
Regulation of Neuronal Chloride Homeostasis by Pro- and Mature Brain-Derived Neurotrophic Factor (BDNF) via KCC2 Cation–Chloride Cotransporters in Rat Cortical Neurons
 ,
,
Abstract
1. Introduction
2. Results
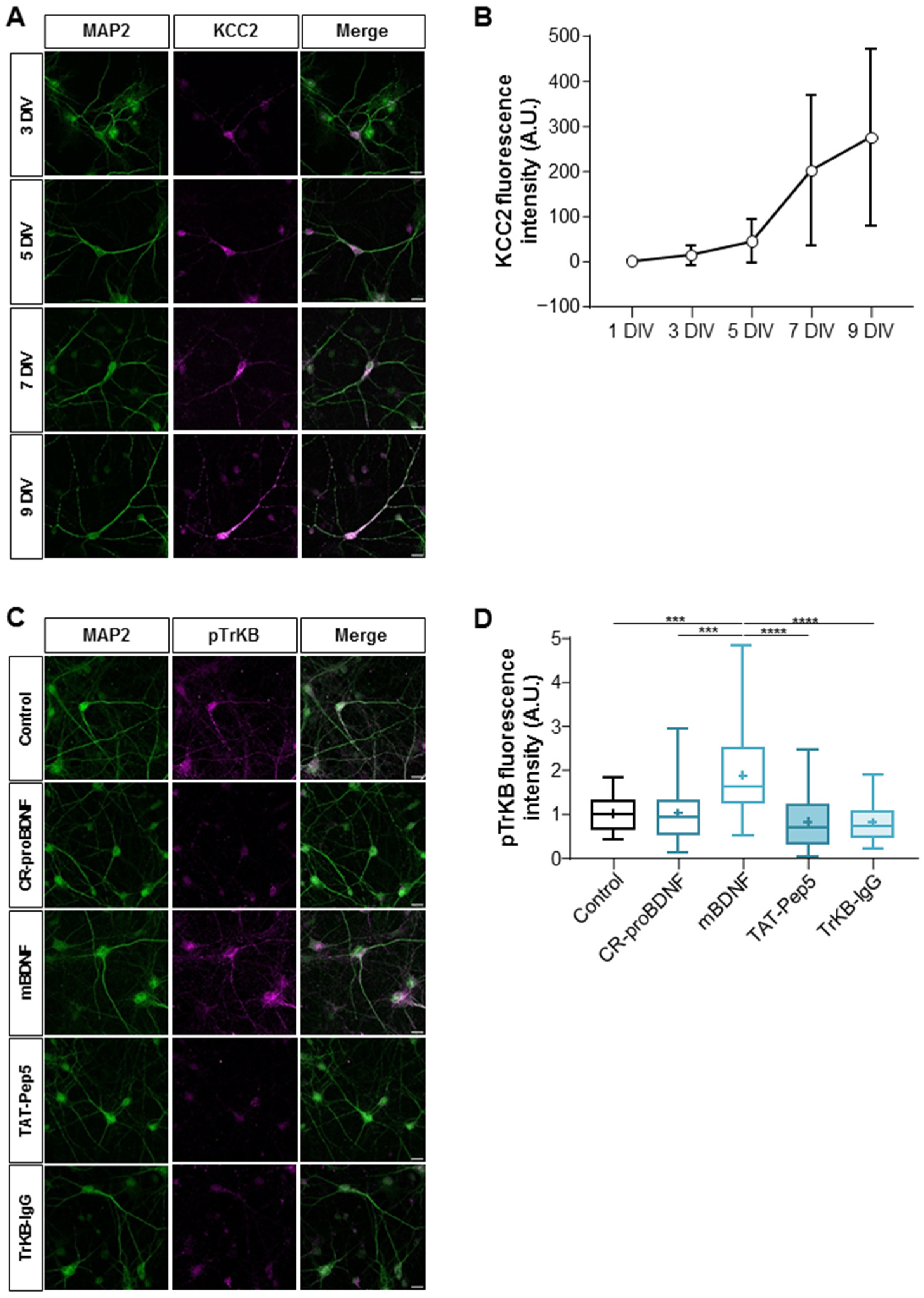
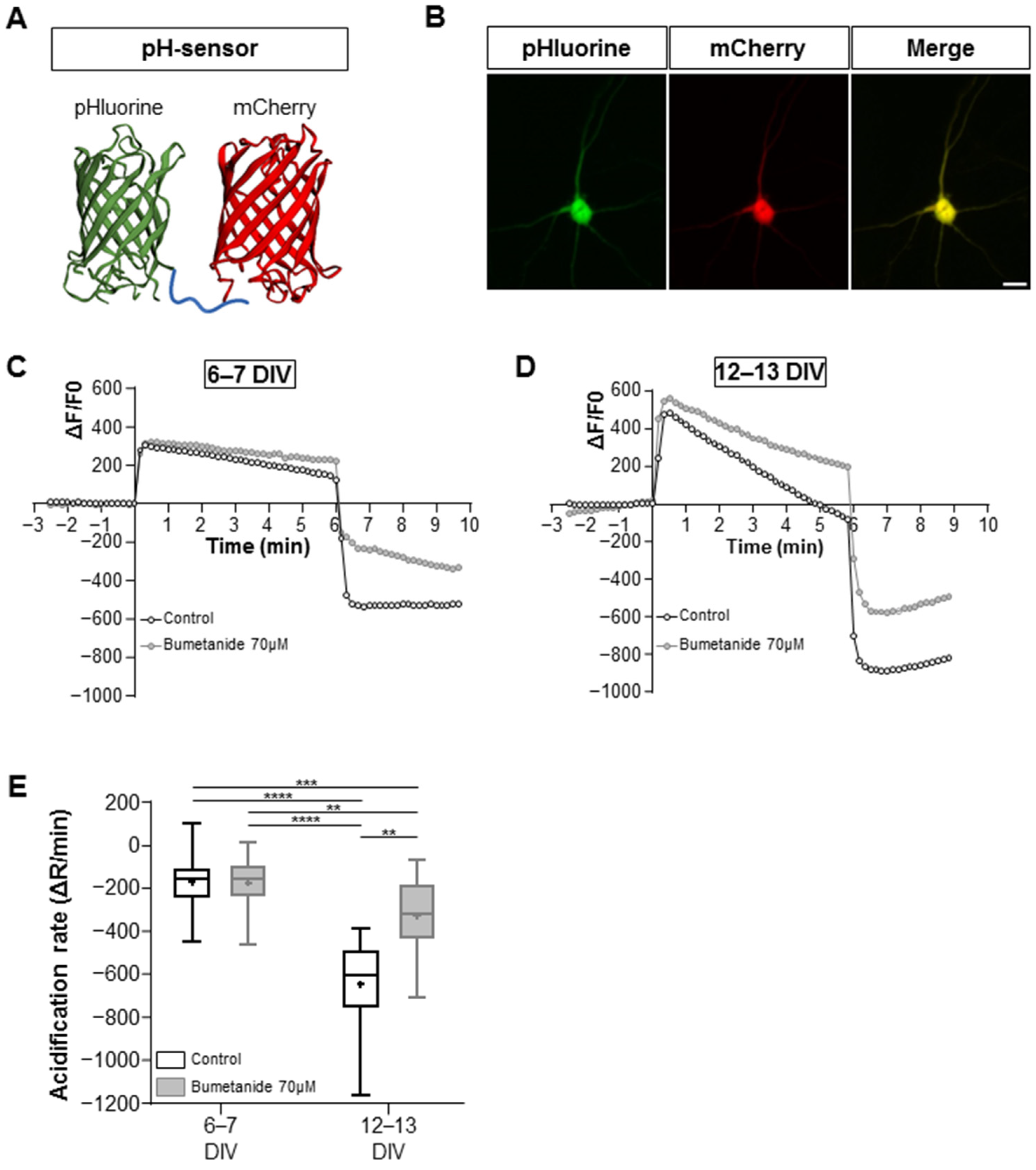
2.1. Functional Activity of KCC2 and BDNF Signaling Pathways in Maturing Cultured Hippocampal Neurons
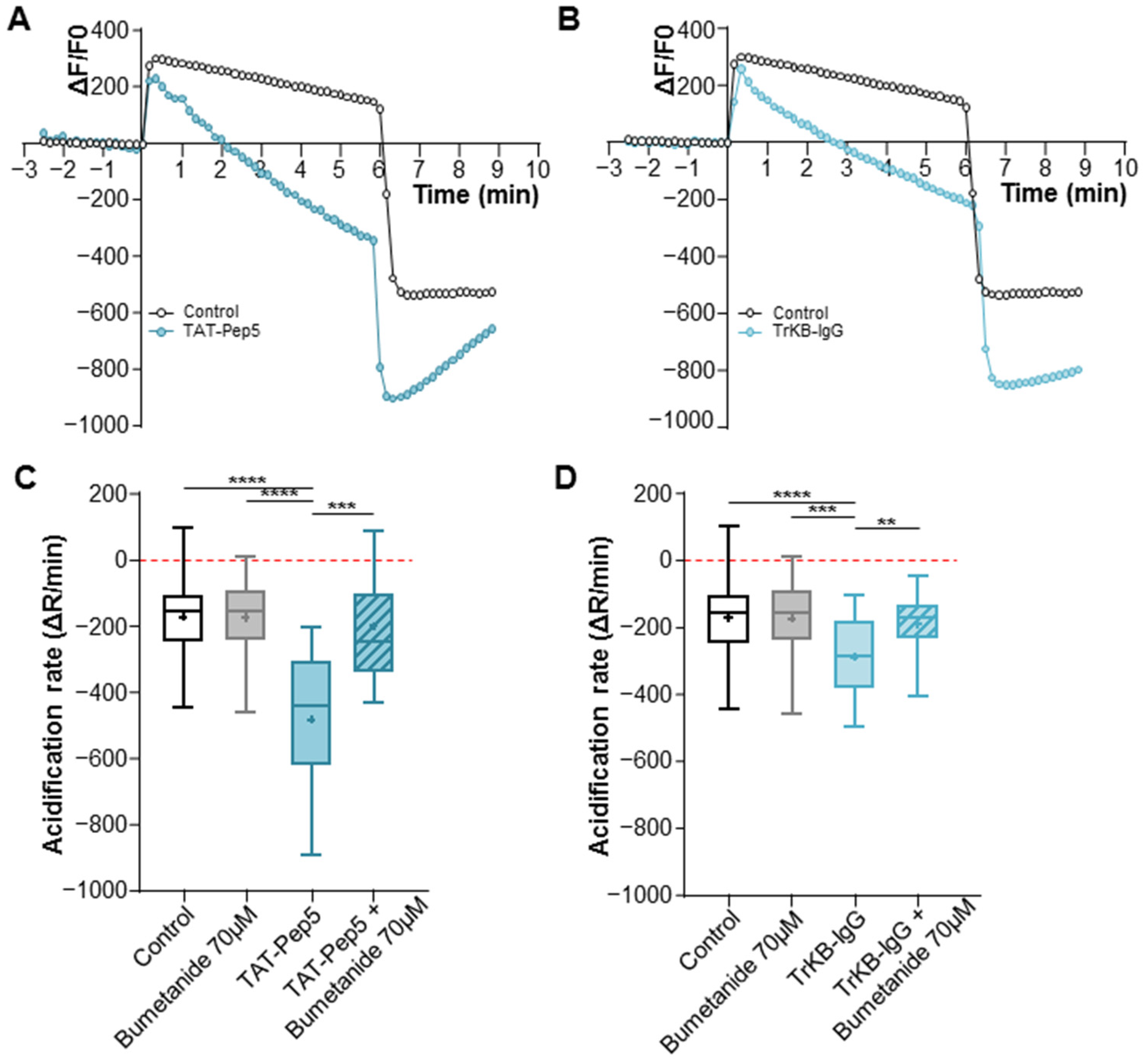
2.2. proBDNF Maintains a Depolarized GABA Response in Hippocampal Neurons
2.3. Effects of Pro- and Mature-BDNF on KCC2 Cell Trafficking
2.4. KCC2-Dependent Ammonium Transport in BDNF-Treated Hippocampal Neurons
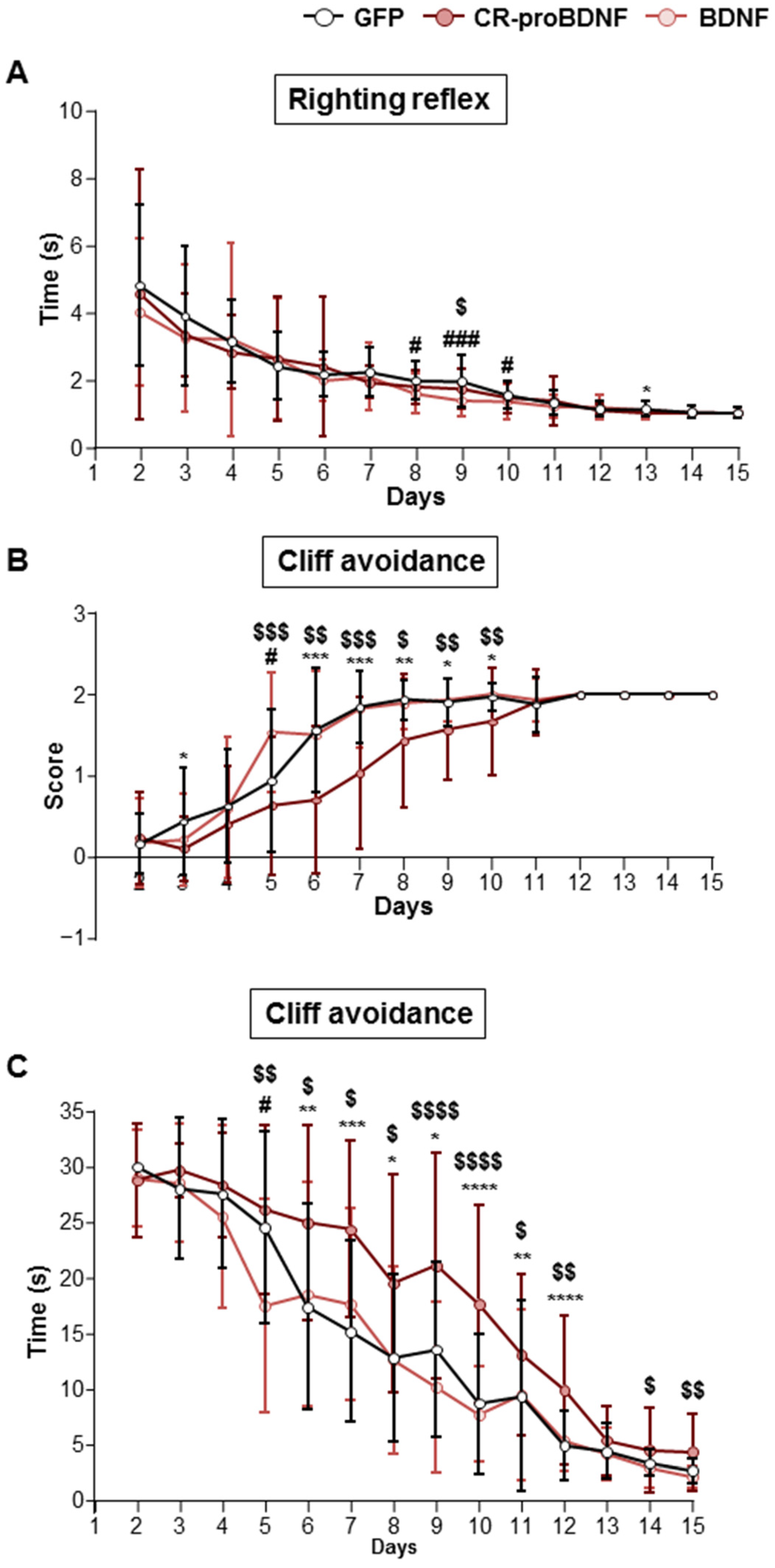
2.5. CR-proBDNF-Electroporated Rat Pups Exhibited Behavioral Deficits
2.6. CR-proBDNF-Electroporated Rats Exhibited Deficits in Sensory–Motor Maturation
3. Discussion
4. Materials and Methods
4.1. Reagents and Treatments
4.2. Primary Cultures of Rat Hippocampal Neurons
4.3. Transfections
4.4. NH+4 Flux Assay
4.5. Gramicidin-Perforated Patch-Clamp Recordings
4.6. Immunocytochemistry and Confocal Microscopy
4.7. Surface Immunolabeling on Living Neurons and Analysis of KCC2-pHext Proteins
4.8. In Utero Electroporation
4.9. Developmental Landmarks and Behavioral Tests
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ben-Ari, Y. Excitatory Actions of Gaba during Development: The Nature of the Nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-Chloride Cotransporters and Neuronal Function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Voipio, J.; Kaila, K. Two Developmental Switches in GABAergic Signalling: The K+–Cl− Cotransporter KCC2 and Carbonic Anhydrase CAVII. J. Physiol. 2005, 562, 27–36. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Gaiarsa, J.-L.; Tyzio, R.; Khazipov, R. GABA: A Pioneer Transmitter That Excites Immature Neurons and Generates Primitive Oscillations. Physiol. Rev. 2007, 87, 1215–1284. [Google Scholar] [CrossRef]
- Medina, I.; Friedel, P.; Rivera, C.; Kahle, K.T.; Kourdougli, N.; Uvarov, P.; Pellegrino, C. Current View on the Functional Regulation of the Neuronal K+-Cl− Cotransporter KCC2. Front. Cell. Neurosci. 2014, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Khalilov, I.; Minlebaev, M.; Mukhtarov, M.; Khazipov, R. Dynamic Changes from Depolarizing to Hyperpolarizing GABAergic Actions during Giant Depolarizing Potentials in the Neonatal Rat Hippocampus. J. Neurosci. 2015, 35, 12635–12642. [Google Scholar] [CrossRef] [PubMed]
- Kirmse, K.; Kummer, M.; Kovalchuk, Y.; Witte, O.W.; Garaschuk, O.; Holthoff, K. GABA Depolarizes Immature Neurons and Inhibits Network Activity in the Neonatal Neocortex in Vivo. Nat. Commun. 2015, 6, 7750. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, M.A.; Uvarov, P.; Hübner, C.A.; Kaila, K. NKCC1, an Elusive Molecular Target in Brain Development: Making Sense of the Existing Data. Cells 2020, 9, E2607. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554. [Google Scholar] [CrossRef]
- Lam, P.; Newland, J.; Faull, R.L.M.; Kwakowsky, A. Cation-Chloride Cotransporters KCC2 and NKCC1 as Therapeutic Targets in Neurological and Neuropsychiatric Disorders. Molecules 2023, 28, 1344. [Google Scholar] [CrossRef]
- Keramidis, I.; McAllister, B.B.; Bourbonnais, J.; Wang, F.; Isabel, D.; Rezaei, E.; Sansonetti, R.; Degagne, P.; Hamel, J.P.; Nazari, M.; et al. Restoring Neuronal Chloride Extrusion Reverses Cognitive Decline Linked to Alzheimer’s Disease Mutations. Brain 2023, 146, 4903–4915. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, J.; Jiang, J.; Zheng, X.; Justice, N.J.; Wang, K.; Ran, X.; Li, Y.; Huo, Q.; Zhang, J.; et al. APP Modulates KCC2 Expression and Function in Hippocampal GABAergic Inhibition. eLife 2017, 6, e20142. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Li, H.; Thomas-Crusells, J.; Lahtinen, H.; Viitanen, T.; Nanobashvili, A.; Kokaia, Z.; Airaksinen, M.S.; Voipio, J.; Kaila, K.; et al. BDNF-Induced TrkB Activation down-Regulates the K+-Cl- Cotransporter KCC2 and Impairs Neuronal Cl- Extrusion. J. Cell Biol. 2002, 159, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Aguado, F.; Carmona, M.A.; Pozas, E.; Aguiló, A.; Martínez-Guijarro, F.J.; Alcantara, S.; Borrell, V.; Yuste, R.; Ibañez, C.F.; Soriano, E. BDNF Regulates Spontaneous Correlated Activity at Early Developmental Stages by Increasing Synaptogenesis and Expression of the K+/Cl- Co-Transporter KCC2. Dev. Camb. Engl. 2003, 130, 1267–1280. [Google Scholar] [CrossRef]
- Porcher, C.; Medina, I.; Gaiarsa, J.-L. Mechanism of BDNF Modulation in GABAergic Synaptic Transmission in Healthy and Disease Brains. Front. Cell. Neurosci. 2018, 12, 273. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.E.; Xu, B.; Lu, B.; Hempstead, B.L. Symposium: New Insights in the Biology of BDNF Synthesis and Release: Implications in CNS Function. J. Neurosci. 2009, 29, 12764. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Cheng, H.; Yang, Y.; Tang, D.; Li, X.; An, L. Requirements of Postnatal proBDNF in the Hippocampus for Spatial Memory Consolidation and Neural Function. Front. Cell Dev. Biol. 2021, 9, 678182. [Google Scholar] [CrossRef] [PubMed]
- Menshanov, P.N.; Lanshakov, D.A.; Dygalo, N.N. proBDNF Is a Major Product of Bdnf Gene Expressed in the Perinatal Rat Cortex. Physiol. Res. Acad. Sci. Bohemoslov. 2015, 64, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Dincheva, I.; Lynch, N.B.; Lee, F.S. The Role of Bdnf in The Development of Fear Learning. Depress. Anxiety 2016, 33, 907–916. [Google Scholar] [CrossRef]
- Riffault, B.; Kourdougli, N.; Dumon, C.; Ferrand, N.; Buhler, E.; Schaller, F.; Chambon, C.; Rivera, C.; Gaiarsa, J.-L.; Porcher, C. Pro-Brain-Derived Neurotrophic Factor (proBDNF)-Mediated p75NTR Activation Promotes Depolarizing Actions of GABA and Increases Susceptibility to Epileptic Seizures. Cereb. Cortex 2016, 28, 510–527. [Google Scholar] [CrossRef]
- Kourdougli, N.; Pellegrino, C.; Renko, J.-M.; Khirug, S.; Chazal, G.; Kukko-Lukjanov, T.-K.; Lauri, S.E.; Gaiarsa, J.-L.; Zhou, L.; Peret, A.; et al. Depolarizing γ-Aminobutyric Acid Contributes to Glutamatergic Network Rewiring in Epilepsy. Ann. Neurol. 2017, 81, 251–265. [Google Scholar] [CrossRef]
- Ludwig, A.; Uvarov, P.; Soni, S.; Thomas-Crusells, J.; Airaksinen, M.S.; Rivera, C. Early Growth Response 4 Mediates BDNF Induction of Potassium Chloride Cotransporter 2 Transcription. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 644–649. [Google Scholar] [CrossRef]
- Mapplebeck, J.C.S.; Lorenzo, L.-E.; Lee, K.Y.; Gauthier, C.; Muley, M.M.; De Koninck, Y.; Prescott, S.A.; Salter, M.W. Chloride Dysregulation through Downregulation of KCC2 Mediates Neuropathic Pain in Both Sexes. Cell Rep. 2019, 28, 590–596.e4. [Google Scholar] [CrossRef]
- Patel, D.C.; Thompson, E.G.; Sontheimer, H. Brain-Derived Neurotrophic Factor Inhibits the Function of Cation-Chloride Cotransporter in a Mouse Model of Viral Infection-Induced Epilepsy. Front. Cell Dev. Biol. 2022, 10, 961292. [Google Scholar] [CrossRef]
- Shulga, A.; Magalhães, A.C.; Autio, H.; Plantman, S.; di Lieto, A.; Nykjær, A.; Carlstedt, T.; Risling, M.; Arumäe, U.; Castrén, E.; et al. The Loop Diuretic Bumetanide Blocks Posttraumatic p75NTR Upregulation and Rescues Injured Neurons. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 1757–1770. [Google Scholar] [CrossRef]
- Medina, I.; Pisella, L.I. Methods for Investigating the Activities of Neuronal Chloride Transporters. In Neuronal Chloride Transporters in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2020; pp. 21–41. ISBN 978-0-12-815318-5. [Google Scholar]
- Hershfinkel, M.; Kandler, K.; Knoch, M.E.; Dagan-Rabin, M.; Aras, M.A.; Abramovitch-Dahan, C.; Sekler, I.; Aizenman, E. Intracellular Zinc Inhibits KCC2 Transporter Activity. Nat. Neurosci. 2009, 12, 725–727. [Google Scholar] [CrossRef]
- Järvelä, V.; Hamze, M.; Komulainen-Ebrahim, J.; Rahikkala, E.; Piispala, J.; Kallio, M.; Kangas, S.M.; Nickl, T.; Huttula, M.; Hinttala, R.; et al. A Novel Pathogenic SLC12A5 Missense Variant in Epilepsy of Infancy with Migrating Focal Seizures Causes Impaired KCC2 Chloride Extrusion. Front. Mol. Neurosci. 2024, 17, 1372662. [Google Scholar] [CrossRef]
- Miesenböck, G.; De Angelis, D.A.; Rothman, J.E. Visualizing Secretion and Synaptic Transmission with pH-Sensitive Green Fluorescent Proteins. Nature 1998, 394, 192–195. [Google Scholar] [CrossRef]
- Eftekhari, S.; Mehrabi, S.; Soleimani, M.; Hassanzadeh, G.; Shahrokhi, A.; Mostafavi, H.; Hayat, P.; Barati, M.; Mehdizadeh, H.; Rahmanzadeh, R.; et al. BDNF Modifies Hippocampal KCC2 and NKCC1 Expression in a Temporal Lobe Epilepsy Model. Acta Neurobiol. Exp. 2014, 74, 276–287. [Google Scholar] [CrossRef]
- Ishibashi, M.; Egawa, K.; Fukuda, A. Diverse Actions of Astrocytes in GABAergic Signaling. Int. J. Mol. Sci. 2019, 20, 2964. [Google Scholar] [CrossRef]
- Friedel, P.; Kahle, K.T.; Zhang, J.; Hertz, N.; Pisella, L.I.; Buhler, E.; Schaller, F.; Duan, J.; Khanna, A.R.; Bishop, P.N.; et al. WNK1-Regulated Inhibitory Phosphorylation of the KCC2 Cotransporter Maintains the Depolarizing Action of GABA in Immature Neurons. Sci. Signal. 2015, 8, ra65. [Google Scholar] [CrossRef]
- Lee, H.H.C.; Walker, J.A.; Williams, J.R.; Goodier, R.J.; Payne, J.A.; Moss, S.J. Direct Protein Kinase C-Dependent Phosphorylation Regulates the Cell Surface Stability and Activity of the Potassium Chloride Cotransporter KCC2. J. Biol. Chem. 2007, 282, 29777–29784. [Google Scholar] [CrossRef]
- Kahle, K.T.; Merner, N.D.; Friedel, P.; Silayeva, L.; Liang, B.; Khanna, A.; Shang, Y.; Lachance-Touchette, P.; Bourassa, C.; Levert, A.; et al. Genetically Encoded Impairment of Neuronal KCC2 Cotransporter Function in Human Idiopathic Generalized Epilepsy. EMBO Rep. 2014, 15, 766–774. [Google Scholar] [CrossRef]
- Friedel, P.; Ludwig, A.; Pellegrino, C.; Agez, M.; Jawhari, A.; Rivera, C.; Medina, I. A Novel View on the Role of Intracellular Tails in Surface Delivery of the Potassium-Chloride Cotransporter KCC2. eNeuro 2017, 4, 1–19. [Google Scholar] [CrossRef]
- Baeza-Raja, B.; Sachs, B.D.; Li, P.; Christian, F.; Vagena, E.; Davalos, D.; Le Moan, N.; Ryu, J.K.; Sikorski, S.L.; Chan, J.P.; et al. P75 Neurotrophin Receptor Regulates Energy Balance in Obesity. Cell Rep. 2016, 14, 255–268. [Google Scholar] [CrossRef]
- Wilson, S.P. Modelling the Emergence of Rodent Filial Huddling from Physiological Huddling. R. Soc. Open Sci. 2017, 4, 170885. [Google Scholar] [CrossRef]
- Naskar, S.; Narducci, R.; Balzani, E.; Cwetsch, A.W.; Tucci, V.; Cancedda, L. The Development of Synaptic Transmission Is Time-Locked to Early Social Behaviors in Rats. Nat. Commun. 2019, 10, 1195. [Google Scholar] [CrossRef]
- Hübner, C.A.; Holthoff, K. Anion Transport and GABA Signaling. Front. Cell. Neurosci. 2013, 7, 177. [Google Scholar] [CrossRef]
- Inoue, K.; Furukawa, T.; Kumada, T.; Yamada, J.; Wang, T.; Inoue, R.; Fukuda, A. Taurine Inhibits K+-Cl− Cotransporter KCC2 to Regulate Embryonic Cl- Homeostasis via with-No-Lysine (WNK) Protein Kinase Signaling Pathway. J. Biol. Chem. 2012, 287, 20839–20850. [Google Scholar] [CrossRef]
- Pol, E.; Côme, E.; Merlaud, Z.; Gouhier, J.; Russeau, M.; Scotto-Lomassese, S.; Moutkine, I.; Marques, X.; Lévi, S. NKCC1 and KCC2 Chloride Transporters Have Different Membrane Dynamics on the Surface of Hippocampal Neurons. Cells 2023, 12, 2363. [Google Scholar] [CrossRef]
- KOURDOUGLI, N.; Varpula, S.; Chazal, G.; Rivera, C. Detrimental Effect of Post Status Epilepticus Treatment with ROCK Inhibitor Y-27632 in a Pilocarpine Model of Temporal Lobe Epilepsy. Front. Cell. Neurosci. 2015, 9, 413. [Google Scholar] [CrossRef]
- Porcher, C.; Hatchett, C.; Longbottom, R.E.; McAinch, K.; Sihra, T.S.; Moss, S.J.; Thomson, A.M.; Jovanovic, J.N. Positive Feedback Regulation between Gamma-Aminobutyric Acid Type A (GABA(A)) Receptor Signaling and Brain-Derived Neurotrophic Factor (BDNF) Release in Developing Neurons. J. Biol. Chem. 2011, 286, 21667–21677. [Google Scholar] [CrossRef]
- de Los Heros, P.; Alessi, D.R.; Gourlay, R.; Campbell, D.G.; Deak, M.; Macartney, T.J.; Kahle, K.T.; Zhang, J. The WNK-Regulated SPAK/OSR1 Kinases Directly Phosphorylate and Inhibit the K+-Cl− Co-Transporters. Biochem. J. 2014, 458, 559–573. [Google Scholar] [CrossRef]
- McMoneagle, E.; Zhou, J.; Zhang, S.; Huang, W.; Josiah, S.S.; Ding, K.; Wang, Y.; Zhang, J. Neuronal K+-Cl− Cotransporter KCC2 as a Promising Drug Target for Epilepsy Treatment. Acta Pharmacol. Sin. 2023, 45, 1–22. [Google Scholar] [CrossRef]
- Gagnon, K.B.; Delpire, E. On the Substrate Recognition and Negative Regulation of SPAK, a Kinase Modulating Na+-K+-2Cl− Cotransport Activity. Am. J. Physiol. Cell Physiol. 2010, 299, C614–C620. [Google Scholar] [CrossRef]
- Zanin, J.P.; Montroull, L.E.; Volosin, M.; Friedman, W.J. The P75 Neurotrophin Receptor Facilitates TrkB Signaling and Function in Rat Hippocampal Neurons. Front. Cell. Neurosci. 2019, 13, 485. [Google Scholar] [CrossRef]
- Lu, B. BDNF and Activity-Dependent Synaptic Modulation. Learn. Mem. 2003, 10, 86–98. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Lee, F.S.; Hempstead, B.L. New Roles for an Ancient Factor. Trends Neurosci. 2018, 41, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.-J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF Negatively Regulates Neuronal Remodeling, Synaptic Transmission, and Synaptic Plasticity in Hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Ernfors, P.; Lee, K.-F.; Jaenisch, R. Mice Lacking Brain-Derived Neurotrophic Factor Develop with Sensory Deficits. Nature 1994, 368, 147–150. [Google Scholar] [CrossRef]
- Jones, D.L. A Potential Role for Nerve Growth Factor in Regulating the Maturation of Inhibitory Neurotransmission. J. Neurosci. 2010, 30, 6813–6814. [Google Scholar] [CrossRef]
- Andreska, T.; Lüningschrör, P.; Sendtner, M. Regulation of TrkB Cell Surface Expression—A Mechanism for Modulation of Neuronal Responsiveness to Brain-Derived Neurotrophic Factor. Cell Tissue Res. 2020, 382, 5–14. [Google Scholar] [CrossRef]
- Genoud, C.; Knott, G.; Sakata, K.; Lu, B.; Welker, E. Altered Synapse Formation in the Adult Somatosensory Cortex of Brain-Derived Neurotrophic Factor Heterozygote Mice. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 2394–2400. [Google Scholar] [CrossRef]
- Buerli, T.; Pellegrino, C.; Baer, K.; Lardi-Studler, B.; Chudotvorova, I.; Fritschy, J.-M.; Medina, I.; Fuhrer, C. Efficient Transfection of DNA or shRNA Vectors into Neurons Using Magnetofection. Nat. Protoc. 2007, 2, 3090–3101. [Google Scholar] [CrossRef]
- Kriegstein, A.R.; Noctor, S.C. Patterns of Neuronal Migration in the Embryonic Cortex. Trends Neurosci. 2004, 27, 392–399. [Google Scholar] [CrossRef]
- Heyser, C.J. Assessment of Developmental Milestones in Rodents. Curr. Protoc. Neurosci. 2004, 25, 8–18. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Days/Condition | GFP | CR-proBDNF | BDNF |
|---|---|---|---|
| 2 | 0.16 ± 0.37 | 0.23 ± 0.57 | 0.18 ± 0.55 |
| 3 | 0.44 ± 0.67 | 0.1 ± 0.4 * | 0.21 ± 0.57 |
| 4 | 0.63 ± 0.7 | 0.4 ± 0.72 | 0.61 ± 0.88 |
| 5 | 0.94 ± 0.88 | 0.63 ± 0.85 | 1.54 ± 0.74 # |
| 6 | 1.56 ± 0.76 | 0.7 ± 0.92 *** | 1.5 ± 0.79 $$ |
| 7 | 1.84 ± 0.45 | 1.03 ± 0.93 *** | 1.82 ± 0.48 $$$ |
| 8 | 1.94 ± 0.25 | 1.43 ± 0.82 ** | 1.9 ± 0.31 $ |
| 9 | 1.91 ± 0.3 | 1.6 ± 0.62 * | 1.93 ± 0.26 $$ |
| 10 | 1.97 ± 0.18 | 1.7 ± 0.66 * | 2 ± 0 $$ |
| 11 | 1.88 ± 0.34 | 1.9 ± 0.4 | 1.93 ± 0.26 |
| 12 | 2 ± 0 | 2 ± 0 | 2 ± 0 |
| 13 | 2 ± 0 | 2 ± 0 | 2 ± 0 |
| 14 | 2 ± 0 | 2 ± 0 | 2 ± 0 |
| 15 | 2 ± 0 | 2 ± 0 | 2 ± 0 |
| Days/Condition | GFP | CR-proBDNF | BDNF |
|---|---|---|---|
| 2 | 30 ± 0 | 28.7 ± 5.08 | 28.96 ± 4.31 |
| 3 | 28 ± 6.37 | 29.6 ± 2.37 | 28.54 ± 5.39 |
| 4 | 27.53 ± 6.7 | 28.23 ± 4.75 | 25.54 ± 8.24 |
| 5 | 24.5 ± 8.67 | 26.03 ± 7.62 | 17.5 ± 9.68 $$ # |
| 6 | 17.4 ± 9.23 | 24.9 ± 8.79 ** | 18.54 ± 10.11 |
| 7 | 15.16 ± 8.15 | 24.3 ± 7.95 *** | 17.64 ± 8.63 $ |
| 8 | 12.78 ± 7.51 | 19.4 ± 9.77 * | 12.61 ± 8.45 $ |
| 9 | 13.5 ± 7.93 | 21.03 ± 10.16 * | 10.18 ± 7.65 $$$$ |
| 10 | 8.7 ± 6.3 | 17.5 ± 8.95 **** | 7.71 ± 4.31 $$$$ |
| 11 | 9.31 ± 8.59 | 13 ± 7.27 ** | 9.5 ± 7.71 $ |
| 12 | 4.9 ± 3.07 | 9.83 ± 6.74 **** | 5.32 ± 2.68 $$ |
| 13 | 4.4 ± 2.47 | 5.3 ± 3.01 | 4.14 ± 2.37 |
| 14 | 3.31 ± 1.18 | 4.4 ± 3.78 | 2.9 ± 1.75 $ |
| 15 | 2.63 ± 1.16 | 4.23 ± 3.45 | 2.11 ± 0.95 $$ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamze, M.; Brier, C.; Buhler, E.; Zhang, J.; Medina, I.; Porcher, C. Regulation of Neuronal Chloride Homeostasis by Pro- and Mature Brain-Derived Neurotrophic Factor (BDNF) via KCC2 Cation–Chloride Cotransporters in Rat Cortical Neurons. Int. J. Mol. Sci. 2024, 25, 6253. https://doi.org/10.3390/ijms25116253
Hamze M, Brier C, Buhler E, Zhang J, Medina I, Porcher C. Regulation of Neuronal Chloride Homeostasis by Pro- and Mature Brain-Derived Neurotrophic Factor (BDNF) via KCC2 Cation–Chloride Cotransporters in Rat Cortical Neurons. International Journal of Molecular Sciences. 2024; 25(11):6253. https://doi.org/10.3390/ijms25116253
Chicago/Turabian StyleHamze, Mira, Cathy Brier, Emmanuelle Buhler, Jinwei Zhang, Igor Medina, and Christophe Porcher. 2024. "Regulation of Neuronal Chloride Homeostasis by Pro- and Mature Brain-Derived Neurotrophic Factor (BDNF) via KCC2 Cation–Chloride Cotransporters in Rat Cortical Neurons" International Journal of Molecular Sciences 25, no. 11: 6253. https://doi.org/10.3390/ijms25116253
APA StyleHamze, M., Brier, C., Buhler, E., Zhang, J., Medina, I., & Porcher, C. (2024). Regulation of Neuronal Chloride Homeostasis by Pro- and Mature Brain-Derived Neurotrophic Factor (BDNF) via KCC2 Cation–Chloride Cotransporters in Rat Cortical Neurons. International Journal of Molecular Sciences, 25(11), 6253. https://doi.org/10.3390/ijms25116253