
In Vitro Hypoxia/Reoxygenation Induces Mitochondrial Cardiolipin Remodeling in Human Kidney Cells

 ,
, 

Abstract
:1. Introduction
2. Results
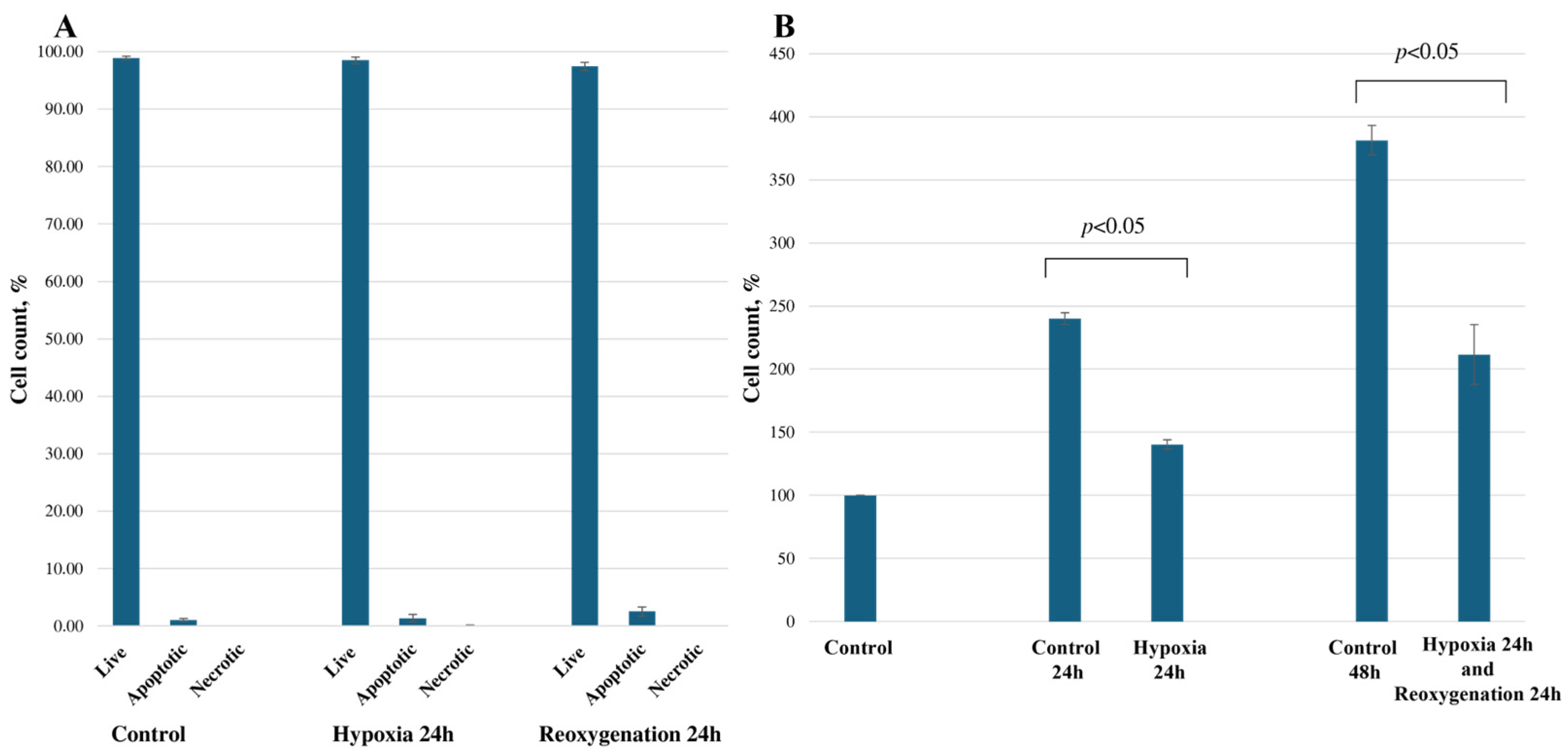
2.1. Hypoxia/Reoxygenation Reduces RPTEC Cell Proliferation
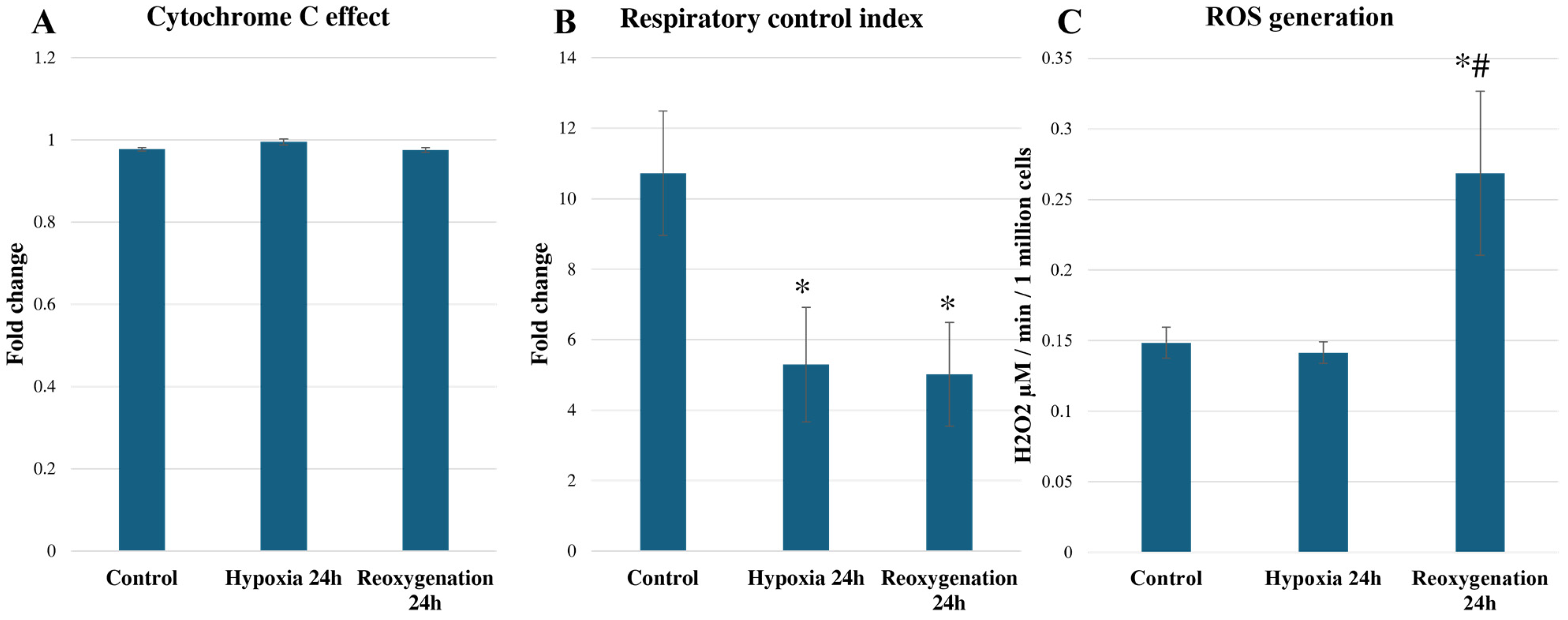
2.2. Hypoxia/Reoxygenation Reduces RPTEC Mitochondrial Respiration and Increases ROS Generation
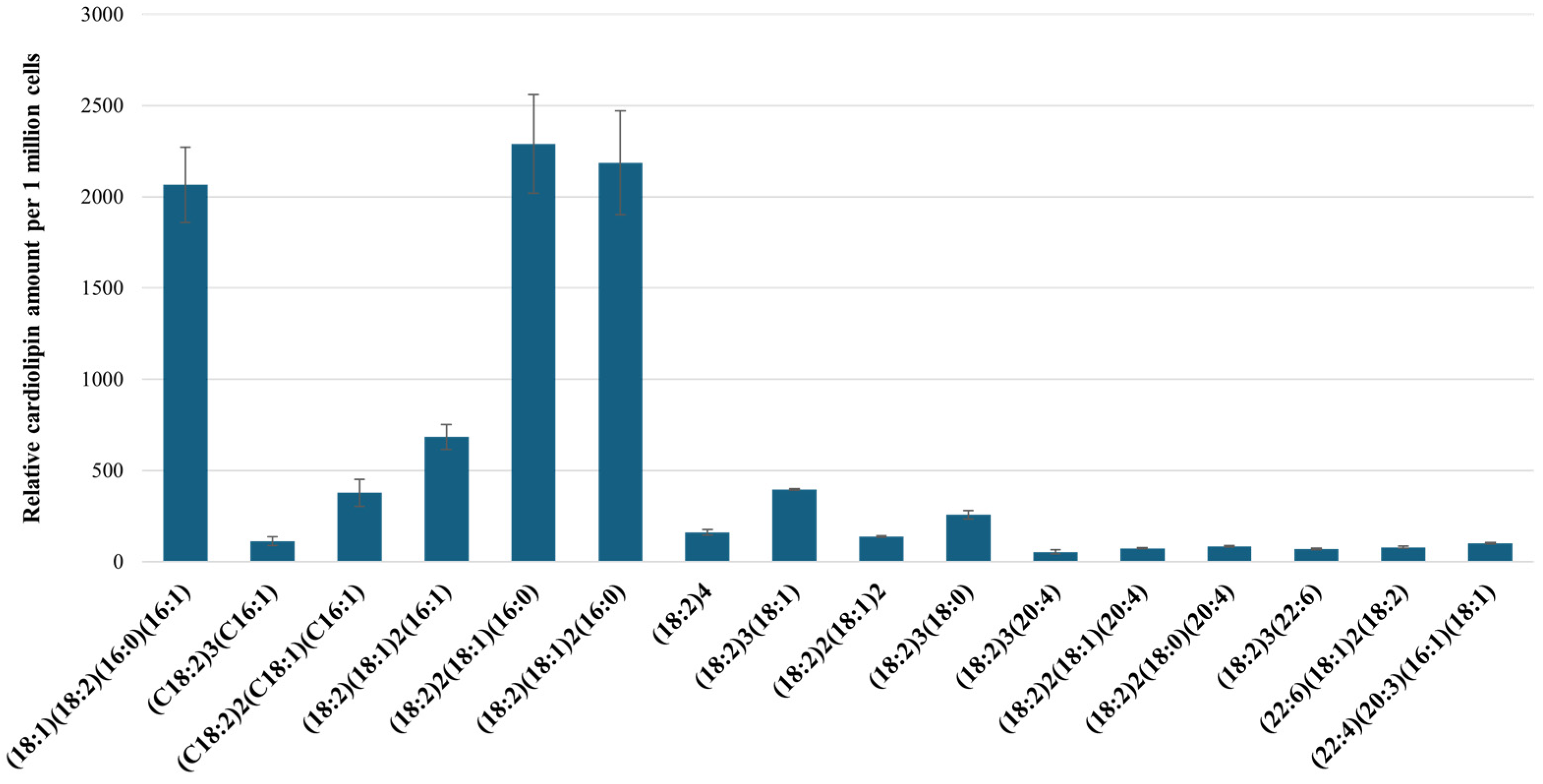
2.3. Characterization of RPTEC Cardiolipins Using UPLC-MS/MS
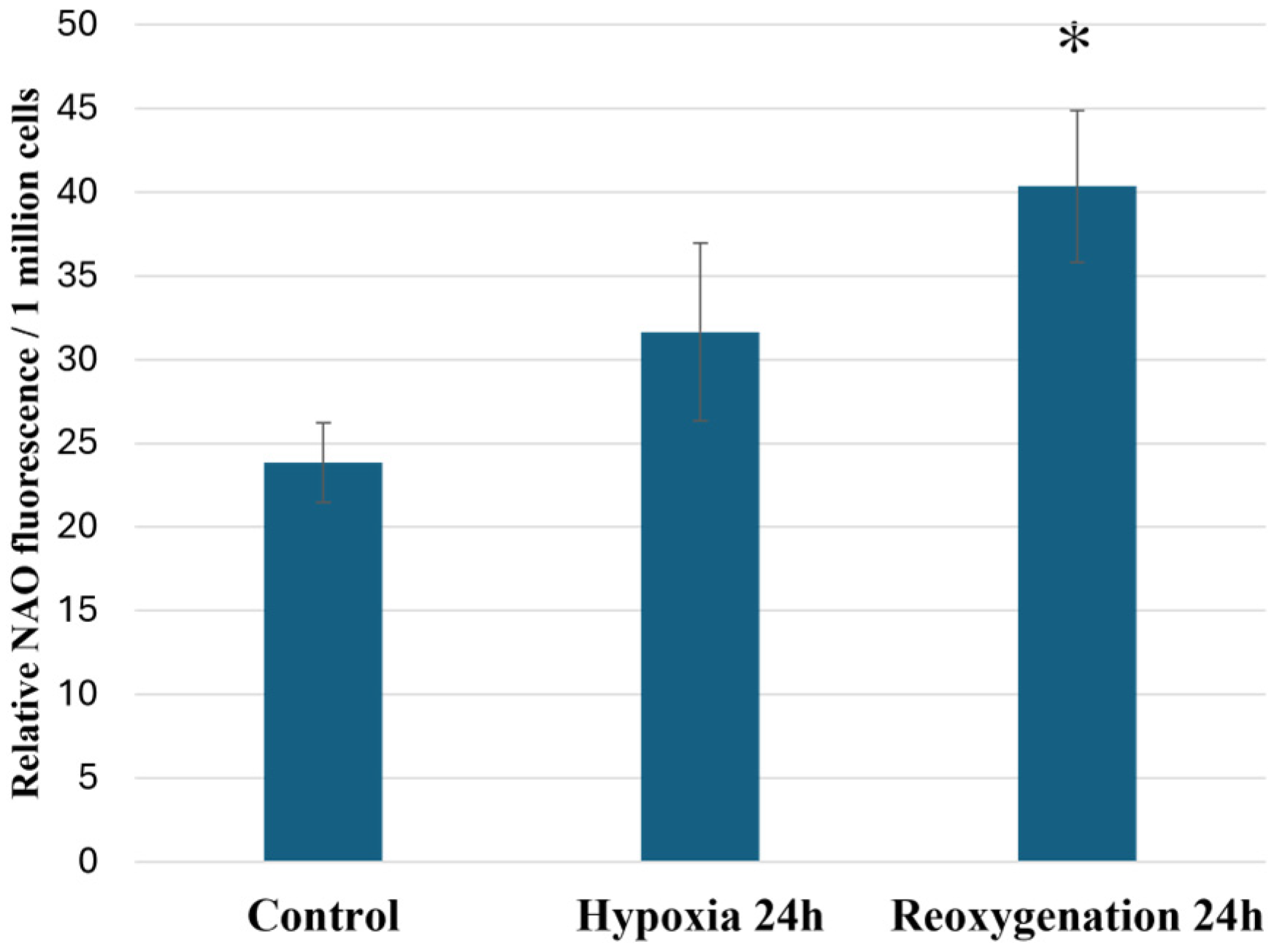
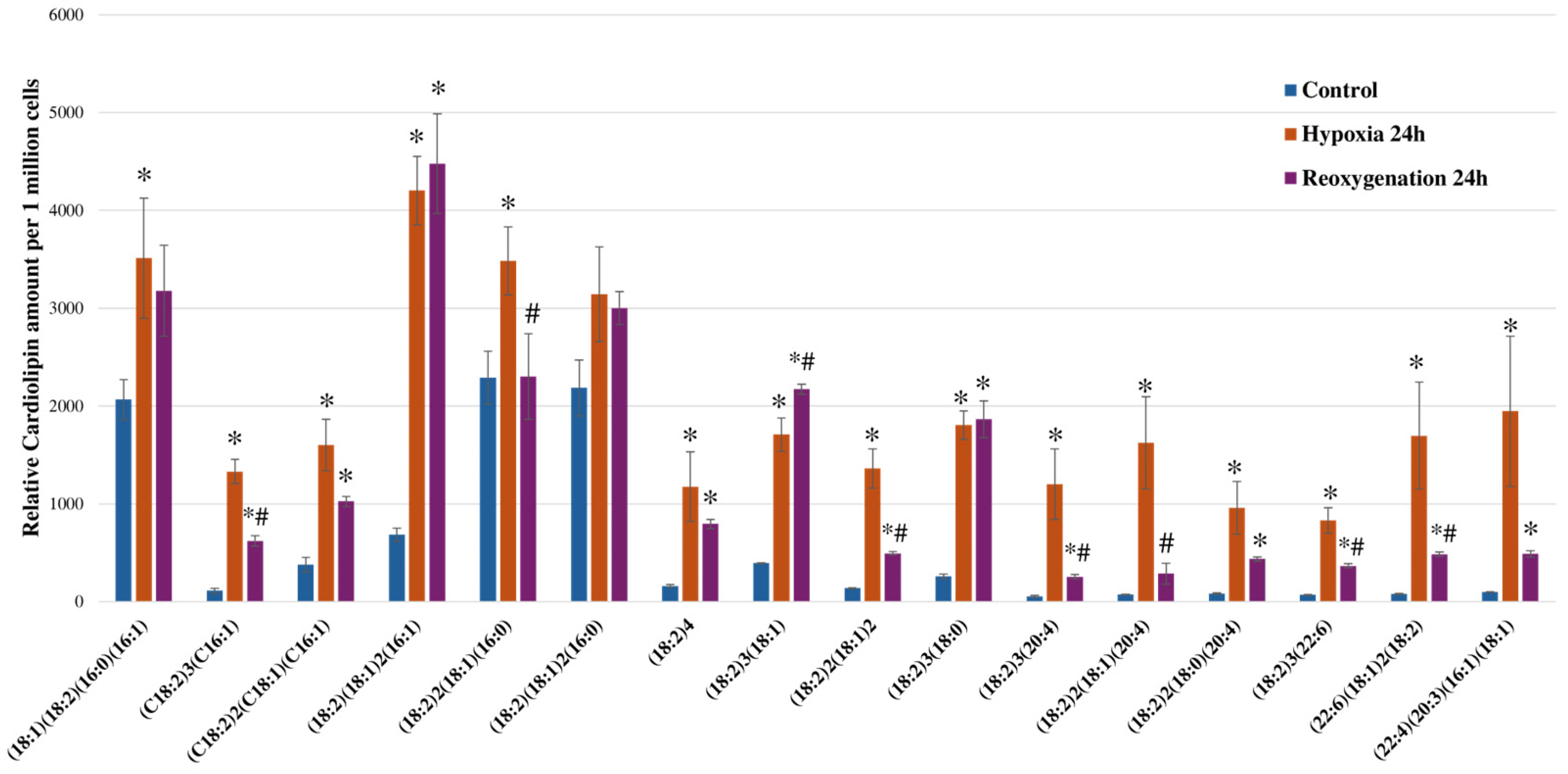
2.4. Hypoxia and Reoxygenation Increases Cardiolipin Amounts in RPTEC Cells
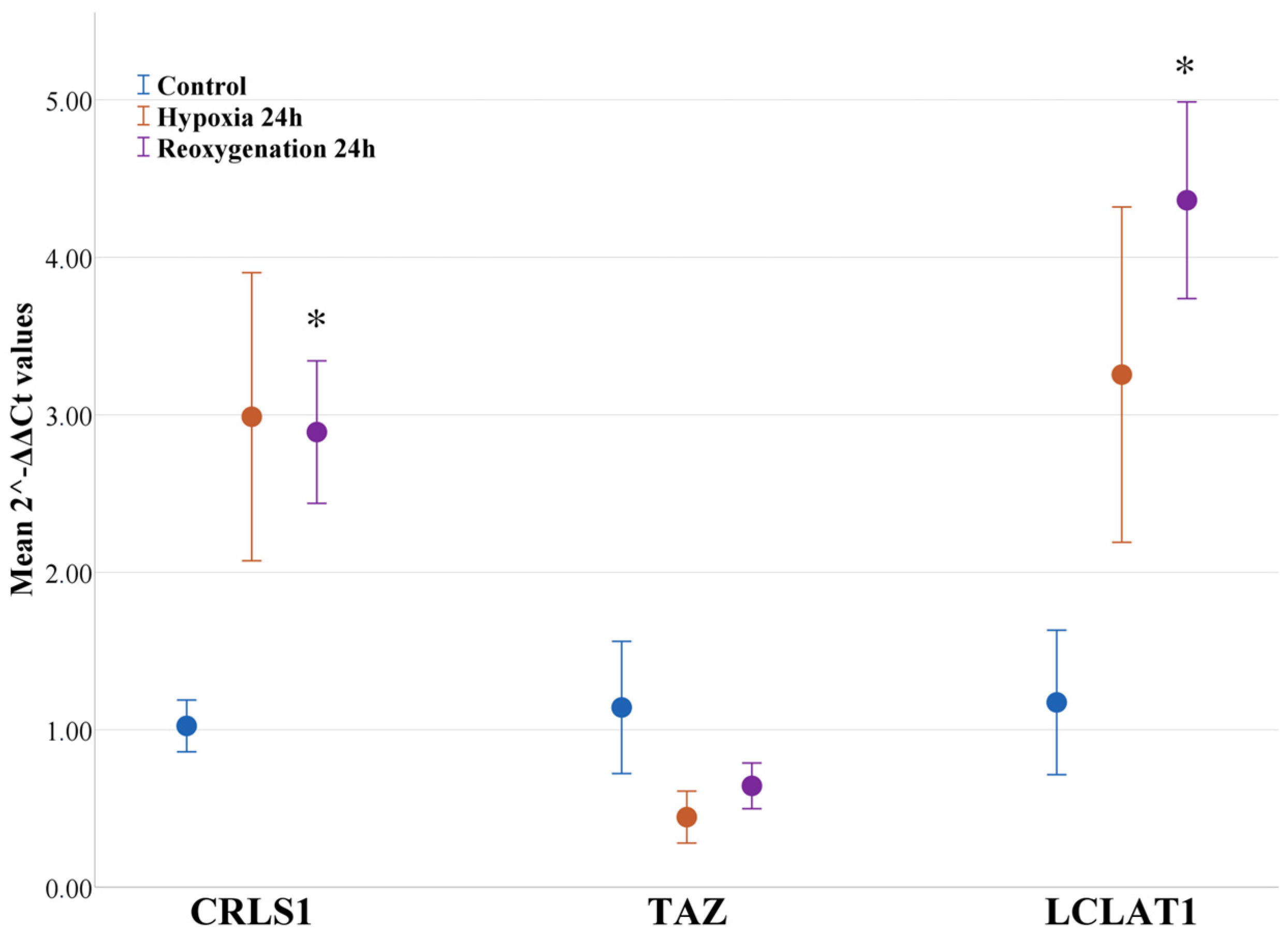
2.5. Hypoxia and Reoxygenation Increases the Expression of Genes and the Respective Enzymes Involved in Cardiolipin Remodeling
3. Discussion
4. Materials and Method
4.1. Cell Culture
4.2. Induction of Hypoxia/Reoxygenation
4.3. Cell Viability Assessment
4.4. Mitochondrial Respiration Assessment
4.5. Analysis of H2O2 Production
4.6. Total Cardiolipin Content Fluorometric Analysis
4.7. Lipid Extraction
4.8. Cardiolipin Analysis Using UPLC-MS/MS
4.9. Gene Expression Analysis Using RT-qPCR
4.10. Analysis of Cardiolipin Synthesis/Remodeling Enzyme Levels in RPTEC Cells
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar] [PubMed]
- Turgut, F.; Awad, A.S.; Abdel-Rahman, E.M. Acute Kidney Injury: Medical Causes and Pathogenesis. J. Clin. Med. 2023, 12, 375. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuijs-Moeke, G.J.; Pischke, S.E.; Berger, S.P.; Sanders, J.S.F.; Pol, R.A.; Struys, M.M.R.F.; Ploeg, R.J.; Leuvenink, H.G.D. Ischemia and reperfusion injury in kidney transplantation: Relevant mechanisms in injury and repair. J. Clin. Med. 2020, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Huang, M.; Zeng, X.; Zhang, Y.; Sun, A.; Wu, Q.; Zhu, L.; Zhao, H.; Liao, Y. The Role of TRPC6 in Renal Ischemia/Reperfusion and Cellular Hypoxia/Reoxygenation Injuries. Front. Mol. Biosci. 2021, 8, 698975. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhu, J.; Miao, C.; Wang, S.; Zhang, L.; Li, P.; Wang, Z.; Shao, P. Protective Effects of the Segmental Renal Artery Clamping Technique on Ischemia-Reperfusion Injury in db/db Diabetic Mice. BioMed Res. Int. 2017, 2017, 4763828. [Google Scholar] [CrossRef] [PubMed]
- Miguel, V.; Rojo, A. Hypoxia-Driven Responses in Chronic Kidney Disease. Oxygen 2023, 3, 300–321. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 2015, 4, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.; Chicco, A.J.; Ball, D.; Chen, T.W. Analysis of mitochondrial oxygen consumption and hydrogen peroxide release from cardiac mitochondria using electrochemical multi-sensors. Sens. Actuators B Chem. 2022, 360, 131641. [Google Scholar] [CrossRef]
- Wefers Bettink, M.A.; Arbous, M.S.; Raat, N.J.H.; Mik, E.G. Mind the mitochondria! J. Emerg. Crit. Care Med. 2019, 3, 45. [Google Scholar] [CrossRef]
- Martens, J.C.; Keilhoff, G.; Gardemann, A.; Schild, L. Oxidation of cardiolipin is involved in functional impairment and disintegration of liver mitochondria by hypoxia/ reoxygenation in the presence of increased Ca2+ concentrations. Mol. Cell. Biochem. 2014, 394, 119–127. [Google Scholar] [CrossRef]
- Adzigbli, L.; Sokolov, E.P.; Wimmers, K.; Sokolova, I.M.; Ponsuksili, S. Effects of hypoxia and reoxygenation on mitochondrial functions and transcriptional profiles of isolated brain and muscle porcine cells. Sci. Rep. 2022, 12, 19881. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Filippidis, G.; Liakopoulos, V.; Stefanidis, I. Reoxygenation induces reactive oxygen species production and ferroptosis in renal tubular epithelial cells by activating aryl hydrocarbon receptor. Mol. Med. Rep. 2021, 23, 41. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta—Bioenerg. 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Dudek, J. Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.E.; Pennington, E.R.; Perry, J.B.; Dadoo, S.; Makrecka-Kuka, M.; Dambrova, M.; Moukdar, F.; Patel, H.D.; Han, X.; Kidd, G.K.; et al. The cardiolipin-binding peptide elamipretide mitigates fragmentation of cristae networks following cardiac ischemia reperfusion in rats. Commun. Biol. 2020, 3, 389. [Google Scholar] [CrossRef] [PubMed]
- Planas-Iglesias, J.; Dwarakanath, H.; Mohammadyani, D.; Yanamala, N.; Kagan, V.E.; Klein-Seetharaman, J. Cardiolipin Interactions with Proteins. Biophys. J. 2015, 109, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Wasmus, C.; Dudek, J. Metabolic alterations caused by defective cardiolipin remodeling in inherited cardiomyopathies. Life 2020, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Gao, J.; Siira, S.J.; Shearwood, A.M.; Ermer, J.A.; Hofferek, V.; Mathews, J.C.; Zheng, M.; Reid, G.E.; Rackham, O.; et al. Cardiolipin is required for membrane docking of mitochondrial ribosomes and protein synthesis. J. Cell Sci. 2020, 133, jcs240374. [Google Scholar] [CrossRef]
- Chao, H.; Lin, C.; Zuo, Q.; Liu, Y.; Xiao, M.; Xu, X.; Li, Z.; Bao, Z.; Chen, H.; You, Y.; et al. Cardiolipin-dependent mitophagy guides outcome after traumatic brain injury. J. Neurosci. 2019, 39, 1930–1943. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Li, X.X.; Tsoi, B.; Li, Y.F.; Kurihara, H.; He, R.R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta—Biomembr. 2009, 1788, 2022–2031. [Google Scholar] [CrossRef]
- Oemer, G.; Koch, J.; Wohlfarter, Y.; Alam, M.T.; Lackner, K.; Sailer, S.; Neumann, L.; Lindner, H.H.; Watschinger, K.; Haltmeier, M.; et al. Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Rep. 2020, 30, 4281–4291. [Google Scholar] [CrossRef] [PubMed]
- Raja, V.; Greenberg, M.L. The functions of cardiolipin in cellular metabolism-potential modifiers of the Barth syndrome phenotype. Chem. Phys. Lipids 2014, 179, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.C.; Keilhoff, G.; Halangk, W.; Wartmann, T.; Gardemann, A.; Päge, I.; Schild, L. Lipidomic analysis of molecular cardiolipin species in livers exposed to ischemia/reperfusion. Mol. Cell. Biochem. 2015, 400, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Ziemann, M.; Thallas-Bonke, V.; Snelson, M.; Kumar, V.; Laskowski, A.; Nguyen, T.-V.; Huynh, K.; Clarke, M.V.; Libianto, R.; et al. Complement C5a induces renal injury in diabetic kidney disease by disrupting mitochondrial metabolic agility. Diabetes 2020, 69, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, B.; Miller, S.; Yuan, H.; Zhang, H.; Tian, L.; Nie, J.; Imae, R.; Arai, H.; Li, Y.; et al. Ablation of ALCAT1 Mitigates Hypertrophic Cardiomyopathy through Effects on Oxidative Stress and Mitophagy. Mol. Cell. Biol. 2012, 32, 4493–4504. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Baart, S.; Vikulina, A.S.; Clark, R.S.B.; Anthonymuthu, T.S.; Tyurin, V.A.; Du, L.; Croix, C.M.S.; Tyurina, Y.Y.; Lewis, J.; et al. Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion. J. Cereb. Blood Flow Metab. 2015, 35, 319–328. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Minkler, P.; Hoppel, C.L. Enhanced modification of cardiolipin during ischemia in the aged heart. J. Mol. Cell. Cardiol. 2009, 46, 1008–1015. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: Implications for pharmacological cardioprotection. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef] [PubMed]
- Heerdt, P.M.; Schlame, M.; Jehle, R.; Barbone, A.; Burkhoff, D.; Blanck, T.J.J. Disease-specific remodeling of cardiac mitochondria after a left ventricular assist device. Ann. Thorac. Surg. 2002, 73, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Rieger, B.; Krajčová, A.; Duwe, P.; Busch, K.B. ALCAT1 Overexpression Affects Supercomplex Formation and Increases ROS in Respiring Mitochondria. Oxid. Med. Cell. Longev. 2019, 2019, 9186469. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Zhang, J.; Nie, J.; Andersen, J.P.; Rendon, S.; Zheng, Y.; Liu, X.; Tian, Z.; Shi, Y. Cardiolipin remodeling by ALCAT1 links hypoxia to coronary artery disease by promoting mitochondrial dysfunction. Mol. Ther. 2021, 29, 3498–3511. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Fan, Y.; Feng, J.; Zhu, Z.; Luo, Z.; Hu, H.; Li, W.; Yang, H.; Ding, G. ALCAT1-mediated abnormal cardiolipin remodelling promotes mitochondrial injury in podocytes in diabetic kidney disease. Cell Commun. Signal. 2024, 22, 26. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hajnóczky, G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Díaz, A.G.; Cardona-Muñoz, E.G.; Pacheco-Moisés, F.P. The Role of Cardiolipin and Mitochondrial Damage in Kidney Transplant. Oxid. Med. Cell. Longev. 2019, 2019, 3836186. [Google Scholar] [CrossRef] [PubMed]
- Jassem, W.; Heaton, N.D. The role of mitochondria in ischemia/reperfusion injury in organ transplantation. Kidney Int. 2004, 66, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol.-Ren. Physiol. 2012, 302, 853–864. [Google Scholar] [CrossRef]
- Gan, E.S.; Ooi, E.E. Oxygen: Viral friend or foe? Virol. J. 2020, 17, 115. [Google Scholar] [CrossRef]
- Zheng, W.; Gu, X.; Sun, X.; Hu, D. Effects of hypoxia-inducible factor-1α on the proliferation and apoptosis of human synovial mesenchymal stem cells. Mol. Med. Rep. 2019, 20, 4315–4322. [Google Scholar] [CrossRef] [PubMed]
- Shiva, N.; Sharma, N.; Kulkarni, Y.A.; Mulay, S.R.; Gaikwad, A.B. Renal ischemia/reperfusion injury: An insight on in vitro and in vivo models. Life Sci. 2020, 256, 117860. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Vunjak-Novakovic, G. In Vitro Models of Ischemia-Reperfusion Injury. Regen. Eng. Transl. Med. 2018, 4, 142–153. [Google Scholar] [CrossRef]
- Giraud, S.; Thuillier, R.; Cau, J.; Hauet, T. In vitro/ex vivo models for the study of ischemia reperfusion injury during kidney perfusion. Int. J. Mol. Sci. 2020, 21, 8156. [Google Scholar] [CrossRef]
- Vormann, M.K.; Tool, L.M.; Ohbuchi, M.; Gijzen, L.; Van Vught, R.; Hankemeier, T.; Kiyonaga, F.; Kawabe, T.; Goto, T.; Fujimori, A.; et al. Modelling and Prevention of Acute Kidney Injury through Ischemia and Reperfusion in a Combined Human Renal Proximal Tubule/Blood Vessel-on-a-Chip. Kidney360 2022, 3, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhang, X.; Ma, C.; Chen, Y. TGF-β/smad3 pathway enhances the cardio-protection of S1R/SIPR1 in in vitro ischemia-reperfusion myocardial cell model. Exp. Ther. Med. 2018, 16, 178–184. [Google Scholar] [CrossRef]
- Sebastião, M.J.; Serra, M.; Pereira, R.; Palacios, I.; Gomes-Alves, P.; Alves, P.M. Human cardiac progenitor cell activation and regeneration mechanisms: Exploring a novel myocardial ischemia/reperfusion in vitro model. Stem Cell Res. Ther. 2019, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Prather, E.R.; Garrison, D.E.; Zuo, L. Interplay between ROS and antioxidants during ischemia-reperfusion injuries in cardiac and skeletal muscle. Int. J. Mol. Sci. 2018, 19, 417. [Google Scholar] [CrossRef] [PubMed]
- Pedriali, G.; Ramaccini, D.; Bouhamida, E.; Wieckowski, M.R.; Giorgi, C.; Tremoli, E.; Pinton, P. Perspectives on mitochondrial relevance in cardiac ischemia/reperfusion injury. Front. Cell Dev. Biol. 2022, 10, 1082095. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, J.; Zhou, Q.; He, X.; Zheng, Z.; Wei, Y.; Zhou, K.; Lin, Y.; Yu, H.; Zhang, H.; et al. Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation. Cell Res. 2024, 34, 13–30. [Google Scholar] [CrossRef]
- Yu, F.; Meng, Y.; Chen, K.; He, Y. Wcn23-0490 Protein Lactylation Leads to the Chronic Transition of Acute Kidney Injury by Activating Inflammasomes. Kidney Int. Rep. 2023, 8 (Suppl. S3), S32. [Google Scholar] [CrossRef]
- Liss, K.H.H.; Mousa, M.; Bucha, S.; Lutkewitte, A.; Allegood, J.; Cowart, L.A.; Finck, B.N. Dynamic changes in the mouse hepatic lipidome following warm ischemia reperfusion injury. Sci. Rep. 2024, 14, 3584. [Google Scholar] [CrossRef]
- Wu, D.; An, Q.; Ji, H.; Dai, J.; Suo, L.; Zhang, C. Retinal ischemia-reperfusion injury induces intense lipid synthesis and remodeling. Biochem. Biophys. Res. Commun. 2023, 689, 149232. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J. Biomed. Res. 2010, 24, 6–15. [Google Scholar] [CrossRef]
- Bradley, R.M.; Stark, K.D.; Duncan, R.E. Influence of tissue, diet, and enzymatic remodeling on cardiolipin fatty acyl profile. Mol. Nutr. Food Res. 2016, 60, 1804–1818. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Javadov, S. Unraveling the mechanisms of cardiolipin function: The role of oxidative polymerization of unsaturated acyl chains. Redox Biol. 2023, 64, 102774. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in Mitochondrial Complex I Activity in Ischemic/Reperfused Rat Heart: Involvement of Reactive Oxygen Species and Cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef]
- Petrosillo, G.; Di Venosa, N.; Pistolese, M.; Casanova, G.; Tiravanti, E.; Colantuono, G.; Federici, A.; Paradies, G.; Ruggiero, F.M. Protective effect of melatonin against mitochondrial dysfunction associated with cardiac ischemia-reperfusion: Role of cardiolipin. FASEB J. 2006, 20, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Strazdauskas, A.; Trumbeckaite, S.; Jakstas, V.; Kamarauskaite, J.; Ivanauskas, L.; Baniene, R. Ischemia In Vivo Induces Cardiolipin Oxidation in Rat Kidney Mitochondria. Biology 2022, 11, 541. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proposed CL FA Composition | CL Mass, m/z | Diacylglycerol Phosphate Fragment, m/z | Fatty Acid Fragment, m/z | Retention Factor (CL Retention Time/IS Retention Time) |
|---|---|---|---|---|
| (18:1)(18:2)(16:0)(16:1) | 1400 | 671 | 281, 253 | 1.16 |
| (C18:2)3(C16:1) | 1422 | 695 | 279, 253 | 0.99 |
| (C18:2)2(C18:1)(C16:1) | 1424 | 669, 671, 697 | 279, 253 | 1.06 |
| (18:2)(18:1)2(16:1) | 1426 | 671, 697, 728 | 253 | 1.15 |
| (18:2)2(18:1)(16:0) | 1426 | 671, 695 | 279, 255 | 1.16 |
| (18:2)(18:1)2(16:0) | 1428 | 697 | 281, 279, 255 | 1.23 |
| (18:2)4 | 1448 | 695 | 279 | 0.995 |
| (18:2)3(18:1) | 1450 | 697 | 279, 281 | 1.08 |
| (18:2)2(18:1)2 | 1452 | 695 | 281, 279 | 0.998 |
| (18:2)3(18:0) | 1452 | 695, 699 | 283, 279 | 1.07 |
| (18:2)3(20:4) | 1472 | 695, 719 | 279, 303 | 0.92 |
| (18:2)2(18:1)(20:4) | 1474 | 719 | 279, 281, 303 | 0.98 |
| (18:2)2(C18:0)(20:3) | 1476 | 723, 699, 695 | 279, 283, 303 | 0.981 |
| (18:2)3(22:6) | 1496 | 695 | 279, 327 | 0.98 |
| (22:6)(18:1)2(18:2) | 1500 | 697 | 279, 281, 327 | 1.057 |
| (22:4)(20:3)(16:1)(18:1) | 1502 | 721, 723 | 281, 305, 331 | 1.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strazdauskas, A.; Trumbeckaite, S.; Jakstas, V.; Dambrauskiene, J.; Mieldazyte, A.; Klimkaitis, K.; Baniene, R. In Vitro Hypoxia/Reoxygenation Induces Mitochondrial Cardiolipin Remodeling in Human Kidney Cells. Int. J. Mol. Sci. 2024, 25, 6223. https://doi.org/10.3390/ijms25116223
Strazdauskas A, Trumbeckaite S, Jakstas V, Dambrauskiene J, Mieldazyte A, Klimkaitis K, Baniene R. In Vitro Hypoxia/Reoxygenation Induces Mitochondrial Cardiolipin Remodeling in Human Kidney Cells. International Journal of Molecular Sciences. 2024; 25(11):6223. https://doi.org/10.3390/ijms25116223
Chicago/Turabian StyleStrazdauskas, Arvydas, Sonata Trumbeckaite, Valdas Jakstas, Justina Dambrauskiene, Ausra Mieldazyte, Kristupas Klimkaitis, and Rasa Baniene. 2024. "In Vitro Hypoxia/Reoxygenation Induces Mitochondrial Cardiolipin Remodeling in Human Kidney Cells" International Journal of Molecular Sciences 25, no. 11: 6223. https://doi.org/10.3390/ijms25116223
APA StyleStrazdauskas, A., Trumbeckaite, S., Jakstas, V., Dambrauskiene, J., Mieldazyte, A., Klimkaitis, K., & Baniene, R. (2024). In Vitro Hypoxia/Reoxygenation Induces Mitochondrial Cardiolipin Remodeling in Human Kidney Cells. International Journal of Molecular Sciences, 25(11), 6223. https://doi.org/10.3390/ijms25116223