Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease with no effective treatments, not least due to the lack of authentic animal models. Typically, rodent models recapitulate the effects but not causes of AD, such as cholinergic neuron loss: lesioning of cholinergic neurons mimics the cognitive decline reminiscent of AD but not its neuropathology. Alternative models rely on the overexpression of genes associated with familial AD, such as amyloid precursor protein, or have genetically amplified expression of mutant tau. Yet transgenic rodent models poorly replicate the neuropathogenesis and protein overexpression patterns of sporadic AD. Seeding rodents with amyloid or tau facilitates the formation of these pathologies but cannot account for their initial accumulation. Intracerebral infusion of proinflammatory agents offer an alternative model, but these fail to replicate the cause of AD. A novel model is therefore needed, perhaps similar to those used for Parkinson’s disease, namely adult wildtype rodents with neuron-specific (dopaminergic) lesions within the same vulnerable brainstem nuclei, ‘the isodendritic core’, which are the first to degenerate in AD. Site-selective targeting of these nuclei in adult rodents may recapitulate the initial neurodegenerative processes in AD to faithfully mimic its pathogenesis and progression, ultimately leading to presymptomatic biomarkers and preventative therapies.
1. Introduction
Animal models are vital for furthering our understanding of disease mechanisms and for prompting new therapeutic strategies. A field with a clear need for a valid and translatable animal model is neurodegeneration, particularly Alzheimer’s disease (AD), which constitutes 60–70% of all dementia cases worldwide and is characterised by progressive and catastrophic neuronal degeneration, leading to cognitive and memory deficits [1]. Despite its prevalence, there are no disease-modifying treatments for AD. Current medications include donepezil, galantamine and memantine, which increase cholinergic synaptic transmission to ameliorate the cognitive deficits in mild AD, but do not improve the disease’s prognosis [2]. Emerging therapies, notably anti-amyloid beta (Aβ) monoclonal antibodies such as Lecanemab, may slow disease progression, but have also not been shown to cure AD [3,4]. There are also no biomarkers for the presymptomatic detection of AD. This deficiency is due to a poor understanding of the neuropathological mechanisms of AD, in turn resulting from a lack of authentic animal models. To understand the pathogenesis of AD and how it progresses through the brain, we need an animal model that reproduces the causal mechanism driving the disease. This review will outline the various apparent causes of AD and evaluate the success or otherwise of their reproduction in rodents. We shall then consider new insights into the pathological mechanisms of neurodegeneration with the potential for alternative, and likely more valid, rodent models.
2. Cholinergic Hypothesis
An early theory of the origin of cognitive impairments in AD was the loss of cholinergic signalling [5,6,7]. Post mortem studies show that AD brains have 75% fewer cholinergic neurons in the basal forebrain compared to healthy controls [8,9,10]. These cells also have significantly lower concentrations of acetylcholine (ACh) and its synthesising enzyme, choline acetyltransferase, in the hippocampus and cortex [5,11,12,13,14]. Given ACh plays a key role in cognition and memory [15], loss of cholinergic signalling may account for the memory deficits seen in mild to moderate AD [16,17]. To compensate for the loss of cholinergic neurons, drugs that enhance cholinergic transmission were developed as treatments for mild cognitive impairment (see [18]); the most common have been acetylcholinesterase (AChE) inhibitors, starting with tacrine in 1989 and now superseded by drugs such as Aricept (donepezil) and galantamine [19], which prolong the availability of ACh by impeding its metabolism.
Several approaches have been used to model cholinergic neuron loss in AD, typically in rats. The muscarinic receptor antagonist scopolamine, for example, induces memory deficits when injected into the rat basal forebrain [20,21]. Specific cholinergic neuron degeneration can also be produced by adding carrier molecules to non-specific toxins, such as 192 IgG-saporin, which result in degeneration of cholinergic cell bodies and terminals via apoptotic cell death [22,23,24]. Intraventricular infusion of 192 IgG-saporin into rats causes working memory impairments on the Morris water maze, a gold-standard test for spatial memory in rodents (behavioural deficits seen in rats with cholinergic lesions, see [23]). Alternative approaches to the selective destruction of cholinergic neurons include mechanical lesions to cholinergic nuclei in the brainstem, brain trauma and hyperthermia after post-ischemic brain injury [25].
While cholinergic models successfully identified AChE inhibitors as cognitive enhancers for the early stages of AD, their ability to model the disease in its entirety is questionable. For instance, the effects of neurotoxic lesions on behaviour are highly variable and not always detected [26,27]. Extensive cholinergic neuron loss, potentially as great as 75%, is needed to detect behavioural effects in rodents [28]. In contrast, post mortem imaging shows that even patients with severe AD (Braak stages IV-VI) only lost approximately one third of the volume of the nucleus basalis of Meynert (NbM), a midbrain cholinergic nucleus that is particularly susceptible to degeneration in AD [29]. The critical threshold for neuronal loss to become apparent as cognitive deficits [30] may therefore be much lower in humans than in these rodent models, meaning they do not replicate the initial stages of neurodegeneration. Furthermore, most experimental models use neurotoxins infused via the ventricles, meaning cell loss is brain-wide and not targeted to the cholinergic nuclei [23]. These models also do not mimic the classic molecular hallmarks of AD: amyloid plaques and neurofibrillary tau tangles (NFTs). If anything, these models demonstrate the importance of cholinergic transmission in cognition, but poorly recapitulate the disease phenotype in humans. Finally, not all cholinergic cells degenerate in AD, and not all cells that degenerate are cholinergic—for example, cholinergic interneurons in the striatum are spared even in patients with AD [31]. This double dissociation between AD and cholinergic transmission suggests that the latter is not the primary mechanism mediating pathogenesis of AD.
3. Amyloid Hypothesis
The ‘amyloid hypothesis’, or the ‘amyloid cascade hypothesis’, was first proposed in 1991 following the discovery of a mutation in amyloid precursor protein (APP) in the post mortem brains of patients with AD [32,33]. This theory proposes that the accumulation of a toxic form of Aβ, through abnormal cleavage of the precursor molecule APP [34] and/or reduced clearance [35], leads to the downstream aggregation of tau and cell death, thus precipitating neurodegeneration in AD. This hypothesis was supported by genetic studies that identified several causative familial AD mutations in the amyloid production pathway, including to APP, presenilin 1 (PS1) and presenilin 2 (PS2) [36,37,38]. Common human polymorphisms in the apolipoprotein E (APOE) gene, namely APOE ε4, also elevate Aβ levels and increase the risk of sporadic AD [39,40]. Increased plasma concentrations of Aβ correlate with increased risk of AD, with levels elevated years before the onset of symptoms [41]. Indeed, the presence of Aβ plaques is a criterion for a neuropathologic diagnosis of AD [42].
3.1. Amyloid Transgenic Rodents
The most common approach to reproduce amyloid dysregulation in AD is transgenic mice. Mice do not spontaneously develop amyloid pathology [43,44]: hence, transgenic mice express human genes associated with familial AD mutations, namely in APP and PS1 [45], under the assumption that the same pathways are involved in sporadic AD. The first successful AD transgenic mouse was developed by Games et al., (1995): researchers genetically modified mice to overexpress platelet-derived APP (PDAPP) with a human mutation. These mice exhibit increased Aβ and plaque formation, as well as cognitive and memory impairment with age [46,47]. Other APP-targeting mice models express two mutations of the same type (e.g., Tg2576; Table 1) or two different mutations (e.g., J20, PDGF-APPSw, Ind; Table 1). Bi-genic mice, such as the PSAPP strain, express mutations to both APP and PS1 (APP/PS1), whilst the most extensive amyloid model to date is the 5xFAD mouse, which contains five familial AD mutations (Table 1; [48]). Other strains combine mutations to other AD-associated pathways, for example overexpressing human APOE ε4 [18,39,49,50,51] or knocking out β-secretase 1 [52,53,54].
Most studies of amyloid-overexpressing mice show at least some degree of Aβ accumulation and deposition, but the various mutations produce different phenotypes. In general, the more mutations the strain contains, or the more genes targeted, the more aggressive and earlier are the AD-like neuropathology and behavioural impairments. Interestingly, pathology also appears to be more pronounced in female than in male mice [55]. Table 1 shows examples of the different amyloid transgenics and their phenotypes.

Table 1.
Summary of amyloid transgenic rodents. Aβ, amyloid beta; APP, amyloid precursor protein; LC, locus coeruleus; MWM, Morris Water Maze; NOR, novel object recognition; PSEN1, presenilin 1.
Table 1.
Summary of amyloid transgenic rodents. Aβ, amyloid beta; APP, amyloid precursor protein; LC, locus coeruleus; MWM, Morris Water Maze; NOR, novel object recognition; PSEN1, presenilin 1.
| Model | Modification | Pathology | Behaviour | References |
|---|---|---|---|---|
| Mice | ||||
| Single APP mutations e.g., PDAPP, APP(V717L), APP23 and APP E693Δ. | Overexpression of human mutations to APP (e.g., Swedish K670N/M671L, London V717L and Indiana V717F). |
|
| [46,56,57,58,59] |
| Double APP mutations (same mutation), e.g., Tg2576 (double Swedish). | Overexpression of two of the same mutations, e.g., Tg2567 have double Swedish mutation. |
|
| [60,61,62] |
| Double APP mutations (different mutations), e.g., J20, A7 APP, PDGF-APPSw, Ind and TgCRND8. | Overexpression of two different human APP mutations (e.g., J20 have Swedish and Indiana). |
|
| [62,63,64,65] |
| Single PS1 mutations e.g., PS1(A246E), PS1(M146L), PS1(M146V). | Overexpression of mutations to the human PSEN1 gene. |
|
| [66,67,68,69,70] |
| APP/PS1 bi-genic strains, e.g., PSAPP, PS/APP, APP23 x PS1-R278I, PS/APP (Tg2576 x PS1 M146L) and APP(V717I) x PS1 (A246E). | Crossed APP-mutant and PS1 mutant mice. |
|
| [41,69,71,72,73,74,75,76,77,78,79] |
| Triple mutant strains e.g., 3xTg-AD. | Mice overexpressing APP, PSEN1 and tau mutations. |
|
| [80,81,82] |
| 5xFAD. | 5xFAD on B6SJL or C57BL6 backgrounds (APP Swedish, Florida I716V and London; PSEN1 M146L and L286V). |
|
| [48,83] |
| APP Knock-in e.g., APPNL-F and APPNL-G-F. | Crispr/Cas9 used to humanise Aβ sequence in endogenous APP gene and add human mutations. |
|
| [84,85,86,87,88] |
| Rats | ||||
| Double APP mutations, e.g., APP21, McGill-R-Thy1-APP. | Viral vectors introduced human mutations to zygotes containing two different human APP mutations, e.g., McGill-R-Thy1-APP have Swedish and Indiana. |
|
| [89,90,91] |
| APP/PS1 bi-genic, e.g., APP + PS1, Tg478, Tg1116 Tg344-AD (APP Swedish, PS1 ΔE9). | Viral vectors introduced human mutations to zygotes containing mutations to APP and PS1. |
|
| [92,93,94,95,96,97,98] |
| APP knock-in strains. | Crispr/Cas9 used to humanise Aβ sequence in endogenous APP gene; others include human mutations to APP gene. |
|
| [88,99,100,101] |
Despite their extensive use, transgenic models have fundamental limitations for studying AD. Firstly, they are based on familial mutations, which only 5–10% of patients with familial AD express [38]. For patients with sporadic AD, there is little indication that disruption to these genes plays any role in the pathogenesis of the disease [102]. Moreover, unlike transgenic mice that can express up to five different mutations [42,48], there are strikingly few examples of patients having multiple AD-associated mutations, even in familial AD [103]. A single mutant transgenic may therefore model specific familial AD mutations, but this does not reflect the causal mechanisms of sporadic disease.
Furthermore, the overexpression of mutant human genes in addition to the endogenous mouse genes leads to non-physiological levels of protein expression. Even the PDAPP mouse with just one mutation to APP has ten-fold more APP than wildtype controls [46]. This excessive protein expression is not representative of patients with familial AD mutations, let alone sporadic AD. While knock-in models humanise existing rodent genes to induce mutations and therefore have more physiological levels of Aβ (e.g., [84,104]), they are still often bred onto APP-transgenic lines to accelerate the appearance of AD-like phenotypes, and thus still rely on transgene overexpression (e.g., [84]).
While familial AD patients express mutations throughout life, AD-associated gene expression varies greatly across age and disease stage [105,106]. Particularly relevant to sporadic AD is that there may be changes in gene expression or activity of proteins that does not become apparent until adulthood. Transgenic rodents, on the other hand, will exhibit brain-wide changes in protein expression throughout development and in the postnatal period. This potentially alters normal brain development and therefore the progression of AD-like pathology in adulthood. A better model of AD would therefore trigger changes in gene expression or alter protein expression in adult animals, to recapitulate the age-dependent nature of the disease.
3.2. Seeded Amyloid Models
More recently, “seeded” models have been used to localise AD-like pathology in specific brain structures and to accelerate the onset and severity of amyloid deposition in adult animals [107]. This procedure involves injecting Aβ, whether that be synthetic oligomers or brain homogenates from transgenic mice or patients with AD, into the brains of transgenic mice. Typically, injections are targeted to the hippocampus and the overlaying cortex [107]; for example, bilateral hippocampal injections of Aβ into the of 5-month-old APP mice produced amyloid plaques in the hippocampus one month earlier than in sham-injected APP23 mice [108]. Seeding Aβ into the olfactory bulb in 2-month-old 5xFAD mice (before amyloid deposits are detectable) increased Aβ expression 12 weeks later compared to non-injected 5xFAD mice. This resulted in reduced neurogenesis and activity of the targeted cells, as well as olfactory function [109]. Seeding Aβ into the hippocampus of APP-mutant mice reduced hippocampal neurogenesis and proliferation, and increased cell death compared to controls [110].
Nonetheless, the effect of seeding on amyloid pathology is highly variable and depends on both the source of Aβ and the host. For instance, APP/PS1 mice injected with APP23 homogenates develop hippocampal plaques that are more diffuse than APP/PS1 injected with APP/PS1 homogenates [108,111]. These discrepancies make it difficult to compare results between strains and constructs. Moreover, it has not yet been possible to induce AD-like pathology by injecting mutant Aβ into wildtype mice, nor can non-transgenic homogenates produce amyloid pathology in young transgenic hosts [107,109]. This is likely because endogenous Aβ expression is too low to maintain seeded Aβ in wildtype animals, and aggregated Aβ is needed to further misfold endogenous Aβ in transgenics. Overall, these failures demonstrate that simply increasing Aβ levels in the rodent brain is not sufficient to trigger downstream AD pathology, thereby questioning the validity of amyloid seeding in modelling the causative mechanisms of AD.
3.3. Limitations of Amyloid-Targeting AD Models
Rodent models overexpressing amyloid are predicated on the assumption that the accumulation of Aβ in the brain is the cause of AD. Yet, it is becoming increasingly apparent that this peptide is not likely to be the primary agent driving neurodegeneration. For example, post mortem brains of elderly patients without dementia can have extensive amyloid deposits, suggesting that Aβ alone is not sufficient to produce AD [112]. Moreover, amyloid deposits are absent in the early stages of AD, yet other AD-associated pathology is already present, such as NFTs (see following section). Models based on amyloid overlook vital neuropathological changes that occur before Aβ accumulation, meaning they do not simulate the true cause of the disease. The lack of tau pathology in amyloid transgenics (e.g., [48]) in these models is perhaps their most important limitation for authentic modelling of AD.
Amyloid-targeting models also fail to replicate the onset of AD-like pathology. In transgenic mice, abnormalities appear much earlier than even in early-onset AD: for example, 5xFAD mice display increased Aβ levels at 1.5 months old [48]. In contrast, Aβ levels start to rise as early as twenty years before the onset of cognitive symptoms in patients [113]. Not only does this disparity suggest that these models are not completely authentic, but the accelerated emergence of Aβ pathology also restricts the window for the potential detection of presymptomatic markers.
Despite these drawbacks, APP-transgenic mice have been central in the development of anti-Aβ therapies. For instance, immunisation of PDAPP mice with Aβ decreased amyloid pathology [114] and prevented cognitive deficits in other mouse strains [64,115,116]. Studies such as these have led to drugs such as Lecanemab and Donanemab (monoclonal antibodies against Aβ), which show some efficacy in reducing plaque burden in patients [3,4]. Nonetheless, Aβ-targeting drugs still routinely underperform in clinical trials in that they reduce amyloid burden but do not ameliorate cognitive deficits [117] and have not been shown to improve life expectancy [3,118]. The lack of translatability between encouraging results in rodents and failures in humans further suggests that amyloid-targeting animal models have poor predictive validity for AD. Other potential mechanisms should therefore be explored in models with the aim of producing more successful treatments in the future.
4. Tau Hypothesis
Tau is a microtubule-associated protein synthesised from the MAPT gene that stabilises microtubules [119]. In AD, tau becomes hyperphosphorylated and aggregates into NFTs [120], destabilising microtubules and thus leading to cell damage and eventual loss, although the exact mechanisms remain unclear [121]. While historically thought to be a consequence of Aβ accumulation [112], tau pathology can appear in the AD brain before, and independently of, Aβ deposits [122,123]. Moreover, the highly predictable spread of hyperphosphorylated tau correlates better with cognitive decline than amyloid pathology [124,125], suggesting that tau may be the initiating agent for neurodegeneration in AD [126].
4.1. Tau Transgenic Rodents
The induction of tauopathies in mice is difficult as endogenous murine tau does not aggregate into NFTs [44]. Thus, like amyloid-targeting transgenic models, human tau with one or more familial AD-associated mutations, predominantly P301L or P301S mutations, are instead overexpressed to induce high levels of hyperphosphorylated tau and consequently neurodegeneration [127,128,129,130,131,132]. Overexpression of mutant tau in h2N4P301L mice accordingly produced a four-fold increase in tau expression at 6–7 months old, which aggregated into NFTs. There was also evidence of tau aggregation and neuronal loss in the hippocampus and neocortex of hAPP-3RTau mice, an APP-mutant line with three additional mutations to tau [133]. This cellular phenotype was associated with motor and cognitive deficits, comparable to those seen in advanced AD [134]. Interestingly, hyperphosphorylated and aggregated tau has also been detected in mice expressing non-mutant human tau [135]. Human tau mutations also increase phosphorylated tau and produce NFTs when expressed in rats, although generally in the absence of significant neuronal loss (reviewed in [25]).
Nonetheless, the phenotypes of different tau transgenics vary depending on the human mutations they express and under which promoters. For instance, the P301L mutation under Thy-1 regulation produced a 0.7-fold increase in tau expression in 3-month-old mice, whereas under the CaMKII promoter, tau expression was 13-fold greater from 1 month old [130,134,136,137]. It is therefore difficult to generalise findings to other transgenics, let alone to the human condition.
The lack of amyloid plaques in tau-only mutants also challenges the authenticity of these models for AD. To overcome this issue, Oddo et al. developed the 3xTg-AD strain, which expresses human mutations to APP (Swedish), PS1 (M146V) and tau (MAPT P301L). This mouse exhibits brain-wide, age-dependent expression of amyloid plaques, tau hyperphosphorylation and progressive cognitive deficits [80,138,139]. Unlike other transgenic mice that exhibit hippocampal neurodegeneration from an early age [140,141], this strain shows limited neuronal loss in the hippocampus even up to 17 months old. This resembles the hippocampal degeneration seen in later stages of human AD (where hippocampal volume loss occurs after degeneration of other structures [123] and correlates with cognitive decline, i.e., is more severe in later stages of the disease [142,143]). Nonetheless, the concurrent presence of Aβ and tau throughout development and maturation in 3xTg-AD mice does not reflect the progressive and potentially sequential appearance of these markers in AD. The 3xTg-AD strain may therefore be a good model of late-stage AD pathology, but poorly recapitulates its early pathogenesis.
4.2. Seeded Tau Models
Tau can also be ‘seeded’ into the brain to induce neurodegenerative pathology. This process involves the stereotaxic injection of tau protein into the hippocampus and/or overlying cortex. For example, injecting synthetic preformed fibrils of tau into the hippocampus led to the spread of NFT-like inclusions to anatomically connected structures, including the thalamus and cortex [144,145]. Injections of human brain extracts from patients with high levels of tau also produced similar spread in P301L tau transgenic and 5xFAD mice [146,147], as well as in mice expressing wildtype human tau (without AD mutations; [148]). Although these seeding models, by definition, localise tau pathology to specific brain regions, the spread of aggregated tau to other structures differs from the clinical progression of AD [144,149]. Indeed, one study found that the transentorhinal cortex was spared from tau pathology after injection of synthetic preformed tau fibrils, yet this is the first cortical structure to show tau aggregation in human AD [123,149].
An alternative approach is to trigger the conditional and inducible overexpression of mutant tau genes in wildtype rodents via AAV-virus transfection. In one example, AAV-hTau-P301L injected into the cortex of wildtype mice produced NFTs in the hippocampus in 10 weeks [150]. This was associated with cognitive impairment, evident as a reduction in novel object recognition compared to controls [151]. This AAV-mediated approach allows for tau overexpression to be induced in adult animals, removing the disruption tau causes during development in constitutive transgenics.
However, the mutations used to induce tauopathies in rodents are predominantly comparable to those seen in frontotemporal dementia (FTD) and parkinsonism, not in AD [152,153]. Indeed, the tau tangles detected in these rodents more closely resemble Pick bodies of FTD than the NFTs of AD [132]. Thus, while tau-only transgenics and seeded models may be useful in understanding the mechanisms by which tau hyperphosphorylation and aggregation spread and cause neuronal loss, they are not reflective of the pathogenesis of AD. One reason for the lack of authenticity of tau models could be that tau hyperphosphorylation and aggregation is not the underlying driver of AD, but rather a consequence of other degenerative processes. We must first identify the key mechanism underlying neurodegeneration in AD before we can reproduce it in rodents.
5. Neuroinflammation Hypothesis
There is substantial evidence that amyloid depositions and NFTs trigger inflammatory mechanisms in the CNS [154], including the production of proinflammatory cytokines and the activation of microglia and astrocytes [92,155,156,157]. Yet, there is emerging evidence that inflammation itself may drive the early pathological processes in AD [158,159,160,161]. For example, microglial activation has been reported in patients with mild cognitive impairment in the absence of amyloid pathology [162]. Moreover, individuals with amyloid plaques that lacked evidence of microglial activation were asymptomatic, suggesting that neuroinflammatory responses are required for AD-related cognitive deficits [163]. The discovery of AD risk factors associated with innate immune functions further implicates neuroinflammation as a contributor to sporadic AD [164].
Neuroinflammatory mouse models typically use the intracerebral infusion of chemicals to trigger inflammatory responses and the downstream markers of AD [165,166]. For example, four months of intracerebroventricular infusions of okadaic acid, a toxic inhibitor of serine/threonine phosphatases [167], increased tau hyperphosphorylation, induced Aβ deposition and triggered apoptotic cell death in the rat cortex compared to untreated controls [168,169]. While this model did not recapitulate all the neurochemical markers of AD, for instance there were no NFTs [170], okadaic acid infusion induced significant hippocampal pathology and spatial memory deficits on the Morris water maze [168,169].
Inducing neuroinflammation throughout life can also produce AD-like pathology in aging. The polyriboinosinic–polyribocytidylic acid-induced mouse model involves injecting pregnant mice with this inflammatory agent, producing age-dependent increases in tau expression in the offspring. Critically, tau progresses in the spatiotemporal patterns seen in human AD and causes spatial memory impairments in aged animals compared to offspring from untreated mothers [18,171]. Other constitutive changes to neuroinflammation can be produced via the overexpression of cyclin-dependent kinase 5 and p25, which induce tau hyperphosphorylation [172,173], or by knocking out triggering receptor expressed on myeloid cells 2 (TREM2) from APP-mutant mice, which accelerates Aβ pathology and neuronal atrophy [174]. Neuroinflammation may therefore be upstream of amyloid and tau pathology and its models thus better recapitulate the temporal order over which pathology appears in patients. The independence from the overexpression of familial AD genes also suggests neuroinflammatory models may better mimic sporadic AD. Moreover, single nucleotide polymorphisms in neuroinflammatory pathways have also been linked to AD [175] and could therefore inspire future animal models.
Nonetheless, it is difficult to compare findings between constructs as the method to induce pathology varies so greatly. Introduction of inflammatory agents into the brain may not parallel the underlying causes of neuroinflammation in AD, which may arise from a wide variety of insults ranging from vascular abnormalities, mitochondrial dysfunction, to head trauma [164]. Hence, the most fundamental issue with these models is the assumption that inflammation is the first step in the neurodegenerative pathway, rather than a natural response to neuronal injury and damage. The endogenous primary trigger for inflammation in AD remains elusive such that we must resort to injecting pro-inflammatory chemicals into rodents. It is also not clear whether these compounds activate the same inflammatory processes as the human disease. As such, a better model would induce neuroinflammation via the same mechanism as in patients, although we must first understand these processes before such models can be developed. Furthermore, a genetic predisposition to altered neuroinflammation does not circumvent the need for an initial trigger, meaning transgenic models of neuroinflammation are again modelling a response, not a cause, of AD.
In summary, the current approaches to modelling AD in rodents (listed in Table 2) do not faithfully replicate all the features of the disease. They are not based on the underlying mechanisms driving AD, particularly in its initial stages. Consequently, these models do not recapitulate the full neuropathological phenotype and have yielded few effective treatments and no disease-modifying therapies. We therefore need a completely different approach to modelling AD.
Table 2.
Summary of approaches to induce AD-like pathology in rodents.
6. Lessons Learned from Modelling Parkinson’s Disease
A related disorder in which animal models have been used successfully is Parkinson’s disease (PD). PD is a progressive neurodegenerative disease characterised by movement dysfunctions such as bradykinesia, tremor and rigidity [176]. In the later stages of the disease, PD is also associated with dementia and cognitive dysfunction [176]. Only 5–10% of patients have a family history of PD, with just 5% having an autosomal mutation [177]. In these cases, mutations occur in the gene for α-synuclein (SNCA), or in leucine-rich repeat kinase 2 (LRRK2), vacuolar protein sorting ortholog 35 (VPS35) and in genes linked to mitochondrial function [178]. Interestingly, there is a frequently observed co-pathology with AD and sporadic PD [179].
The classical neuropathological hallmark of PD is the presence of cytoplasmic deposits in cell bodies, known as Lewy bodies (LBs), that contain misfolded α-synuclein [180,181]. The progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SN) and the degeneration of its projections to the striatum cause the characteristic motor dysfunction in PD, which correlates with disease severity and latency of disease onset [182,183,184]. When motor symptoms emerge, a significant number of dopaminergic neurons in the SN and terminals in the striatum have been lost [184,185,186,187,188], with up to 90% of dopaminergic neurons having degenerated by the later stages of PD [182,184,189].
Drug-induced parkinsonian movement dysfunction has classically been used to study PD in rodents. The most widely used model has been to lesion dopaminergic neurons with the neurotoxin 6-hydroxydopamine (6-OHDA). 6-OHDA is taken up selectively into catecholaminergic neurons, where it is oxidised to yield hydrogen peroxide and reactive oxygen species, resulting in cell death [190,191]. When injected unilaterally into the midbrain or striatum, 6-OHDA kills dopaminergic neurons and causes rats to rotate towards the contralateral side [192]. These animals also show deficits on the rotarod test, in that rodents with dopamine lesions have a reduced latency to fall off the apparatus compared to controls (e.g., [193]).
An alternative method of inducing dopaminergic neurodegeneration is by infusing the mitochondrial complex I inhibitor 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) into the rodent brain [11,194]. Dopaminergic cells are particularly sensitive to this toxin due to their high metabolic demand [195]. Immunohistochemical staining showed that chronic MPTP infusion in rats destroys dopaminergic neurons [196] and produces LB-like structures in striatal and nigral neurons [197,198]. This degeneration is associated with bradykinesia and akinesia (see [199]), resembling the movement deficits seen in PD. Neurochemical lesions such as those produced by 6-OHDA and MPTP produce more selective and thorough lesions than historical electrolytic or radiofrequency lesioning of the SN, and thus produce behavioural effects more similar to Parkinsonian symptoms [200].
Recently, transgenic mice have been used to model PD. These include mice overexpressing mutant α-synuclein, which exhibit fibrillar α-synuclein and brain-wide neurodegeneration [132]. Unlike pharmacological models, however, constitutive α-synuclein mice do not show evidence of dopaminergic neuron degeneration (reviewed in [201,202,203,204]), suggesting that they are poor models of PD-like pathology, at least regarding the mechanisms underlying the movement deficits. In contrast, conditional or inducible cell-type specific overexpression of α-synuclein, for example by injecting viral vectors containing wildtype or mutant SNCA into the nigrostriatal pathway, produces robust and extensive degeneration of dopaminergic neurons [205,206].
There are several lessons that can be learned from rodent models of PD for the study of AD. Firstly, targeting dopaminergic neurons, either by the infusion of selective neurotoxins or by the cell-type specific overexpression of mutated α-synuclein, is more effective than constitutively overexpressing genes. The ability to induce neurodegeneration in adulthood, compared to developmental overexpression in transgenic models, also better models the development of PD with age, which is perhaps unsurprising given that most cases of PD, as with AD, do not have a strong genetic basis.
Secondly, these PD models highlight how modelling specific aspects of a disease can be valuable for identifying therapies for specific symptoms, rather than trying to tackle the entire disease.
The 6-OHDA model has been used to demonstrate the efficacy of L-dopa (a dopaminergic precursor) and carbidopa (a peripheral decarboxylase inhibitor that increases the bioavailability of L-dopa) for the amelioration of movement deficits produced by dopaminergic cell loss (reviewed in [207]). This is also a good model for the demonstration of basic pharmacological principles, such as the supersensitivity to dopamine and dopamine receptor agonists that emerges after dopaminergic denervation, itself potentially evidence of the compensatory mechanisms early in PD [208]. Nonetheless, the construct used in these models does not reflect the underlying pathological processes in PD; dopaminergic degeneration, as measured by cell loss and α-synuclein immunoreactivity in post mortem PD brains, does not occur until the middle stages of PD (Braak stage 3; [209]). As such, this model has not yielded compounds that can reverse, or even stabilise, dopaminergic cell loss [132].
Clearly, we need models for neurodegenerative diseases that reproduce the pivotal, driving neuropathological mechanisms. Models should be inducible in adulthood and lead to the characteristic progression of neurodegeneration throughout the brain without relying on mutations associated with the familial form of the disease. They should also target the specific nuclei most susceptible to the disease.
7. A Novel Approach to AD Highlighting Primarily Vulnerable Nuclei: The Isodendritic Core
Neurodegeneration in AD begins several decades before cognitive symptoms emerge and appears to precede the accumulation of Aβ [210]. Psychological symptoms such as anxiety, depression, and sleep disturbances, so-called “pre-cognitive” symptoms, are common in the early stages of AD [210,211] and suggest that there may be disruption to subcortical structures related to these dysfunctions, before cognitive deficits emerge. Parallels have been drawn between AD and PD regarding their mechanisms, frequent co-pathology, and the overlap in brain regions implicated in the early pathogenesis. A persuasive hypothesis of AD pathogenesis is that a group of interconnecting subcortical nuclei are the first to degenerate in AD, which initially impacts emotionality and sleep and only in later stages causes cognitive decline [212]. Perhaps these selectively vulnerable nuclei should instead be the focus for animal models of AD.
7.1. The Isodendritic Core in AD
The isodendritic core (IC) is an evolutionarily conserved network of nuclei derived from the basal plate of the ectoderm in embryonic development. IC extends from the brainstem to the basal forebrain in the adult mammalian brain [213,214], and is so named because it constitutes a region of overlapping dendritic fields [214]. These nuclei are the principal sites of monoaminergic neurotransmitter synthesis, namely noradrenaline in the locus coeruleus (LC), serotonin in the raphe nuclei, dopamine in the SN and ventral tegmental area, and ACh in the basal forebrain (Figure 1A) [117]. Nuclei in the IC send extensive ascending projections to the cortex [215,216], where monoaminergic transmission modulates homeostatic and behavioural responses such as cognition, the sleep/wake cycle, and mood. Due to their broad modulatory effects, these IC cells have also been dubbed ‘global neurons’ [213].
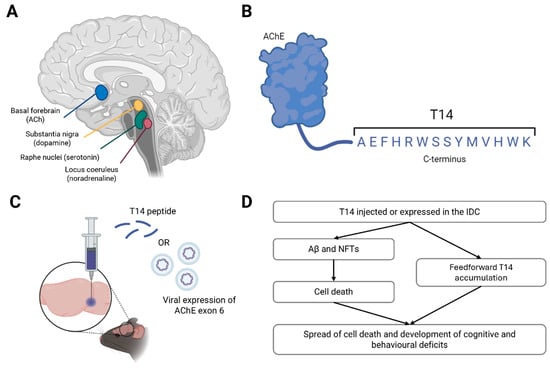
Figure 1.
A novel animal model of AD. Schematic of the isodendritic core (IC) in the human brain, highlighting the main cholinergic, serotonergic, dopaminergic and noradrenergic nuclei (A). The structure and sequence of T14 at the C-terminus of acetylcholinesterase (AChE) (B). Proposed mechanisms of delivery of T14 to the IC in laboratory rodents, either via direct infusion of the peptide or the viral overexpression of exon 6 of AChE (which encodes T14), into the target nuclei (C). Schematic of the hypothesised effects of T14 administration into the rodent brain, including amyloid beta (Aβ) and neurofibrillary tau tangles (NFTs), mimicking AD progression, based on data published in [217,218] (D). Figures made using BioRender.
The IC retains its sensitivity to growth and plasticity factors throughout life (not just during neurodevelopment) and is selectively vulnerable to the early neurodegenerative processes in AD [212,219]. NFTs are found across brainstem nuclei in AD [122,220], including in the LC, SN, dorsal raphe nucleus (DRN) and basal forebrain in post mortem samples of patients with early-stage AD and MCI [221,222,223,224,225,226,227]. Critically, IC pathology appears before that of other AD-associated brain regions; the LC shows evidence of NFTs in the absence of Aβ ten years before the onset of cognitive changes [123,228,229,230,231]. The same is true for the DRN, where lesions were observed in all Braak stage I (BB I) patients, and even in 20% of BB 0 individuals [223,232].
Cell loss also occurs in the IC and increases with disease severity [225,233,234]. As much as 88% of LC neurons were lost in the post mortem brains of individuals with AD compared to healthy controls [229,235,236]. Severe cell loss has also been reported in the SN [237] and basal forebrain [9,10]. Such extensive neurodegeneration has functional implications for monoaminergic neurotransmitters, with studies showing that cortical and limbic levels of dopamine, noradrenaline and serotonin were depleted in AD patients compared to controls [222,238,239].
Further supporting the notion that IC disruption is critical in the development of AD, the extent of IC neuron damage and loss correlates with the extent of AD pathology and symptoms. A greater number of NFTs in the LC was associated with lower cognitive (Mini-Mental State Exam, MMSE) scores, indicating more advanced degeneration [224]. DRN pathology and serotonergic denervation of the cortex also correlated with behavioural changes in patients, such as altered emotionality and psychosis [223,240,241], further linking IC damage to the precognitive symptoms of AD.
7.2. Evidence of IC Degeneration in Current AD Models
Despite the clear link between IC degeneration and the initial stages of AD, current animal models have not yet attempted to reproduce AD-related phenotypes by targeting these subcortical cell groups. Constitutive transgenics, for example, express human mutations throughout the brain. Similarly, neuroinflammatory models typically infuse compounds into the ventricles. Meanwhile, when seeded models introduce Aβ or tau into specific brain regions, it is generally into the hippocampus and cortex, rather than into the IC. Consequently, pathology in these models is initiated in a structure that does not exhibit neurodegeneration until later stages of AD [123] and so fails to replicate the characteristic spatiotemporal patterns of pathology that occur clinically. In APP transgenic mice, for example, there was no evidence of a reduction in the number of cholinergic neurons in the basal forebrain even at 24 months of age [242,243,244,245,246,247].
Established AD models can lead, almost as a side effect, to limited neurodegeneration to the IC. By chance, there is some evidence of age-dependent neurodegeneration of LC noradrenergic neurons in 3xTg-AD and APP/PS1 mice, and in TgF344-AD rats [92,248,249]. Reductions in cortical and hippocampal catecholamine concentrations have also been noted [250,251,252,253]. Nonetheless, these effects have not been detected in all strains [254,255], and evidence suggests that changes in terminal neurotransmitter levels may be driven by local degeneration rather than cell body loss [243,246,256,257,258]. Accordingly, these experimental interventions do not really model AD neuropathology in the IC.
Most studies neglect to investigate possible pathology in the brainstem [259,260], and this holds true even in approaches that claim to fully phenotype a strain: for example, a recent behavioural and neurochemical study of 5xFAD mice did not measure amyloid or tau pathology in the midbrain or IC cell loss [261]. Similarly, studies of seeded models focus on pathology in the treated brain regions and subsequently in cortical projections, rather than in the IC [108,146,147]. The lack of evidence in this field should not be confused with a lack of importance; instead, it serves to highlight the need for a new animal model that targets the IC.
Of particular importance is that the current animal models poorly replicate the precognitive symptoms of AD (for detailed review, see [262]) that can start 10 to 20 years before cognitive symptoms and are a central feature of the early disease stages. Taking the emotional disorder of anxiety as an example, some transgenic mice exhibit increased anxiety on tests such as the elevated plus maze and light dark box (e.g., 3xTg-AD [263], Tg2576 [264]), and increased anxiety has been detected in aged TgF344-AD rats [92,94]. Yet, there is a lot of variability between models; many studies report no change in anxiety-like behaviour [252,265,266], and others see a reduction [267,268,269]. Opposing changes are often seen in males and females of the same strain [270] and altered anxiety-like behaviour tends to emerge with age [271]. Some mice strains do see changes to sleep/wake behaviour—for example, altered sleep bout durations and disruption to circadian rhythms [272,273] that are evident in young mice [274]. Yet, it is unclear whether these changes precede cognitive deficits or are present concurrently. Albeit-limited evidence therefore suggests that transgenic mice do not exhibit the behavioural changes associated with IC degeneration that occur in AD. We therefore need a model that exhibits these early behavioural changes, not just deficits in memory and cognition, to identify prodromal biomarkers that may be useful for the development of preventative therapies.
7.3. A Novel Approach to Modelling the IC in AD
To replicate AD-like neurodegeneration, novel animal models should aim to induce dysfunction in the IC. Mechanical or electrolytic lesions, for example, may be able to destroy individual brainstem nuclei; however, these neurons mediate essential basic functions, such as thermoregulation, breathing and key survival behaviours, such that their destruction would result in the death of the animal. Chemical lesions could be used to target specific neurotransmitters, an approach that resembles the dopamine depletion studies in PD. Yet, the IC consists of heterogeneous transmitter systems of dopamine, noradrenaline, acetylcholine, and serotonin. Targeting just one of these neurotransmitter systems, as demonstrated with the lesions used to investigate the cholinergic hypothesis, will not recapitulate the entire disease or its progression. We would therefore need to identify a bioactive agent common to all nuclei, irrespective of transmitter.
Indicative of their shared evolutionary origin, neurons in the IC express the enzyme AChE, irrespective of their primary transmitter: hence, this familiar protein acts also in an unfamiliar capacity unrelated to is catalytic action [275,276]. The salient part of AChE underlying its bioactivity in relation to AD is a 14mer peptide cleaved from its C-terminus, ‘T14’, generated from exon 6 of the AChE gene (Figure 1B). T14 is proposed to cause AD by recapitulating developmental mechanisms in the ageing brain, where events such as large increases in intracellular calcium or neuronal outgrowth are excitotoxic and thus cause cell death [218,277]. T14 is elevated in the human midbrain as early as the presymptomatic Braak stage II [217,218,278], and in vitro studies have shown it to induce excitotoxicity and reduce cell viability [279]. Critically, T14 appears to operate upstream of Aβ and tau in that it triggers production of both these markers [280]. In fact, pharmacologically blocking T14 binding to its receptor, an allosteric site on the α7 nicotinic receptor [279], reduced Aβ and tau load in the brains of 5xFAD mice, suggesting T14 is vital in their accumulation [217].
Manipulation of IC function via the upregulation of the T14 system, for example by directly injecting the peptide or viral overexpression of T14 in AChE-expressing neurons in the rodent basal forebrain (analogous to the NbM in humans [281]) or LC, is therefore proposed as a novel model of AD (Figure 1C). This model would circumvent the previous difficulties posed by uncertainty of both anatomical and chemical targets in other models by directly targeting the cells proposed to underly AD, not those that simply degenerate with its progression, like the hippocampus. In this case, it would be inappropriate to simply target cholinergic transmission systemically (e.g., pharmacologically), as this would not differentiate between the vulnerable midbrain AChE-containing neurons (not all of which release ACh) and cholinergic interneurons and lower motor neurons that do not readily degenerate in AD [31]. Cholinergic pharmacology would also not affect other monoaminergic transmitters such as noradrenaline and serotonin, whereas T14 is posited to be equally efficacious across neuronal subtypes.
Consequently, we propose that an increase in T14 levels would trigger Aβ accumulation and tau hyperphosphorylation, ultimately leading to cell death and cognitive deficits in these animals (Figure 1D). Given T14 levels start to rise before Aβ, it may be possible for this model to recapitulate the early symptoms of AD, including anxiety and sleep disturbances. This model could also be used for drug discovery and lead to the identification of related novel, potentially druggable targets, which would not be possible without such an authentic model. Of course, a T14 model would still have its limitations—for example, it would not address the range of initial insults that could trigger the accumulation of T14, namely trauma (such as head injury or ischemia [218]). Moreover, we do not yet know how much T14 is generated by such an event, nor the timeframe in which it accrues, to reproduce a quantitative parallel in rodents. Nonetheless, this novel construct would be a major advance towards modelling an upstream cause of AD without the need for FAD-associated mutations.
8. Conclusions
For decades, researchers have attempted to model AD by recapitulating downstream markers of its pathology, including cholinergic neuron loss, Aβ and tau aggregation, and neuroinflammation, rather than exploring its true underlying mechanism. Insights gained from the study of PD show us that we must model the actual neurodegenerative mechanisms of AD in adult, wildtype animals by specifically targeting the neural populations first affected by a disease. A model, by definition, should capture the salient features of its subject, at the expense of the extraneous ones: in AD, anatomy and neurochemistry both need to be considered as salient factors. We propose an alternative model, based on the aberrant accumulation of the novel peptide T14: we suggest that this novel preparation would produce a more authentic and fuller profile of AD progression. Only once we understand the initial causes of the disease can we hope to fully understand progression, identify biomarkers, and ultimately find an effective treatment for AD.
Author Contributions
Conceptualisation, H.M.C. and S.G.; Writing—Original Draft Preparation, H.M.C.; Writing—Review and Editing, H.M.C. and S.G.; Supervision, S.G.; Project Administration, H.M.C.; Funding Acquisition, S.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Neuro-Bio Ltd.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analysed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare the following competing financial interests: Susan Greenfield is the founder and CEO of Neuro-Bio Ltd. (www.neuro-bio.com), a privately owned Company, and holds shares in the company. Helen M Collins was an employee of Neuro-Bio Ltd. during manuscript preparation.
References
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015. The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2022, 388, 9–21. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Fotiou, D.; Kaltsatou, A.; Tsiptsios, D.; Nakou, M. Evaluation of the cholinergic hypothesis in Alzheimer’s disease with neuropsychological methods. Aging Clin. Exp. Res. 2015, 27, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Bigl, V.; Arendt, A.; Tennstedt, A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s Disease. Acta Neuropathol. 1983, 61, 101–108. [Google Scholar] [CrossRef]
- Vogels, O.J.; Broere, C.A.; ter Laak, H.J.; ten Donkelaar, H.J.; Nieuwenhuys, R.; Schulte, B.P. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol. Aging 1990, 11, 3–13. [Google Scholar] [CrossRef]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Ikonomovic, M.D.; Styren, S.D.; Beckett, L.; Wisniewski, S.; Bennett, D.A.; Cochran, E.J.; Kordower, J.H.; Mufson, E.J. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2002, 51, 145–155. [Google Scholar] [CrossRef]
- Mufson, E.J.; Bothwell, M.; Kordower, J.H. Loss of nerve growth factor receptor-containing neurons in Alzheimer’s disease: A quantitative analysis across subregions of the basal forebrain. Exp. Neurol. 1989, 105, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.A.; Perry, E.K.; Tomlinson, B.E. Acetylcholine and choline levels in post-mortem human brain tissue: Preliminary observations in Alzheimer’s disease. Life Sci. 1980, 26, 1683–1689. [Google Scholar] [CrossRef]
- Vnek, N.; Kromer, L.F.; Wiley, R.G.; Rothblat, L.A. The basal forebrain cholinergic system and object memory in the rat. Brain Res. 1996, 710, 265–270. [Google Scholar] [CrossRef]
- Bierer, L.M.; Haroutunian, V.; Gabriel, S.; Knott, P.J.; Carlin, L.S.; Purohit, D.P.; Perl, D.P.; Schmeidler, J.; Kanof, P.; Davis, K.L. Neurochemical correlates of dementia severity in Alzheimer’s disease: Relative importance of the cholinergic deficits. J. Neurochem. 1995, 64, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.R.; Huang, J.B.; Yang, S.L.; Hong, F.F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Neuendorff, M.; Roy, B.; Albensi, B.C. A review of clinical treatment considerations of donepezil in severe Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Pazzagli, A.; Pepeu, G. Amnesic properties of scopolamine and brain acetylcholine in the rat. Int. J. Neuropharmacol. 1965, 4, 291–299. [Google Scholar] [CrossRef]
- Nitta, A.; Katono, Y.; Itoh, A.; Hasegawa, T.; Nabeshima, T. Nicotine reverses scopolamine-induced impairment of performance in passive avoidance task in rats through its action on the dopaminergic neuronal system. Pharmacol. Biochem. Behav. 1994, 49, 807–812. [Google Scholar] [CrossRef]
- Book, A.A.; Wiley, R.G.; Schweitzer, J.B. 192 IgG-saporin: I. Specific lethality for cholinergic neurons in the basal forebrain of the rat. J. Neuropathol. Exp. Neurol. 1994, 53, 95–102. [Google Scholar] [CrossRef] [PubMed]
- McGaughy, J.; Everitt, B.J.; Robbins, T.W.; Sarter, M. The role of cortical cholinergic afferent projections in cognition: Impact of new selective immunotoxins. Behav. Brain Res. 2000, 115, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Holley, L.A.; Wiley, R.G.; Lappi, D.A.; Sarter, M. Cortical cholinergic deafferentation following the intracortical infusion of 192 IgG-saporin: A quantitative histochemical study. Brain Res. 1994, 663, 277–286. [Google Scholar] [CrossRef]
- Do Carmo, S.; Cuello, A.C. Modeling Alzheimer’s disease in transgenic rats. Mol. Neurodegener. 2013, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.G.; Bucci, D.J.; Gorman, L.K.; Wiley, R.G.; Gallagher, M. Selective immunotoxic lesions of basal forebrain cholinergic cells: Effects on learning and memory in rats. Behav. Neurosci. 1995, 109, 714. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.G.; Bucci, D.J.; Sobel, T.J.; Williams, M.J.; Gorman, L.K.; Gallagher, M. Intact spatial learning following lesions of basal forebrain cholinergic neurons. Neuroreport 1996, 7, 1417–1420. [Google Scholar] [CrossRef]
- Wrenn, C.C.; Lappi, D.A.; Wiley, R.G. Threshold relationship between lesion extent of the cholinergic basal forebrain in the rat and working memory impairment in the radial maze. Brain Res. 1999, 847, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Frigerio, I.; Boon, B.D.C.; Zhou, Z.; Rozemuller, A.J.M.; Bouwman, F.H.; Schoonheim, M.M.; van de Berg, W.D.J.; Jonkman, L.E. Structural (dys)connectivity associates with cholinergic cell density in Alzheimer’s disease. Brain 2022, 145, 2869–2881. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Bigl, V. Alzheimer’s disease as a presumptive threshold phenomenon. Neurobiol. Aging 1987, 8, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Tokuno, H.; Hersh, L.; Marsel Mesulam, M. Human striatal cholinergic neurons in development, aging and Alzheimer’s disease. Brain Res. 1990, 508, 310–312. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. Embo J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by β-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.; Kim, S. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Bales, K.R.; Wu, S.; Bhat, P.; Parsadanian, M.; Fagan, A.M.; Chang, L.K.; Sun, Y.; Paul, S.M. Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer’s disease. J. Clin. Investig. 1999, 103, R15–R21. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef]
- Andrade-Moraes, C.H.; Oliveira-Pinto, A.V.; Castro-Fonseca, E.; da Silva, C.G.; Guimarães, D.M.; Szczupak, D.; Parente-Bruno, D.R.; Carvalho, L.R.; Polichiso, L.; Gomes, B.V.; et al. Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain 2013, 136, 3738–3752. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Bodea, L.-G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Chen, G.; Chen, K.S.; Knox, J.; Inglis, J.; Bernard, A.; Martin, S.J.; Justice, A.; McConlogue, L.; Games, D.; Freedman, S.B.; et al. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature 2000, 408, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.M.; Thorngate, F.E.; Katoh-Fukui, Y.; Hamanaka, H.; Williams, D.L.; Fujita, S.; Lamb, B.T. Independent effects of APOE on cholesterol metabolism and brain Abeta levels in an Alzheimer disease mouse model. Hum. Mol. Genet. 2004, 13, 1959–1968. [Google Scholar] [CrossRef]
- Huynh, T.V.; Wang, C.; Tran, A.C.; Tabor, G.T.; Mahan, T.E.; Francis, C.M.; Finn, M.B.; Spellman, R.; Manis, M.; Tanzi, R.E.; et al. Lack of hepatic apoE does not influence early Aβ deposition: Observations from a new APOE knock-in model. Mol. Neurodegener. 2019, 14, 37. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Xia, Y.; Wu, Z.; Kang, S.S.; Zhang, J.-c.; Liu, P.; Liu, X.; Song, W.; Huin, V.; Dhaenens, C.-M. Neuronal ApoE4 stimulates C/EBPβ activation, promoting Alzheimer’s disease pathology in a mouse model. Prog. Neurobiol. 2022, 209, 102212. [Google Scholar] [CrossRef]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Das, B.; Hou, H.; He, W.; Yan, R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J. Exp. Med. 2018, 215, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, M.-H.; Kurz, J.E.; Nomura, T.; Popović, J.; Rajapaksha, T.W.; Dong, H.; Contractor, A.; Chetkovich, D.M.; Tourtellotte, W.G.; Vassar, R.J. Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci. Transl. Med. 2018, 10, eaao5620. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Maldonado Weng, J.; LaDu, M.J.; Brady, S.T. Relevance of transgenic mouse models for Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2021, 177, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.M.; McConlogue, L.; Tan, H.; Power, M.; Masliah, E.; Mucke, L. Levels and alternative splicing of amyloid beta protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J. Biol. Chem. 1995, 270, 28257–28267. [Google Scholar] [CrossRef] [PubMed]
- Moechars, D.; Dewachter, I.; Lorent, K.; Reversé, D.; Baekelandt, V.; Naidu, A.; Tesseur, I.; Spittaels, K.; Haute, C.V.; Checler, F.; et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999, 274, 6483–6492. [Google Scholar] [CrossRef] [PubMed]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A mouse model of amyloid beta oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Lanz, T.; Carter, D.; Merchant, K. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 2003, 13, 246–253. [Google Scholar] [CrossRef]
- Jacobsen, J.S.; Wu, C.-C.; Redwine, J.M.; Comery, T.A.; Arias, R.; Bowlby, M.; Martone, R.; Morrison, J.H.; Pangalos, M.N.; Reinhart, P.H. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5161–5166. [Google Scholar] [CrossRef] [PubMed]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Yabuki, C.; Seubert, P.; Schenk, D.; Hori, Y.; Ohtsuki, S.; Terasaki, T.; Hashimoto, T.; Iwatsubo, T. Abeta immunotherapy: Intracerebral sequestration of Abeta by an anti-Abeta monoclonal antibody 266 with high affinity to soluble Abeta. J. Neurosci. 2009, 29, 11393–11398. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed]
- Schneider, I.; Reverse, D.; Dewachter, I.; Ris, L.; Caluwaerts, N.; Kuiperi, C.; Gilis, M.; Geerts, H.; Kretzschmar, H.; Godaux, E.; et al. Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J. Biol. Chem. 2001, 276, 11539–11544. [Google Scholar] [CrossRef]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.M.; Perez-tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D.; et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Siman, R.; Reaume, A.G.; Savage, M.J.; Trusko, S.; Lin, Y.-G.; Scott, R.W.; Flood, D.G. Presenilin-1 P264L Knock-In Mutation: Differential Effects on Aβ Production, Amyloid Deposition, and Neuronal Vulnerability. J. Neurosci. 2000, 20, 8717–8726. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Flood, D.G.; Reaume, A.G.; Dorfman, K.S.; Lin, Y.G.; Lang, D.M.; Trusko, S.P.; Savage, M.J.; Annaert, W.G.; De Strooper, B.; Siman, R.; et al. FAD mutant PS-1 gene-targeted mice: Increased A beta 42 and A beta deposition without APP overproduction. Neurobiol. Aging 2002, 23, 335–348. [Google Scholar] [CrossRef]
- Saito, T.; Suemoto, T.; Brouwers, N.; Sleegers, K.; Funamoto, S.; Mihira, N.; Matsuba, Y.; Yamada, K.; Nilsson, P.; Takano, J.; et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nat. Neurosci. 2011, 14, 1023–1032. [Google Scholar] [CrossRef]
- Holcomb, L.A.; Gordon, M.N.; Jantzen, P.; Hsiao, K.; Duff, K.; Morgan, D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: Lack of association with amyloid deposits. Behav. Genet. 1999, 29, 177–185. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Ratovitski, T.; van Lare, J.; Lee, M.K.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Price, D.L.; Sisodia, S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Ihalainen, J.; Malm, T.; Matilainen, O.; Keksa-Goldsteine, V.; Goldsteins, G.; Iivonen, H.; Leguit, N.; Glennon, J.; Koistinaho, J.; et al. Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. J. Neurochem. 2008, 105, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener. Dis. 2009, 6, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.-M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, I.; Van Dorpe, J.; Smeijers, L.; Gilis, M.; Kuipéri, C.; Laenen, I.; Caluwaerts, N.; Moechars, D.; Checler, F.; Vanderstichele, H.; et al. Aging increased amyloid peptide and caused amyloid plaques in brain of old APP/V717I transgenic mice by a different mechanism than mutant presenilin1. J. Neurosci. 2000, 20, 6452–6458. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]
- Stover, K.R.; Campbell, M.A.; Van Winssen, C.M.; Brown, R.E. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav. Brain Res. 2015, 289, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; Howland, D.S.; Trusko, S.P.; Savage, M.J.; Lang, D.M.; Greenberg, B.D.; Siman, R.; Scott, R.W. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer’s disease mutations and a “humanized” Abeta sequence. J. Biol. Chem. 1996, 271, 23380–23388. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Mihira, N.; Matsuba, Y.; Sasaguri, H.; Hashimoto, S.; Narasimhan, S.; Zhang, B.; Murayama, S.; Higuchi, M.; Lee, V.M.Y.; et al. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem. 2019, 294, 12754–12765. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Matsuba, Y.; Kamano, N.; Mihira, N.; Sahara, N.; Takano, J.; Muramatsu, S.I.; Saido, T.C.; Saito, T. Tau binding protein CAPON induces tau aggregation and neurodegeneration. Nat. Commun. 2019, 10, 2394. [Google Scholar] [CrossRef] [PubMed]
- Serneels, L.; T’Syen, D.; Perez-Benito, L.; Theys, T.; Holt, M.G.; De Strooper, B. Modeling the β-secretase cleavage site and humanizing amyloid-beta precursor protein in rat and mouse to study Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Agca, C.; Fritz, J.J.; Walker, L.C.; Levey, A.I.; Chan, A.W.; Lah, J.J.; Agca, Y. Development of transgenic rats producing human beta-amyloid precursor protein as a model for Alzheimer’s disease: Transgene and endogenous APP genes are regulated tissue-specifically. BMC Neurosci. 2008, 9, 28. [Google Scholar] [CrossRef]
- Heggland, I.; Storkaas, I.S.; Soligard, H.T.; Kobro-Flatmoen, A.; Witter, M.P. Stereological estimation of neuron number and plaque load in the hippocampal region of a transgenic rat model of Alzheimer’s disease. Eur. J. Neurosci. 2015, 41, 1245–1262. [Google Scholar] [CrossRef]
- Leon, W.C.; Canneva, F.; Partridge, V.; Allard, S.; Ferretti, M.T.; DeWilde, A.; Vercauteren, F.; Atifeh, R.; Ducatenzeiler, A.; Klein, W.; et al. A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J. Alzheimer’s Dis. 2010, 20, 113–126. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef] [PubMed]
- Agca, C.; Klakotskaia, D.; Schachtman, T.R.; Chan, A.W.; Lah, J.J.; Agca, Y. Presenilin 1 transgene addition to amyloid precursor protein overexpressing transgenic rats increases amyloid beta 42 levels and results in loss of memory retention. BMC Neurosci. 2016, 17, 46. [Google Scholar] [CrossRef]
- Pentkowski, N.S.; Berkowitz, L.E.; Thompson, S.M.; Drake, E.N.; Olguin, C.R.; Clark, B.J. Anxiety-like behavior as an early endophenotype in the TgF344-AD rat model of Alzheimer’s disease. Neurobiol. Aging 2018, 61, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Klakotskaia, D.; Agca, C.; Richardson, R.A.; Stopa, E.G.; Schachtman, T.R.; Agca, Y. Memory deficiency, cerebral amyloid angiopathy, and amyloid-β plaques in APP+PS1 double transgenic rat model of Alzheimer’s disease. PLoS ONE 2018, 13, e0195469. [Google Scholar] [CrossRef] [PubMed]
- Flood, D.G.; Lin, Y.G.; Lang, D.M.; Trusko, S.P.; Hirsch, J.D.; Savage, M.J.; Scott, R.W.; Howland, D.S. A transgenic rat model of Alzheimer’s disease with extracellular Abeta deposition. Neurobiol. Aging 2009, 30, 1078–1090. [Google Scholar] [CrossRef]
- Liu, L.; Orozco, I.J.; Planel, E.; Wen, Y.; Bretteville, A.; Krishnamurthy, P.; Wang, L.; Herman, M.; Figueroa, H.; Yu, W.H. A transgenic rat that develops Alzheimer’s disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol. Dis. 2008, 31, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Ducatenzeiler, A.; Alhonen, L.; Janne, J.; Grant, S.M.; Wandosell, F.; Muro, A.; Baralle, F.; Li, H.; Duff, K. Rat transgenic models with a phenotype of intracellular Aβ accumulation in hippocampus and cortex. J. Alzheimer’s Dis. 2004, 6, 209–219. [Google Scholar] [CrossRef]
- Tambini, M.D.; Yao, W.; D’Adamio, L. Facilitation of glutamate, but not GABA, release in Familial Alzheimer’s APP mutant Knock-in rats with increased β-cleavage of APP. Aging Cell 2019, 18, e13033. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Norris, K.A.; D’Adamio, L. Opposite changes in APP processing and human Aβ levels in rats carrying either a protective or a pathogenic APP mutation. Elife 2020, 9, e52612. [Google Scholar] [CrossRef]
- Pang, K.; Jiang, R.; Zhang, W.; Yang, Z.; Li, L.L.; Shimozawa, M.; Tambaro, S.; Mayer, J.; Zhang, B.; Li, M.; et al. An App knock-in rat model for Alzheimer’s disease exhibiting Aβ and tau pathologies, neuronal death and cognitive impairments. Cell Res. 2022, 32, 157–175. [Google Scholar] [CrossRef]
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Børte, S.; Winsvold, B.S.; Drange, O.K.; et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 2021, 53, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Dobricic, V.; Stefanova, E.; Jankovic, M.; Gurunlian, N.; Novakovic, I.; Hardy, J.; Kostic, V.; Guerreiro, R. Genetic testing in familial and young-onset Alzheimer’s disease: Mutation spectrum in a Serbian cohort. Neurobiol. Aging 2012, 33, 1481.e7–1481.e12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [PubMed]
- Marques-Coelho, D.; Iohan, L.d.C.C.; Melo de Farias, A.R.; Flaig, A.; Letournel, F.; Martin-Négrier, M.-L.; Chapon, F.; Faisant, M.; Godfraind, C.; Maurage, C.-A.; et al. Differential transcript usage unravels gene expression alterations in Alzheimer’s disease human brains. NPJ Aging Mech. Dis. 2021, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Guennewig, B.; Lim, J.; Marshall, L.; McCorkindale, A.N.; Paasila, P.J.; Patrick, E.; Kril, J.J.; Halliday, G.M.; Cooper, A.A.; Sutherland, G.T. Defining early changes in Alzheimer’s disease from RNA sequencing of brain regions differentially affected by pathology. Sci. Rep. 2021, 11, 4865. [Google Scholar] [CrossRef] [PubMed]
- Ulm, B.S.; Borchelt, D.R.; Moore, B.D. Remodeling Alzheimer-amyloidosis models by seeding. Mol. Neurodegener. 2021, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous Induction of Cerebral ß-Amyloidogenesis Is Governed by Agent and Host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Ziegler-Waldkirch, S.; Friesen, M.; Loreth, D.; Sauer, J.-F.; Kemna, S.; Hilse, A.; Erny, D.; Helm, C.; d’Errico, P.; Prinz, M.; et al. Seed-induced Aβ deposition alters neuronal function and impairs olfaction in a mouse model of Alzheimer’s disease. Mol. Psychiatry 2022, 27, 4274–4284. [Google Scholar] [CrossRef]
- Ziegler-Waldkirch, S.; Marksteiner, K.; Stoll, J.; d’ Errico, P.; Friesen, M.; Eiler, D.; Neudel, L.; Sturn, V.; Opper, I.; Datta, M. Environmental enrichment reverses Aβ pathology during pregnancy in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 44. [Google Scholar] [CrossRef]
- Heilbronner, G.; Eisele, Y.S.; Langer, F.; Kaeser, S.A.; Novotny, R.; Nagarathinam, A.; Åslund, A.; Hammarström, P.; Nilsson, K.P.R.; Jucker, M. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013, 14, 1017–1022. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Janus, C.; Pearson, J.; McLaurin, J.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Chishti, M.A.; Horne, P.; Heslin, D.; French, J. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 2000, 408, 979–982. [Google Scholar] [CrossRef]
- Morgan, D.; Diamond, D.M.; Gottschall, P.E.; Ugen, K.E.; Dickey, C.; Hardy, J.; Duff, K.; Jantzen, P.; DiCarlo, G.; Wilcock, D. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 2000, 408, 982–985. [Google Scholar] [CrossRef]
- Theofilas, P.; Dunlop, S.; Heinsen, H.; Grinberg, L.T. Turning on the Light Within: Subcortical Nuclei of the Isodentritic Core and their Role in Alzheimer’s Disease Pathogenesis. J. Alzheimer’s Dis. 2015, 46, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Smith, J.; Donohue, M.C.; Delmar, P.; Abbas, R.; Salloway, S.; Wojtowicz, J.; Blennow, K.; Bittner, T.; Black, S.E.; et al. Two Phase 3 Trials of Gantenerumab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 389, 1862–1876. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of tau aggregates and neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Uematsu, M.; Nakamura, A.; Ebashi, M.; Hirokawa, K.; Takahashi, R.; Uchihara, T. Brainstem tau pathology in Alzheimer’s disease is characterized by increase of three repeat tau and independent of amyloid β. Acta Neuropathol. Commun. 2018, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Berg, L.; McKeel, D.W.; Miller, J.P.; Storandt, M.; Rubin, E.H.; Morris, J.C.; Baty, J.; Coats, M.; Norton, J.; Goate, A.M. Clinicopathologic studies in cognitively healthy aging and Alzheimer disease: Relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch. Neurol. 1998, 55, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Herrmann, F.; Bussière, T.; Bouras, C.; Kovari, E.; Perl, D.; Morrison, J.; Gold, G.; Hof, P. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Götz, J.; Ittner, L.M. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci. 2008, 9, 532–544. [Google Scholar] [CrossRef]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Arner, A.; Rockenstein, E.; Mante, M.; Florio, J.; Masliah, D.; Salehi, B.; Adame, A.; Overk, C.; Masliah, E.; Rissman, R.A. Increased Vulnerability of the Hippocampus in Transgenic Mice Overexpressing APP and Triple Repeat Tau. J. Alzheimer’s Dis. 2018, 61, 1201–1219. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Colin, M.; Buée, L. Invited review: Animal models of tauopathies and their implications for research/translation into the clinic. Neuropathol. Appl. Neurobiol. 2015, 41, 59–80. [Google Scholar] [CrossRef]
- Andorfer, C.; Kress, Y.; Espinoza, M.; de Silva, R.; Tucker, K.L.; Barde, Y.A.; Duff, K.; Davies, P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 2003, 86, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Chen, F.; Barmettler, R.; Nitsch, R.M. Tau filament formation in transgenic mice expressing P301L tau. J. Biol. Chem. 2001, 276, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Pennanen, L.; Welzl, H.; D’Adamo, P.; Nitsch, R.M.; Götz, J. Accelerated extinction of conditioned taste aversion in P301L tau transgenic mice. Neurobiol. Dis. 2004, 15, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Oddo, S.; Billings, L.M.; Green, K.N.; Martinez-Coria, H.; Fisher, A.; LaFerla, F.M. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 2006, 49, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.C.; Rosario, E.R.; Chang, L.; Stanczyk, F.Z.; Oddo, S.; LaFerla, F.M.; Pike, C.J. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 2007, 27, 13357–13365. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vassar, R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol. Neurodegener. 2013, 8, 2. [Google Scholar] [CrossRef]
- Neuman, K.M.; Molina-Campos, E.; Musial, T.F.; Price, A.L.; Oh, K.J.; Wolke, M.L.; Buss, E.W.; Scheff, S.W.; Mufson, E.J.; Nicholson, D.A. Evidence for Alzheimer’s disease-linked synapse loss and compensation in mouse and human hippocampal CA1 pyramidal neurons. Brain Struct. Funct. 2015, 220, 3143–3165. [Google Scholar] [CrossRef]
- Ball, M.J.; Fisman, M.; Hachinski, V.; Blume, W.; Fox, A.; Kral, V.A.; Kirshen, A.J.; Fox, H.; Merskey, H. A new definition of Alzheimer’s disease: A hippocampal dementia. Lancet 1985, 1, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Hanseeuw, B.J.; Jacobs, H.I.L.; Schultz, A.P.; Buckley, R.F.; Farrell, M.E.; Guehl, N.J.; Becker, J.A.; Properzi, M.; Sanchez, J.S.; Quiroz, Y.T.; et al. Association of Pathologic and Volumetric Biomarker Changes With Cognitive Decline in Clinically Normal Adults. Neurology 2023, 101, e2533–e2544. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Peeraer, E.; Bottelbergs, A.; Van Kolen, K.; Stancu, I.C.; Vasconcelos, B.; Mahieu, M.; Duytschaever, H.; Ver Donck, L.; Torremans, A.; Sluydts, E.; et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 2015, 73, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Boluda, S.; Iba, M.; Zhang, B.; Raible, K.M.; Lee, V.M.; Trojanowski, J.Q. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015, 129, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; McBride, J.D.; Guo, J.L.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015, 130, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Bennett, R.E.; Delorme, L.; Robbins, A.B.; Hu, M.; McKenzie, D.; Kirk, M.J.; Schiantarelli, J.; Tunio, N.; Amaral, A.C.; et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019, 5, eaaw6404. [Google Scholar] [CrossRef]
- Bell, B.J.; Malvankar, M.M.; Tallon, C.; Slusher, B.S. Sowing the Seeds of Discovery: Tau-Propagation Models of Alzheimer’s Disease. ACS Chem. Neurosci. 2020, 11, 3499–3509. [Google Scholar] [CrossRef]
- Cuello, A.C.; Hall, H.; Do Carmo, S. Experimental Pharmacology in Transgenic Rodent Models of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 443703. [Google Scholar] [CrossRef]
- Sirkis, D.W.; Geier, E.G.; Bonham, L.W.; Karch, C.M.; Yokoyama, J.S. Recent advances in the genetics of frontotemporal dementia. Curr. Genet. Med. Rep. 2019, 7, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Saido, T.C. Neuroinflammation in mouse models of Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2018, 9, 211–218. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, N.; Liu, Q.; Shin, S.; Singh, R.; Agca, Y.; Agca, C.; Hachinski, V.; Whitehead, S.N. APP21 transgenic rats develop age-dependent cognitive impairment and microglia accumulation within white matter tracts. J. Neuroinflamm. 2018, 15, 241. [Google Scholar] [CrossRef] [PubMed]
- Metaxas, A.; Anzalone, M.; Vaitheeswaran, R.; Petersen, S.; Landau, A.M.; Finsen, B. Neuroinflammation and amyloid-beta 40 are associated with reduced serotonin transporter (SERT) activity in a transgenic model of familial Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; Van Leuven, F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J. Neuroinflamm. 2005, 2, 22. [Google Scholar] [CrossRef]
- Eikelenboom, P.; van Exel, E.; Hoozemans, J.J.; Veerhuis, R.; Rozemuller, A.J.; van Gool, W.A. Neuroinflammation—An early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 38–41. [Google Scholar] [CrossRef]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, M.S.; Wyss-Coray, T. Modelling neuroinflammatory phenotypes in vivo. J. Neuroinflamm. 2004, 1, 10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Z.Y.; Zhang, Y. Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Tapia, R.; Peña, F.; Arias, C. Neurotoxic and synaptic effects of okadaic acid, an inhibitor of protein phosphatases. Neurochem. Res. 1999, 24, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.P.; Tramontina, A.C.; Biasibetti, R.; Batassini, C.; Lopes, M.W.; Wartchow, K.M.; Bernardi, C.; Tortorelli, L.S.; Leal, R.B.; Gonçalves, C.-A. Neuroglial alterations in rats submitted to the okadaic acid-induced model of dementia. Behav. Brain Res. 2012, 226, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Tota, S.; Saxena, G.; Shukla, R.; Nath, C. Okadaic acid (ICV) induced memory impairment in rats: A suitable experimental model to test anti-dementia activity. Brain Res. 2010, 1309, 66–74. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Fruth, R.; Brückner, M.K.; Gärtner, U. Phosphorylation of tau, Abeta-formation, and apoptosis after in vivo inhibition of PP-1 and PP-2A. Neurobiol. Aging 1998, 19, 3–13. [Google Scholar] [CrossRef]
- Krstic, D.; Madhusudan, A.; Doehner, J.; Vogel, P.; Notter, T.; Imhof, C.; Manalastas, A.; Hilfiker, M.; Pfister, S.; Schwerdel, C. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflamm. 2012, 9, 151. [Google Scholar] [CrossRef]
- Ahlijanian, M.K.; Barrezueta, N.X.; Williams, R.D.; Jakowski, A.; Kowsz, K.P.; McCarthy, S.; Coskran, T.; Carlo, A.; Seymour, P.A.; Burkhardt, J.E.; et al. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc. Natl. Acad. Sci. USA 2000, 97, 2910–2915. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, J.R.; Chan, E.S.; Poore, C.P.; Pareek, T.K.; Cheong, W.F.; Shui, G.; Tang, N.; Low, C.M.; Wenk, M.R.; Kesavapany, S. Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J. Neurosci. 2012, 32, 1020–1034. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; LaDu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Schulte, C.; Gasser, T. Genetic basis of Parkinson’s disease: Inheritance, penetrance, and expression. Appl. Clin. Genet. 2011, 4, 67–80. [Google Scholar] [CrossRef]
- Smith, C.; Malek, N.; Grosset, K.; Cullen, B.; Gentleman, S.; Grosset, D.G. Neuropathology of dementia in patients with Parkinson’s disease: A systematic review of autopsy studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Ma, S.Y.; Röyttä, M.; Rinne, J.O.; Collan, Y.; Rinne, U.K. Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J. Neurol. Sci. 1997, 151, 83–87. [Google Scholar] [CrossRef]
- Giguère, N.; Burke Nanni, S.; Trudeau, L.E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Greffard, S.; Verny, M.; Bonnet, A.-M.; Beinis, J.-Y.; Gallinari, C.; Meaume, S.; Piette, F.; Hauw, J.-J.; Duyckaerts, C. Motor Score of the Unified Parkinson Disease Rating Scale as a Good Predictor of Lewy Body–Associated Neuronal Loss in the Substantia Nigra. Arch. Neurol. 2006, 63, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Wuketich, S. Time course of nigrostriatal degeneration in parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J. Neural Transm. 1976, 38, 277–301. [Google Scholar] [CrossRef]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef] [PubMed]
- Luthman, J.; Fredriksson, A.; Sundström, E.; Jonsson, G.; Archer, T. Selective lesion of central dopamine or noradrenaline neuron systems in the neonatal rat: Motor behavior and monoamine alterations at adult stage. Behav. Brain Res. 1989, 33, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Kulich, S.M.; Horbinski, C.; Patel, M.; Chu, C.T. 6-Hydroxydopamine induces mitochondrial ERK activation. Free Radic. Biol. Med. 2007, 43, 372–383. [Google Scholar] [CrossRef]
- Ungerstedt, U. Adipsia and Aphagia after 6-Hydroxydopamine Induced Degeneration of the Nigro-striatal Dopamine System. Acta Physiol. Scand. 1971, 82, 95–122. [Google Scholar] [CrossRef]
- Sun, X.; Li, X.; Zhang, L.; Zhang, Y.; Qi, X.; Wang, S.; Qin, C. Longitudinal assessment of motor function following the unilateral intrastriatal 6-hydroxydopamine lesion model in mice. Front. Behav. Neurosci. 2022, 16, 982218. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Park. Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Djaldetti, R.; Liberatore, G.; Vila, M.; Vukosavic, S.; Almer, G. The parkinsonian toxin MPTP: Action and mechanism. Restor. Neurol. Neurosci. 2000, 16, 135–142. [Google Scholar] [PubMed]
- Yazdani, U.; German, D.C.; Liang, C.L.; Manzino, L.; Sonsalla, P.K.; Zeevalk, G.D. Rat model of Parkinson’s disease: Chronic central delivery of 1-methyl-4-phenylpyridinium (MPP+). Exp. Neurol. 2006, 200, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Forno, L.S.; DeLanney, L.E.; Irwin, I.; Langston, J.W. Similarities and differences between MPTP-induced parkinsonsim and Parkinson’s disease. Neuropathologic considerations. Adv. Neurol. 1993, 60, 600–608. [Google Scholar] [PubMed]
- Fornai, F.; Schlüter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef] [PubMed]
- Sedelis, M.; Schwarting, R.K.; Huston, J.P. Behavioral phenotyping of the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 2001, 125, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Brotchie, J.M.; Kalia, L.V.; Koprich, J.B.; Tandon, A.; Watts, J.C.; Lang, A.E. α-Synuclein-based animal models of Parkinson’s disease: Challenges and opportunities in a new era. Trends Neurosci. 2016, 39, 750–762. [Google Scholar] [CrossRef]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal models of α-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef]
- Hatami, A.; Chesselet, M.-F. Transgenic rodent models to study alpha-synuclein pathogenesis, with a focus on cognitive deficits. Behav. Neurobiol. Huntington’s Dis. Park. Dis. 2015, 22, 303–330. [Google Scholar]
- Bezard, E.; Yue, Z.; Kirik, D.; Spillantini, M.G. Animal models of Parkinson’s disease: Limits and relevance to neuroprotection studies. Mov. Disord. 2013, 28, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Parisiadou, L.; Sgobio, C.; Liu, G.; Yu, J.; Sun, L.; Shim, H.; Gu, X.-L.; Luo, J.; Long, C.-X. Conditional expression of Parkinson’s disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J. Neurosci. 2012, 32, 9248–9264. [Google Scholar] [CrossRef] [PubMed]
- Van der Perren, A.; Van den Haute, C.; Baekelandt, V. Viral vector-based models of Parkinson’s disease. Behav. Neurobiol. Huntington’s Dis. Park. Dis. 2015, 22, 271–301. [Google Scholar]
- Bagga, V.; Dunnett, S.B.; Fricker, R.A. The 6-OHDA mouse model of Parkinson’s disease—Terminal striatal lesions provide a superior measure of neuronal loss and replacement than median forebrain bundle lesions. Behav. Brain Res. 2015, 288, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.F.; Ungerstedt, U. Supersensitivity to apomorphine following destruction of the ascending dopamine neurons: Quantification using the rotational model. Eur. J. Pharmacol. 1977, 41, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: Separating the wheat from the chaff. J. Park. Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [PubMed]
- Ehrenberg, A.J.; Suemoto, C.K.; França Resende, E.P.; Petersen, C.; Leite, R.E.P.; Rodriguez, R.D.; Ferretti-Rebustini, R.E.L.; You, M.; Oh, J.; Nitrini, R.; et al. Neuropathologic Correlates of Psychiatric Symptoms in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 66, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Botto, R.; Callai, N.; Cermelli, A.; Causarano, L.; Rainero, I. Anxiety and depression in Alzheimer’s disease: A systematic review of pathogenetic mechanisms and relation to cognitive decline. Neurol. Sci. 2022, 43, 4107–4124. [Google Scholar] [CrossRef]
- Rossor, M.N. Parkinson’s disease and Alzheimer’s disease as disorders of the isodendritic core. Br. Med. J. (Clin. Res. Ed.) 1981, 283, 1588–1590. [Google Scholar] [CrossRef]
- Woolf, N.J. Global and serial neurons form A hierarchically arranged interface proposed to underlie memory and cognition. Neuroscience 1996, 74, 625–651. [Google Scholar] [CrossRef] [PubMed]
- Ramón-Moliner, E.; Nauta, W.J. The isodendritic core of the brain stem. J. Comp. Neurol. 1966, 126, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Zoli, M.; Jansson, A.; Syková, E.; Agnati, L.F.; Fuxe, K. Volume transmission in the CNS and its relevance for neuropsychopharmacology. Trends Pharmacol. Sci. 1999, 20, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Dahlström, A.; Höistad, M.; Marcellino, D.; Jansson, A.; Rivera, A.; Diaz-Cabiale, Z.; Jacobsen, K.; Tinner-Staines, B.; Hagman, B. From the Golgi–Cajal mapping to the transmitter-based characterization of the neuronal networks leading to two modes of brain communication: Wiring and volume transmission. Brain Res. Rev. 2007, 55, 17–54. [Google Scholar] [CrossRef]
- Greenfield, S.A.; Cole, G.M.; Coen, C.W.; Frautschy, S.; Singh, R.P.; Mekkittikul, M.; Garcia-Ratés, S.; Morrill, P.; Hollings, O.; Passmore, M.; et al. A novel process driving Alzheimer’s disease validated in a mouse model: Therapeutic potential. Alzheimer’s Dement 2022, 8, e12274. [Google Scholar] [CrossRef]
- Garcia-Ratés, S.; Greenfield, S. When a trophic process turns toxic: Alzheimer’s disease as an aberrant recapitulation of a developmental mechanism. Int. J. Biochem. Cell Biol. 2022, 149, 106260. [Google Scholar] [CrossRef] [PubMed]
- Sarah Benton, J.; Bowen, D.M.; Allen, S.; Haan, E.A.; Davison, A.N.; Neary, D.; Murphy, R.P.; Snowden, J. Alzheimer’s Disease as a Disorder of Isodendritic Core. Lancet 1982, 319, 456. [Google Scholar] [CrossRef] [PubMed]
- Parvizi, J.; Van Hoesen, G.W.; Damasio, A. The selective vulnerability of brainstem nuclei to Alzheimer’s disease. Ann. Neurol. 2001, 49, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T. Distribution of Alzheimer’s neurofibrillary changes in the brain stem and hypothalamus of senile dementia. Acta Neuropathol. 1966, 6, 181–187. [Google Scholar] [CrossRef]
- Gibb, W.R.; Mountjoy, C.Q.; Mann, D.M.; Lees, A.J. The substantia nigra and ventral tegmental area in Alzheimer’s disease and Down’s syndrome. J. Neurol. Neurosurg. Psychiatry 1989, 52, 193–200. [Google Scholar] [CrossRef]
- Rüb, U.; Del Tredici, K.; Schultz, C.; Thal, D.R.; Braak, E.; Braak, H. The evolution of Alzheimer’s disease-related cytoskeletal pathology in the human raphe nuclei. Neuropathol. Appl. Neurobiol. 2000, 26, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Grudzien, A.; Shaw, P.; Weintraub, S.; Bigio, E.; Mash, D.C.; Mesulam, M.M. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging 2007, 28, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Sassin, I.; Schultz, C.; Thal, D.R.; Rüb, U.; Arai, K.; Braak, E.; Braak, H. Evolution of Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol. 2000, 100, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Kemper, T. Nucleus raphe dorsalis in dementia of the Alzheimer type: Neurofibrillary changes and neuronal packing density. J. Neuropathol. Exp. Neurol. 1984, 43, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Mesulam, M.M.; Saroff, D.M.; Wu, C.K. Relationship between plaques, tangles, and loss of cortical cholinergic fibers in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998, 57, 63–75. [Google Scholar] [CrossRef]
- Braak, E.; Braak, H.; Mandelkow, E.M. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994, 87, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Zweig, R.M.; Ross, C.A.; Hedreen, J.C.; Steele, C.; Cardillo, J.E.; Whitehouse, P.J.; Folstein, M.F.; Price, D.L. The neuropathology of aminergic nuclei in Alzheimer’s disease. Ann. Neurol. 1988, 24, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.; Shaw, P.; Mash, D.; Weintraub, S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. 2004, 55, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.J.; Mather, M.; Werkle-Bergner, M.; Kennedy, B.L.; Guzman, S.; Hurth, K.; Miller, C.A.; Qiao, Y.; Shi, Y.; Chui, H.C.; et al. Locus coeruleus integrity is related to tau burden and memory loss in autosomal-dominant Alzheimer’s disease. Neurobiol. Aging 2022, 112, 39–54. [Google Scholar] [CrossRef]
- Grinberg, L.; Rüb, U.; Ferretti, R.; Nitrini, R.; Farfel, J.; Polichiso, L.; Gierga, K.; Jacob-Filho, W.; Heinsen, H.; Group, B.B.B.S. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol. Appl. Neurobiol. 2009, 35, 406–416. [Google Scholar] [CrossRef]
- Attems, J.; Quass, M.; Jellinger, K.A. Tau and alpha-synuclein brainstem pathology in Alzheimer disease: Relation with extrapyramidal signs. Acta Neuropathol. 2007, 113, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Zarow, C.; Lyness, S.A.; Mortimer, J.A.; Chui, H.C. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 2003, 60, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, B.E.; Irving, D.; Blessed, G. Cell loss in the locus coeruleus in senile dementia of Alzheimer type. J. Neurol. Sci. 1981, 49, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Chan-Palay, V.; Asan, E. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson’s disease with and without dementia and depression. J. Comp. Neurol. 1989, 287, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Kazee, A.M.; Cox, C.; Richfield, E.K. Substantia nigra lesions in Alzheimer disease and normal aging. Alzheimer Dis. Assoc. Disord. 1995, 9, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Storga, D.; Vrecko, K.; Birkmayer, J.; Reibnegger, G. Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci. Lett. 1996, 203, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Wilcock, G.K.; Esiri, M.M.; Francis, P.T.; Bowen, D.M. Monoaminergic innervation of the frontal and temporal lobes in Alzheimer’s disease. Brain Res. 1987, 401, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Gil-Bea, F.J.; Diez-Ariza, M.; Chen, C.P.; Francis, P.T.; Lasheras, B.; Ramirez, M.J. Cholinergic-serotonergic imbalance contributes to cognitive and behavioral symptoms in Alzheimer’s disease. Neuropsychologia 2005, 43, 442–449. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Noristani, H.N.; Verkhratsky, A. The serotonergic system in ageing and Alzheimer’s disease. Prog. Neurobiol. 2012, 99, 15–41. [Google Scholar] [CrossRef]
- Boncristiano, S.; Calhoun, M.E.; Kelly, P.H.; Pfeifer, M.; Bondolfi, L.; Stalder, M.; Phinney, A.L.; Abramowski, D.; Sturchler-Pierrat, C.; Enz, A.; et al. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J. Neurosci. 2002, 22, 3234–3243. [Google Scholar] [CrossRef]
- Hernandez, D.; Sugaya, K.; Qu, T.; McGowan, E.; Duff, K.; McKinney, M. Survival and plasticity of basal forebrain cholinergic systems in mice transgenic for presenilin-1 and amyloid precursor protein mutant genes. Neuroreport 2001, 12, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Jaffar, S.; Counts, S.E.; Ma, S.Y.; Dadko, E.; Gordon, M.N.; Morgan, D.; Mufson, E.J. Neuropathology of mice carrying mutant APP(swe) and/or PS1(M146L) transgenes: Alterations in the p75(NTR) cholinergic basal forebrain septohippocampal pathway. Exp. Neurol. 2001, 170, 227–243. [Google Scholar] [CrossRef] [PubMed]
- German, D.C.; Yazdani, U.; Speciale, S.G.; Pasbakhsh, P.; Games, D.; Liang, C.L. Cholinergic neuropathology in a mouse model of Alzheimer’s disease. J. Comp. Neurol. 2003, 462, 371–381. [Google Scholar] [CrossRef]
- Perez, S.E.; Dar, S.; Ikonomovic, M.D.; DeKosky, S.T.; Mufson, E.J. Cholinergic forebrain degeneration in the APPswe/PS1DeltaE9 transgenic mouse. Neurobiol. Dis. 2007, 28, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.P.; Debeir, T.; Duff, K.; Cuello, A.C. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J. Neurosci. 1999, 19, 2706–2716. [Google Scholar] [CrossRef]
- Manaye, K.F.; Mouton, P.R.; Xu, G.; Drew, A.; Lei, D.L.; Sharma, Y.; Rebeck, G.W.; Turner, S. Age-related loss of noradrenergic neurons in the brains of triple transgenic mice. Age 2013, 35, 139–147. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.N.; Mouton, P.R.; Tizabi, Y.; Ottinger, M.A.; Lei, D.L.; Ingram, D.K.; Manaye, K.F. Catecholaminergic neuronal loss in locus coeruleus of aged female dtg APP/PS1 mice. J. Chem. Neuroanat. 2007, 34, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, J.; Yang, M.; Ke, X.; Dai, Y.; Lin, H.; Wang, S.; Chen, L.; Tao, J. Chemical genetic activation of the cholinergic basal forebrain hippocampal circuit rescues memory loss in Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, L.Y.; Xu, Y.; Wang, W.Z.; Qiu, N.Z.; Zhang, F.F.; Zhang, F.; Wang, X.D.; Chen, W.; Xu, X.Y.; et al. Imbalance of multiple neurotransmitter pathways leading to depression-like behavior and cognitive dysfunction in the triple transgenic mouse model of Alzheimer disease. Metab. Brain Dis. 2023, 38, 2465–2476. [Google Scholar] [CrossRef]
- Romano, A.; Pace, L.; Tempesta, B.; Lavecchia, A.M.; Macheda, T.; Bedse, G.; Petrella, A.; Cifani, C.; Serviddio, G.; Vendemiale, G. Depressive-like behavior is paired to monoaminergic alteration in a murine model of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2015, 18, pyu020. [Google Scholar] [CrossRef]
- Hartmann, J.; Erb, C.; Ebert, U.; Baumann, K.H.; Popp, A.; König, G.; Klein, J. Central cholinergic functions in human amyloid precursor protein knock-in/presenilin-1 transgenic mice. Neuroscience 2004, 125, 1009–1017. [Google Scholar] [CrossRef]
- German, D.C.; Nelson, O.; Liang, F.; Liang, C.L.; Games, D. The PDAPP mouse model of Alzheimer’s disease: Locus coeruleus neuronal shrinkage. J. Comp. Neurol. 2005, 492, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Balasubramanian, N.; Gaudencio, G.; Wang, R.; Selvakumar, G.P.; Kolling, L.; Pierson, S.; Tadinada, S.M.; Abel, T.; Hefti, M.; et al. Human tau-overexpressing mice recapitulate brainstem involvement and neuropsychiatric features of early Alzheimer’s disease. Acta Neuropathol. Commun. 2023, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wong, T.P.; Côté, S.L.; Bell, K.F.; Cuello, A.C. The impact of Abeta-plaques on cortical cholinergic and non-cholinergic presynaptic boutons in alzheimer’s disease-like transgenic mice. Neuroscience 2003, 121, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.; Ducatenzeiler, A.; Ribeiro-da-Silva, A.; Duff, K.; Bennett, D.A.; Cuello, A.C. The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol. Aging 2006, 27, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Aucoin, J.S.; Jiang, P.; Aznavour, N.; Tong, X.K.; Buttini, M.; Descarries, L.; Hamel, E. Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer’s disease. Neuroscience 2005, 132, 73–86. [Google Scholar] [CrossRef]
- Overk, C.R.; Kelley, C.M.; Mufson, E.J. Brainstem Alzheimer’s-like pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 35, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.Z.; Bayer, T.A.; Wirths, O. Intracellular Aβ triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramár, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci. Data 2021, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Kosel, F.; Pelley, J.M.S.; Franklin, T.B. Behavioural and psychological symptoms of dementia in mouse models of Alzheimer’s disease-related pathology. Neurosci. Biobehav. Rev. 2020, 112, 634–647. [Google Scholar] [CrossRef]
- España, J.; Giménez-Llort, L.; Valero, J.; Miñano, A.; Rábano, A.; Rodriguez-Alvarez, J.; LaFerla, F.M.; Saura, C.A. Intraneuronal β-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer’s disease transgenic mice. Biol. Psychiatry 2010, 67, 513–521. [Google Scholar] [CrossRef]
- Toda, T.; Noda, Y.; Ito, G.; Maeda, M.; Shimizu, T. Presenilin-2 mutation causes early amyloid accumulation and memory impairment in a transgenic mouse model of Alzheimer’s disease. BioMed Res. Int. 2011, 2011, 617974. [Google Scholar] [CrossRef] [PubMed]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; King, D.L.; Gordon, M.N.; Morgan, D.; Hatcher, J.M.; Hope, C.E.; Diamond, D.M. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 2001, 891, 42–53. [Google Scholar] [CrossRef]
- Shruster, A.; Offen, D. Targeting neurogenesis ameliorates danger assessment in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2014, 261, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Filali, M.; Lalonde, R.; Theriault, P.; Julien, C.; Calon, F.; Planel, E. Cognitive and non-cognitive behaviors in the triple transgenic mouse model of Alzheimer’s disease expressing mutated APP, PS1, and Mapt (3xTg-AD). Behav. Brain Res. 2012, 234, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, R.; Higuchi, S.; Nakatsuji, M.; Yasui, M.; Ikedo, T.; Nagata, M.; Hayashi, K.; Yokode, M.; Minami, M. Deficiency in ep4 receptor–associated protein ameliorates abnormal anxiety-like behavior and brain inflammation in a mouse model of alzheimer disease. Am. J. Pathol. 2017, 187, 1848–1854. [Google Scholar] [CrossRef]
- Pietropaolo, S.; Sun, Y.; Li, R.; Brana, C.; Feldon, J.; Yee, B.K. Limited impact of social isolation on Alzheimer-like symptoms in a triple transgenic mouse model. Behav. Neurosci. 2009, 123, 181. [Google Scholar] [CrossRef] [PubMed]
- Pugh, P.L.; Richardson, J.C.; Bate, S.T.; Upton, N.; Sunter, D. Non-cognitive behaviours in an APP/PS1 transgenic model of Alzheimer’s disease. Behav. Brain Res. 2007, 178, 18–28. [Google Scholar] [CrossRef]
- Sethi, M.; Joshi, S.S.; Webb, R.L.; Beckett, T.L.; Donohue, K.D.; Murphy, M.P.; O’Hara, B.F.; Duncan, M.J. Increased fragmentation of sleep–wake cycles in the 5XFAD mouse model of Alzheimer’s disease. Neuroscience 2015, 290, 80–89. [Google Scholar] [CrossRef]
- Duncan, M.J.; Smith, J.T.; Franklin, K.M.; Beckett, T.L.; Murphy, M.P.; Clair, D.K.S.; Donohue, K.D.; Striz, M.; O’Hara, B.F. Effects of aging and genotype on circadian rhythms, sleep, and clock gene expression in APPxPS1 knock-in mice, a model for Alzheimer’s disease. Exp. Neurol. 2012, 236, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Sterniczuk, R.; Dyck, R.H.; LaFerla, F.M.; Antle, M.C. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: Part 1. Circadian changes. Brain Res. 2010, 1348, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, S.A. A noncholinergic action of acetylcholinesterase (AChE) in the brain: From neuronal secretion to the generation of movement. Cell Mol. Neurobiol. 1991, 11, 55–77. [Google Scholar] [CrossRef] [PubMed]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Garcia Ratés, S.; García-Ayllón, M.-S.; Falgàs, N.; Brangman, S.A.; Esiri, M.M.; Coen, C.W.; Greenfield, S.A. Evidence for a novel neuronal mechanism driving Alzheimer’s disease, upstream of amyloid. Alzheimer’s Dement. 2024. Online ahead of print. [Google Scholar] [CrossRef]
- Greenfield, S.A.; Ferrati, G.; Coen, C.W.; Vadisiute, A.; Molnár, Z.; Garcia-Rates, S.; Frautschy, S.; Cole, G.M. Characterization of a Bioactive Peptide T14 in the Human and Rodent Substantia Nigra: Implications for Neurodegenerative Disease. Int. J. Mol. Sci. 2022, 23, 13119. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ratés, S.; Morrill, P.; Tu, H.; Pottiez, G.; Badin, A.S.; Tormo-Garcia, C.; Heffner, C.; Coen, C.W.; Greenfield, S.A. (I) Pharmacological profiling of a novel modulator of the α7 nicotinic receptor: Blockade of a toxic acetylcholinesterase-derived peptide increased in Alzheimer brains. Neuropharmacology 2016, 105, 487–499. [Google Scholar] [CrossRef]
- Brai, E.; Simon, F.; Cogoni, A.; Greenfield, S.A. Modulatory Effects of a Novel Cyclized Peptide in Reducing the Expression of Markers Linked to Alzheimer’s Disease. Front. Neurosci. 2018, 12, 362. [Google Scholar] [CrossRef]
- Mesulam, M.M.; Geula, C. Nucleus basalis (Ch4) and cortical cholinergic innervation in the human brain: Observations based on the distribution of acetylcholinesterase and choline acetyltransferase. J. Comp. Neurol. 1988, 275, 216–240. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).