remedi, M.S.; Nichols, C.G. Glucokinase inhibition: A novel treatment for diabetes? Diabetes 2023, 72, 170–174. [Google Scholar] [CrossRef]
- Omori, K.; Nakamura, A.; Miyoshi, H.; Yamauchi, Y.; Kawata, S.; Takahashi, K.; Kitao, N.; Nomoto, H.; Kameda, H.; Cho, K.Y.; et al. Glucokinase inactivation paradoxically ameliorates glucose intolerance by increasing β-cell mass in db/db mice. Diabetes 2021, 70, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Fortunato, M.; Shyr, Z.A.; Clark, A.L.; Fuess, M.; Nichols, C.G.; Remedi, M.S. Genetic reduction of glucose metabolism preserves functional β-cell mass in KATP-induced neonatal diabetes. Diabetes 2022, 71, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Kefalas, G.; Larose, L. PERK leads a hub dictating pancreatic beta cell homoeostasis. Biol. Cell 2018, 110, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4 amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [PubMed]
- Pucelik, B.; Barzowska, A.; Dąbrowski, J.M.; Czarna, A. Diabetic kinome inhibitors—A new opportunity for β-cells restoration. Int. J. Mol. Sci. 2021, 22, 9083. [Google Scholar] [CrossRef] [PubMed]
- Di Vona, C.; Bezdan, D.; Islam, A.B.; Salichs, E.; López-Bigas, N.; Ossowski, S.; de la Luna, S. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD kinase. Mol. Cell 2015, 57, 506–520. [Google Scholar] [CrossRef]
- Aranda, S.; Alvarez, M.; Turró, S.; Laguna, A.; de la Luna, S. Sprouty2-mediated inhibition of fibroblast growth factor signaling is modulated by the protein kinase DYRK1A. Mol. Cell. Biol. 2008, 28, 5899–5911. [Google Scholar] [CrossRef]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; DeCaprio, J.A. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011, 25, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Ahn, Y.S.; Chung, K.C. Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J. Biol. Chem. 2001, 276, 39819–39824. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Allan, L.A.; Clarke, P.R. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J. 2008, 275, 6268–6280. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Clarke, P.R. p38α-and DYRK1A-dependent phosphorylation of caspase-9 at an inhibitory site in response to hyperosmotic stress. Cell. Signal. 2009, 21, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martinez, J.; Vela, E.M.; Tora-Ponsioen, M.; Ocaña, O.H.; Nieto, M.A.; Galceran, J. Attenuation of Notch signalling by the Down-syndrome-associated kinase DYRK1A. J. Cell Sci. 2009, 122, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Skurat, A.V.; Dietrich, A.D. Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J. Biol. Chem. 2004, 279, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Rachdi, L.; Kariyawasam, D.; Aïello, V.; Herault, Y.; Janel, N.; Delabar, J.M.; Polak, M.; Scharfmann, R. Dyrk1A induces pancreatic β cell mass expansion and improves glucose tolerance. Cell Cycle 2014, 13, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Alvarez-Perez, J.-C.; Felsenfeld, D.P.; Liu, H.; Sivendran, S.; Bender, A.; Kumar, A.; Sanchez, R.; Scott, D.K.; Garcia-Ocaña, A. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat. Med. 2015, 21, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dančík, V.V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A stimulates human β-cell proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef]
- Ackeifi, C.; Swartz, E.; Kumar, K.; Liu, H.; Chalada, S.; Karakose, E.; Scott, D.K.; Garcia-Ocaña, A.; Sanchez, R.; DeVita, R.J.; et al. Pharmacologic and genetic approaches define human pancreatic β cell mitogenic targets of DYRK1A inhibitors. JCI Insight 2020, 5, e132594. [Google Scholar] [CrossRef]
- Kumar, K.; Wang, P.; Sanchez, R.; Swartz, E.A.; Stewart, A.F.; DeVita, R.J. Development of kinase-selective, harmine-based DYRK1A inhibitors that induce pancreatic human β-cell proliferation. J. Med. Chem. 2018, 61, 7687–7699. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Suebsuwong, C.; Wang, P.; Garcia-Ocana, A.; Stewart, A.F.; DeVita, R.J. DYRK1A inhibitors as potential therapeutics for β-cell regeneration for diabetes. J. Med. Chem. 2021, 64, 2901–2922. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Kinases | Inhibitors | Experimental Models and Development Stage | References |
|---|---|---|---|
| Inflammation | |||
| MST1 | Neratinib | In vitro, in vivo animal models, human islets | [29] |
| TAK1 | 5Z-7-oxozeaenol | In vitro, in vivo animal models | [30] |
| TPL2 | 1,7-naphthyridine-3-carbonitriles | In vitro, human islets | [31] |
| c-Abl | Imatinib/Gleevec®/Glivec | In vitro, in vivo animal models, clinical trial Phase II | [32,33] |
| TYK2 | Structure-based drug design | In vitro, human islets | [34] |
| JAK1 | ABT 317, LN3103801 | In vitro, in vivo animal model | [35,36,55,56] |
| Glucotoxicity and glulipotoxicity | |||
| GSK3β | Valproate, TDZD-8, KICG1338, Lithium | In vitro, in vivo animal models | [42,43,44,45] |
| IKKβ | BMS-345541 | In vitro, in vivo animal models | [46] |
| MST1 | Neratinib | In vitro, in vivo animal models, human islets | [29] |
| CK2 kinase | CX4945 | In vitro, human islets | [47,48,49,50] |
| Glucokinase | d-mannoheptulose | In vitro | [51] |
| Glucocorticoids | |||
| GSK3β | LiCl, SB216763 | In vitro | [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalle, S. Targeting Protein Kinases to Protect Beta-Cell Function and Survival in Diabetes. Int. J. Mol. Sci. 2024, 25, 6425. https://doi.org/10.3390/ijms25126425
Dalle S. Targeting Protein Kinases to Protect Beta-Cell Function and Survival in Diabetes. International Journal of Molecular Sciences. 2024; 25(12):6425. https://doi.org/10.3390/ijms25126425
Chicago/Turabian StyleDalle, Stéphane. 2024. "Targeting Protein Kinases to Protect Beta-Cell Function and Survival in Diabetes" International Journal of Molecular Sciences 25, no. 12: 6425. https://doi.org/10.3390/ijms25126425
APA StyleDalle, S. (2024). Targeting Protein Kinases to Protect Beta-Cell Function and Survival in Diabetes. International Journal of Molecular Sciences, 25(12), 6425. https://doi.org/10.3390/ijms25126425