
Triple Blockade of Oncogenic RAS Signaling Using KRAS and MEK Inhibitors in Combination with Irradiation in Pancreatic Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Combination Treatment with KRAS- and MEK-Inhibitor Plus Irradiation Inhibits Cell Growth of KRAS Mutant Pancreatic Cancer Cells
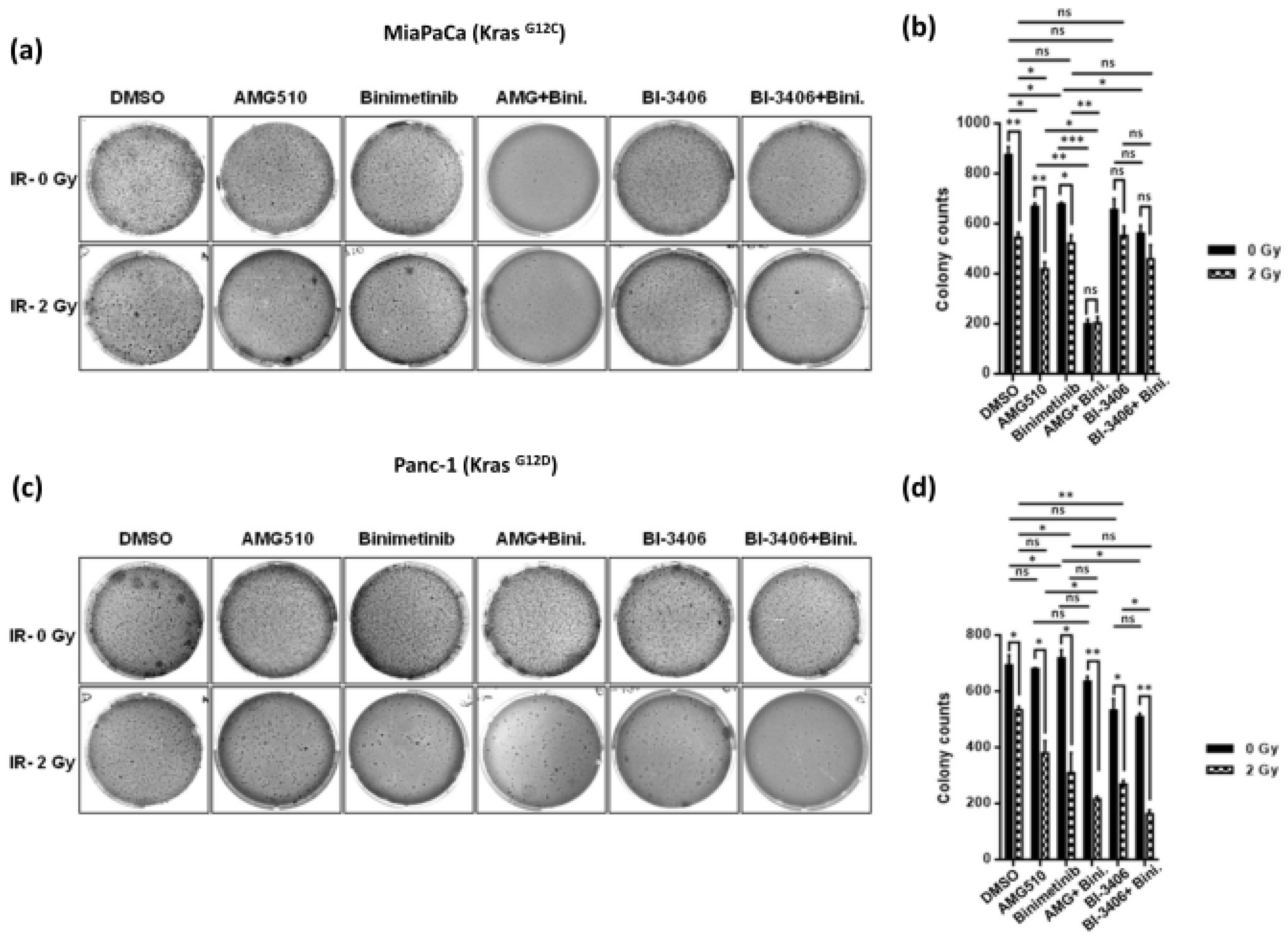
2.2. Combination Treatment Using KRAS- and MEK-Inhibitors Radiosensitizes KRAS Mutant PDAC Cells and Inhibits Clonogenicity
2.3. Combined KRAS and MEK Inhibition plus Irradiation Leads to Enhanced Attenuation of 3D Anchorage Independent Colony Growth in KRAS Mutant PDAC Cells
2.4. KRAS Inhibitors AMG510 and BI-3406 Inhibit KRAS Activation and Signaling through the RAS/MAPK Pathway in KRAS Mutant Pancreatic Cancer Cells
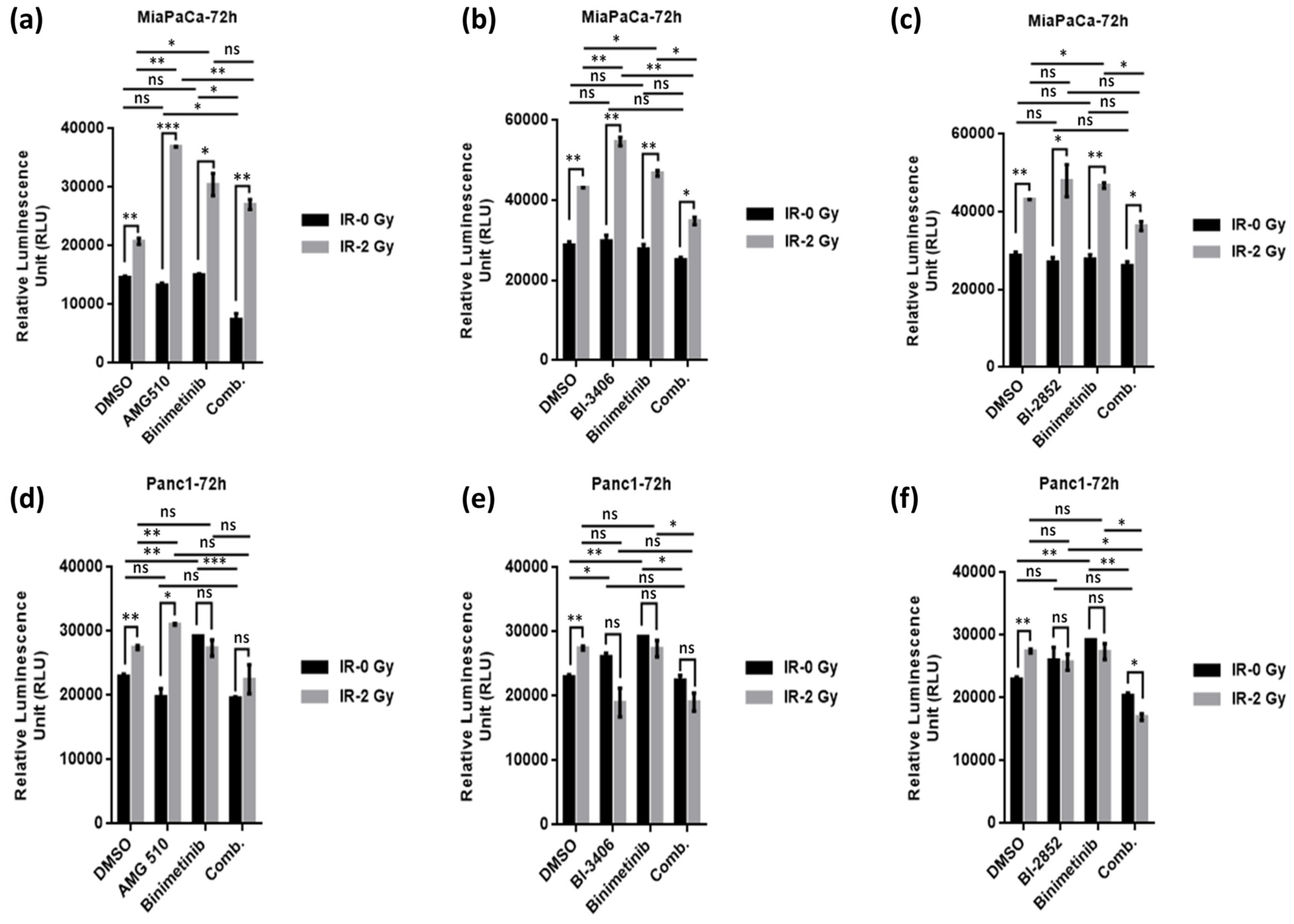
2.5. KRAS/MEK Inhibition Sensitizes KRAS Mutant Pancreatic Cancer Cells to Therapeutic Irradiation and Induces Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell lines and Drugs
4.2. Irradiation
4.3. Cell Viability Assay
4.4. Clonogenicity (2D) Assays
4.5. Anchorage Independent (3D) Soft Agar Clonogenic Assays
4.6. Western Blots
4.7. RealTime-GloTM Annexin V Apoptosis Assay
4.8. Active RAS Pull-Down Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMG-510 | Sotorasib -KRAS G12C inhibitor |
| BI-2852 | KRAS switch I/II pocket inhibitor |
| BI-3406 | SOS1::KRAS inhibitor |
| Binimetinib | MEK inhibitor |
| BRAF | Murine Sarcoma Viral (V-Raf) Oncogene Homolog B1 |
| EGFR | Epidermal Growth Factor Receptor |
| ERK | Extracellular -signal- Regulated Kinase |
| KRAS | Kirsten Rat Sarcoma viral oncogene |
| MAPK (MEK) | Mitogen -Activated Protein Kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| RAF | Rapidly Accelerated Fibrosarcoma |
| RAF-MAPK | RAF-mitogen activated protein kinase |
| RAL | Ras Linked Protein |
| RALGDS | RAL Guanine nucleotide Dissociation Stimulator |
| RAS | Rat Sarcoma |
| SOS1 | Son Of Sevenless homolog 1 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F.; Bsc, M.F.B.; Me, J.F.; Soerjomataram, M.I.; et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-F.; Ye, Z.; Qin, Y.; Xu, X.-W.; Yu, X.-J.; Zhuo, Q.-F.; Ji, S.-R. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741. [Google Scholar] [CrossRef] [PubMed]
- Huffman, B.M.; Ellis, H.; Jordan, A.C.; Freed-Pastor, W.A.; Perez, K.; Rubinson, D.A.; Sethi, N.; Singh, H.; Surana, R.; Wolpin, B.M.; et al. Emerging Role of Targeted Therapy in Metastatic Pancreatic Adenocarcinoma. Cancers 2022, 14, 6223. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hu, K.; Bailey, P.; Springfeld, C.; Roth, S.; Kurilov, R.; Brors, B.; Gress, T.; Buchholz, M.; An, J.; et al. Clinical impact of molecular subtyping of pancreatic cancer. Front. Cell Dev. Biol. 2021, 9, 743908. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef]
- Feldmann, G.; Maitra, A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J. Mol. Diagn. 2008, 10, 111–122. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and Kras G12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepato-Biliary-Pancreat. Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Muratcioglu, S.; Tsai, C.J.; Jang, H.; Gursoy, A.; Keskin, O. The Key Role of Calmodulin in KRAS-Driven Adenocarcinomas. Mol. Cancer Res. 2015, 13, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, A.J.; Solit, D.B. RAF/MEK Dependence of KRAS-Mutant Pancreatic Ductal Adenocarcinomas. Cancer Discov. 2012, 2, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, M.D.; Chen, P.-Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.L.; Kroemer, G.; Kang, R. Oncogenic KRAS blockade therapy: Renewed enthusiasm and persistent challenges. Mol. Cancer 2021, 20, 128. [Google Scholar] [CrossRef]
- Chen, S.-H.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.A.; Whalen, D.M.; Özen, A.; Wongchenko, M.J.; Yin, J.; Yen, I.; Schaefer, G.; Mayfield, J.D.; Chmielecki, J.; Stephens, P.J.; et al. Activation Mechanism of Oncogenic Deletion Mutations in BRAF, EGFR, and HER2. Cancer Cell 2016, 29, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.K.; Neel, N.F.; Hu, C.; Gautam, P.; Chenard, M.; Long, B.; Aziz, M.; Kassner, M.; Bryant, K.L.; Pierobon, M.; et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell 2016, 29, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, E.; Behrens, C.; Lin, H.Y.; Simon, G.; Papadimitrakopoulou, V.; Izzo, J.; Moran, C.; Kalhor, N.; Lee, J.J.; Minna, J.D.; et al. Modulation of EZH2 Expression by MEK-ERK or PI3K-AKT Signaling in Lung Cancer Is Dictated by Different KRAS Oncogene Mutations. Cancer Res. 2016, 76, 675–685. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Zhang, Z.N.; Zhang, H.; Liao, X.; Tsai, H.I. Kras mutation: The booster of pancreatic ductal adenocarcinoma transformation and progression. Front. Cell Dev. Biol. 2023, 11, 1147676. [Google Scholar] [CrossRef]
- Choi, M.; Bien, H.; Mofunanya, A.; Powers, S. Challenges in Ras therapeutics in pancreatic cancer. Semin. Cancer Biol. 2019, 54, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging Ras Back in the Ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.-S.; Janes, M.R.; et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019, 11, eaaw7999. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, K.; Kryzmien, Z.; Koi, L.; Wang, M.; Benes, C.H.; Hering, S.; Willers, H.; Baumann, M.; Krause, M. Radioresistance of KRAS/TP53-mutated lung cancer can be overcome by radiation dose escalation or EGFR tyrosine kinase inhibition in vivo. Int. J. Cancer 2020, 147, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Han, J.; Marcar, L.; Black, J.; Liu, Q.; Li, X.; Nagulapalli, K.; Sequist, L.V.; Mak, R.H.; Benes, C.H.; et al. Radiation Resistance in KRAS-Mutated Lung Cancer Is Enabled by Stem-like Properties Mediated by an Osteopontin-EGFR Pathway. Cancer Res. 2017, 77, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Sklar, M.D. The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science 1988, 239, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, E.J.; McKenna, W.G.; Hamilton, A.D.; Sebti, S.M.; Qian, Y.; Wu, J.M.; Muschel, R.J. Inhibiting Ras prenylation increases the radiosensitivity of human tumor cell lines with activating mutations of ras oncogenes. Cancer Res. 1998, 58, 1754–1761. [Google Scholar] [PubMed]
- McKenna, W.G.; Muschel, R.J.; Gupta, A.K.; Hahn, S.M.; Bernhard, E.J. The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene 2003, 22, 5866–5875. [Google Scholar] [CrossRef] [PubMed]
- Versteijne, E.; van Dam, J.L.; Suker, M.; Janssen, Q.P.; Groothuis, K.; Akkermans-Vogelaar, J.M.; Besselink, M.G.; Bonsing, B.A.; Buijsen, J.; Busch, O.R.; et al. Neoadjuvant Chemoradiotherapy versus Upfront Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Long-Term Results of the Dutch Randomized PREOPANC Trial. J. Clin. Oncol. 2022, 40, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.; Zureikat, A. Neoadjuvant therapy in pancreatic cancer: A review and update on recent trials. Curr. Opin. Gastroenterol. 2022, 38, 521–531. [Google Scholar] [CrossRef]
- Colbert, L.E.; Petrova, A.V.; Fisher, S.B.; Pantazides, B.G.; Madden, M.Z.; Hardy, C.W.; Warren, M.D.; Pan, Y.; Nagaraju, G.P.; Liu, E.A.; et al. CHD7 expression predicts survival outcomes in patients with resected pancreatic cancer. Cancer Res. 2014, 74, 2677–2687. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Brossart, P.; Feldmann, G. Current Therapeutic Options for Pancreatic Ductal Adenocarcinoma. Oncol. Res. Treat. 2018, 41, 590–594. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.; Jain, R.; et al. Open-label, Phase I Study of Nivolumab Combined with nab-Paclitaxel Plus Gemcitabine in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schutz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A.; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2020, 25, e808–e815. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G. Medicinal treatment of pancreatic cancer: Still a domain of chemotherapy? Internist 2020, 61, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef]
- McDonald, E.R.; De Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Xue, J.Y.; Lito, P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell 2020, 183, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.G.; Kopetz, S.; Kuboki, Y.; Kim, T.W.; Munster, P.N.; Krauss, J.C.; Falchook, G.S.; Han, S.W.; Heinemann, V.; Muro, K.; et al. Sotorasib for previously treated colorectal cancers with KRAS(G12C) mutation (CodeBreaK100): A prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022, 23, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.S.; Yaeger, R.; Spira, A.I.; Pelster, M.S.; Sabari, J.K.; Hafez, N.; Barve, M.; Velastegui, K.; Yan, X.; Shetty, A.; et al. Adagrasib in Advanced Solid Tumors Harboring a KRAS(G12C) Mutation. J. Clin. Oncol. 2023, 41, 4097–4106. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.S.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Chafe, S.C.; Schaeffer, D.F.; Li, J.; Renouf, D.J.; Stanger, B.Z.; Dedhar, S. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100131. [Google Scholar] [CrossRef] [PubMed]
- Manchado, E.; Weissmueller, S.; Morris, J.P.; Chen, C.-C.; Wullenkord, R.; Lujambio, A.; de Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016, 534, 647–651. [Google Scholar] [CrossRef]
- Ros, J.; Vaghi, C.; Baraibar, I.; Saoudi González, N.; Rodríguez-Castells, M.; García, A.; Alcaraz, A.; Salva, F.; Tabernero, J.; Elez, E. Targeting KRAS G12C Mutation in Colorectal Cancer, A Review: New Arrows in the Quiver. Int. J. Mol. Sci. 2024, 25, 3304. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Mishra, A.; Hong, S.-M.; Bisht, S.; Strock, C.J.; Ball, D.W.; Goggins, M.; Maitra, A.; Nelkin, B.D. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010, 70, 4460–4469. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, K.K.; McAndrews, K.M.; LeBleu, V.S.; Yang, S.; Lyu, H.; Li, B.; Sockwell, A.M.; Kirtley, M.L.; Morse, S.J.; Diaz, B.A.M.; et al. KRAS(G12D) inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8(+) T cells. Cancer Cell 2023, 41, 1606–1620.e8. [Google Scholar] [CrossRef]
- Dias, E.S.D.; Chung, V. Neoadjuvant treatment for pancreatic cancer: Controversies and advances. Cancer Treat. Res. Commun. 2024, 39, 100804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, W.; Zhu, H.; Chen, X.; Tsai, H.I. Overcoming the limitations of immunotherapy in pancreatic ductal adenocarcinoma: Combining radiotherapy and metabolic targeting therapy. J. Cancer 2024, 15, 2003–2023. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.; Briese, W.; Blieninger, J.; Brossart, P.; Bisht, S.; Feldmann, G. Development of Skin Rash Predicts Outcome of Anti-PD-1- and Anti-CTLA4-Based Immune Checkpoint Inhibitor Therapy in Non-Small Cell Lung Cancer or Squamous Cell Carcinoma of the Head and Neck: A Single-Center Analysis. Oncol. Res. Treat. 2021, 44, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Yousef, A.; Yousef, M.; Chowdhury, S.; Abdilleh, K.; Knafl, M.; Edelkamp, P.; Alfaro-Munoz, K.; Chacko, R.; Peterson, J.; Smaglo, B.G.; et al. Impact of KRAS mutations and co-mutations on clinical outcomes in pancreatic ductal adenocarcinoma. npj Precis. Oncol. 2024, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Li, B.T.; O’Reilly, E.M. Targeting KRAS in cancer. Nat. Med. 2024, 30, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Illert, A.L.; Stenzinger, A.; Bitzer, M.; Horak, P.; Gaidzik, V.I.; Möller, Y.; Beha, J.; Öner, Ö.; Schmitt, F.; Laßmann, S.; et al. The German Network for Personalized Medicine to enhance patient care and translational research. Nat. Med. 2023, 29, 1298–1301. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Miyano, M.; Tsukamoto, S.; Kogai, H.; Yamamoto, A.; Iso, K.; Inoue, S.; Yamane, Y.; Yabe, Y.; Umihara, H.; et al. Delivery of a BET protein degrader via a CEACAM6-targeted antibody-drug conjugate inhibits tumour growth in pancreatic cancer models. Nat. Commun. 2024, 15, 2192. [Google Scholar] [CrossRef]
- Tiriac, H.; Plenker, D.; Baker, L.A.; Tuveson, D.A. Organoid models for translational pancreatic cancer research. Curr. Opin. Genet. Dev. 2019, 54, 7–11. [Google Scholar] [CrossRef]
- van Kuppeveld, F.J.; van der Logt, J.T.; Angulo, A.F.; van Zoest, M.J.; Quint, W.G.; Niesters, H.G.; Galama, J.M.; Melchers, W.J. Genus- and species-specific identification of mycoplasmas by 16S rRNA amplification. Appl. Environ. Microbiol. 1992, 58, 2606–2615. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [PubMed]
- Kessler, D.; Bergner, A.; Boettcher, J.; Fischer, G.; Doebel, S.; Hinkel, M.; Muellauer, B.; Weiss-Puxbaum, A.; McConnell, D.B. Drugging all RAS isoforms with one pocket. Future Med. Chem. 2020, 12, 1911–1923. [Google Scholar] [CrossRef] [PubMed]
- Vlot, A.H.C.; Aniceto, N.; Menden, M.P.; Ulrich-Merzenich, G.; Bender, A. Applying synergy metrics to combination screening data: Agreements, disagreements and pitfalls. Drug Discov. Today 2019, 24, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Feldmann, G.; Soni, S.; Ravi, R.; Karikar, C.; Maitra, A. Polymeric nanoparticle-encapsulated curcumin (“nanocurcumin”): A novel strategy for human cancer therapy. J. Nanobiotechnology 2007, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Nolting, J.; Schütte, U.; Haarmann, J.; Jain, P.; Shah, D.; Brossart, P.; Flaherty, P.; Feldmann, G. Cyclin-Dependent Kinase 5 (CDK5) Controls Melanoma Cell Motility, Invasiveness, and Metastatic Spread-Identification of a Promising Novel therapeutic target. Transl. Oncol. 2015, 8, 295–307. [Google Scholar] [CrossRef]
- Haan, C.; Behrmann, I. A cost effective non-commercial ECL-solution for Western blot detections yielding strong signals and low background. J. Immunol. Methods 2007, 318, 11–19. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Breuer, J.; Garbe, S.; Giordano, F.; Brossart, P.; Feldmann, G.; Bisht, S. Triple Blockade of Oncogenic RAS Signaling Using KRAS and MEK Inhibitors in Combination with Irradiation in Pancreatic Cancer. Int. J. Mol. Sci. 2024, 25, 6249. https://doi.org/10.3390/ijms25116249
Wang X, Breuer J, Garbe S, Giordano F, Brossart P, Feldmann G, Bisht S. Triple Blockade of Oncogenic RAS Signaling Using KRAS and MEK Inhibitors in Combination with Irradiation in Pancreatic Cancer. International Journal of Molecular Sciences. 2024; 25(11):6249. https://doi.org/10.3390/ijms25116249
Chicago/Turabian StyleWang, Xuan, Johanna Breuer, Stephan Garbe, Frank Giordano, Peter Brossart, Georg Feldmann, and Savita Bisht. 2024. "Triple Blockade of Oncogenic RAS Signaling Using KRAS and MEK Inhibitors in Combination with Irradiation in Pancreatic Cancer" International Journal of Molecular Sciences 25, no. 11: 6249. https://doi.org/10.3390/ijms25116249