
Host Cell Death and Modulation of Immune Response against Mycobacterium tuberculosis Infection

 , and
, and 
Abstract
:1. Introduction
2. Immune System Modulation
2.1. Innate and Adaptive Immune System
2.2. TNF-α and Granuloma Formation
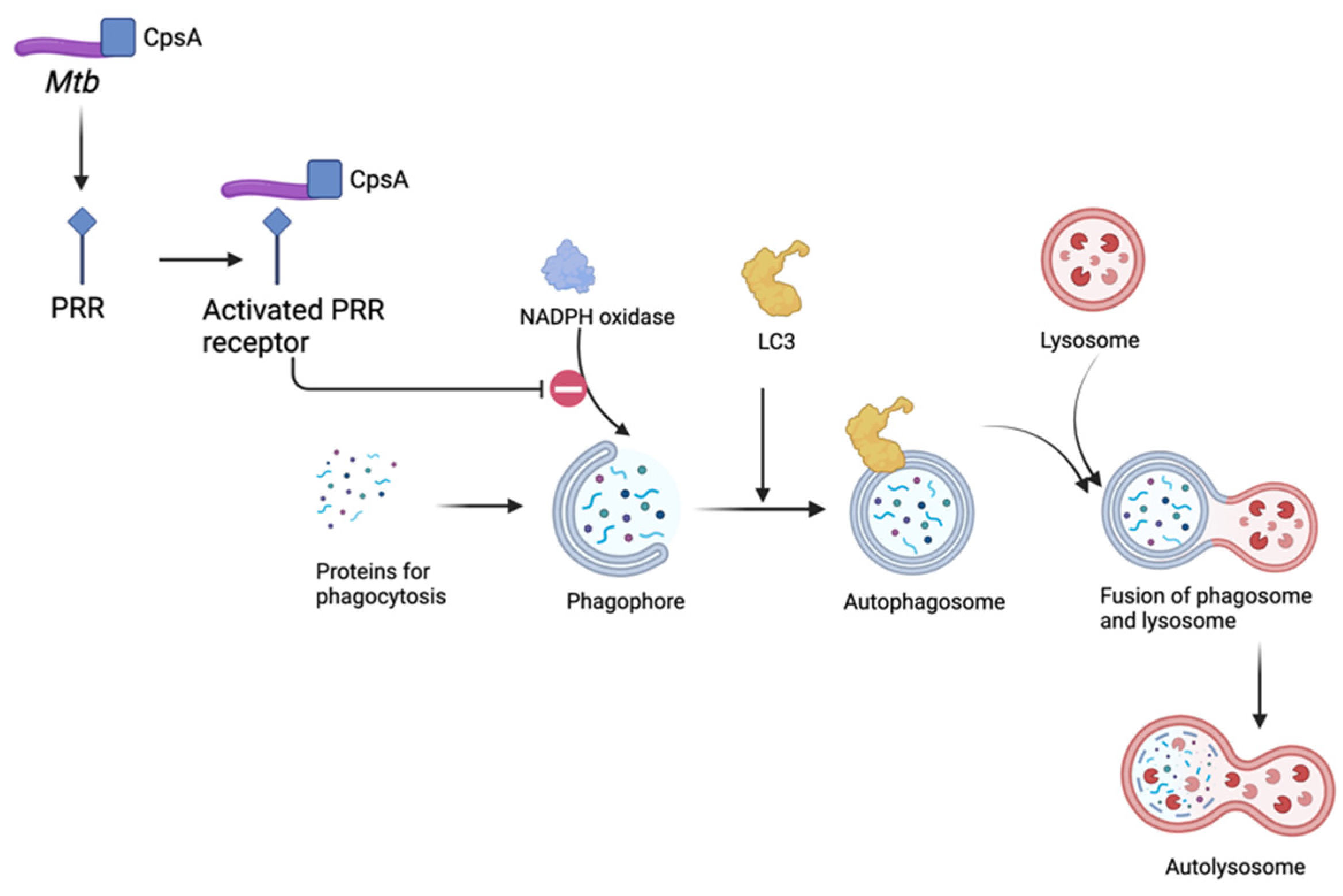
2.3. Role of Autophagy in Mtb Infection
2.4. Foam Cell Macrophages and Mtb-Mediated Modulation
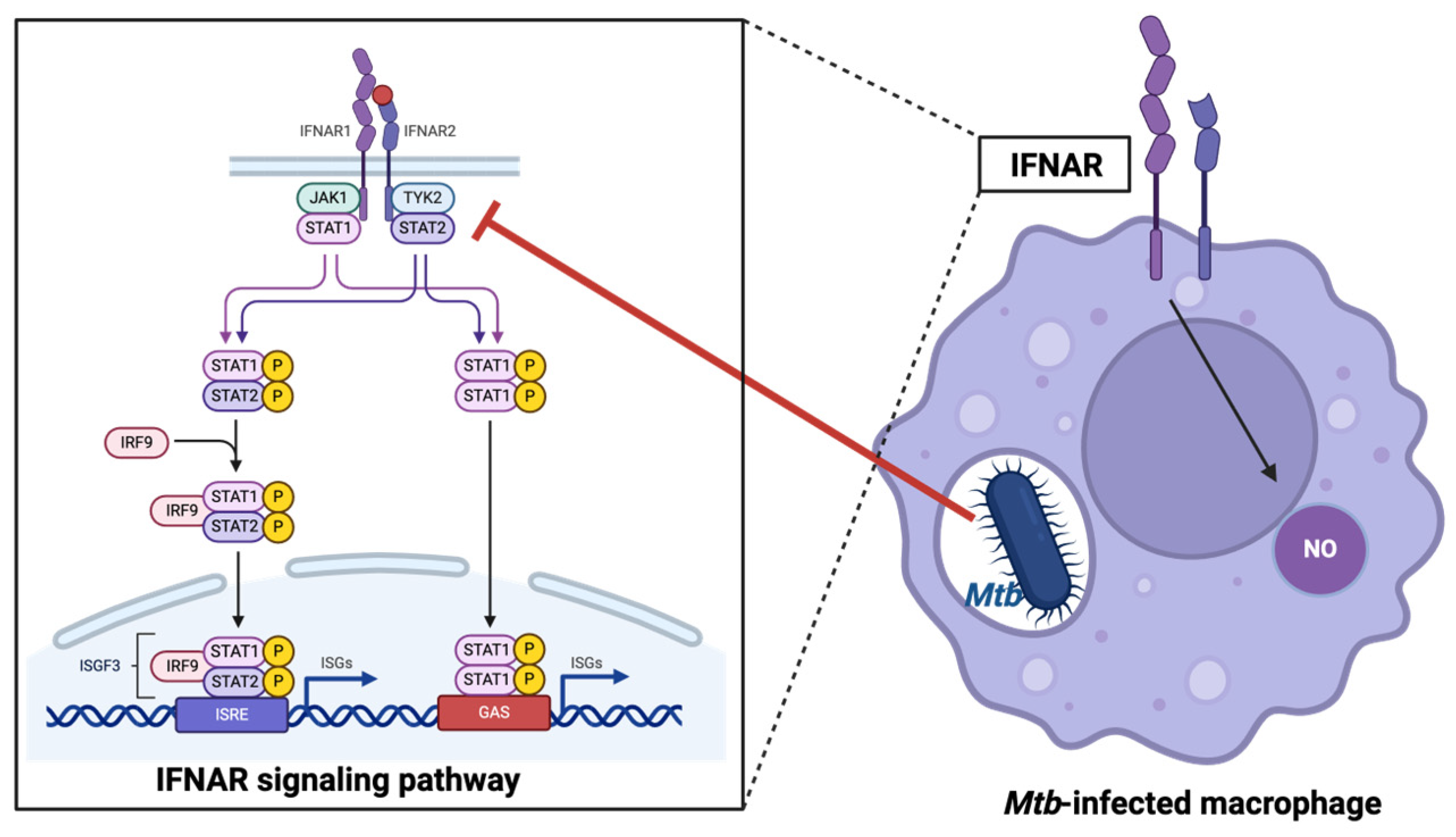
2.5. Role of Type I Interferons
3. Host Cell Death Pathways in Mtb Infection
3.1. Apoptosis: In Favor of the Host
3.2. Caspase-Dependent Pathways
3.3. Caspase-Independent Pathways
4. Necrosis: In Favor of Mtb
5. Therapeutic Considerations Targeting Autophagy, Apoptosis, and Necrosis in Mtb Infection
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, T.; Zhang, J.; Wei, L.; Liang, H.; Zhang, J.; Shi, D.; Wang, Z. The global, regional, and national burden of tuberculosis in 204 countries and territories, 1990–2019. J. Infect. Public Health 2023, 16, 368–375. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2023. 2023. Available online: https://www.who.int/publications/i/item/9789240083851 (accessed on 12 February 2024).
- World Health Organization. Global Tuberculosis Report 2021. 2021. Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 12 February 2024).
- Luies, L.; du Preez, I. The Echo of Pulmonary Tuberculosis: Mechanisms of Clinical Symptoms and Other Disease-Induced Systemic Complications. Clin. Microbiol. Rev. 2020, 33, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Bruchfeld, J.; Correia-Neves, M.; Källenius, G. Tuberculosis and HIV Coinfection. Cold Spring Harb. Perspect. Med. 2015, 5, a017871. [Google Scholar] [CrossRef] [PubMed]
- Menzies, D. Effect of treatment on contagiousness of patients with active pulmonary tuberculosis. Infect. Control Hosp. Epidemiol. 1997, 18, 582–586. [Google Scholar] [CrossRef]
- Gideon, H.P.; Flynn, J.L. Latent tuberculosis: What the host “sees”? Immunol. Res. 2011, 50, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Chan, J. Tuberculosis: Latency and reactivation. Infect. Immun. 2001, 69, 4195–4201. [Google Scholar] [CrossRef] [PubMed]
- Ravimohan, S.; Kornfeld, H.; Weissman, D.; Bisson, G.P. Tuberculosis and lung damage: From epidemiology to pathophysiology. Eur. Respir. Rev. 2018, 27, 170077. [Google Scholar] [CrossRef] [PubMed]
- Vohra, S.; Dhaliwal, H.S. Miliary Tuberculosis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Slane, V.H.; Unakal, C.G. Tuberculous Meningitis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Naicker, K.; Ntsekhe, M. Tuberculous pericardial disease: A focused update on diagnosis, therapy and prevention of complications. Cardiovasc. Diagn. Ther. 2020, 10, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Seung, K.J.; Keshavjee, S.; Rich, M.L. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 5, a017863. [Google Scholar] [CrossRef]
- Bussi, C.; Gutierrez, M.G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol. Rev. 2019, 43, 341–361. [Google Scholar] [CrossRef]
- Augenstreich, J.; Haanappel, E.; Ferre, G.; Czaplicki, G.; Jolibois, F.; Destainville, N.; Guilhot, C.; Milon, A.; Astarie-Dequeker, C.; Chavent, M. The conical shape of DIM lipids promotes Mycobacterium tuberculosis infection of macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 25649–25658. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, L. Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 2012, 12, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G. Who puts the tubercle in tuberculosis? Nat. Rev. Microbiol. 2007, 5, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different modalities of host cell death and their impact on Mycobacterium tuberculosis infection. Am. J. Physiol. Cell Physiol. 2022, 323, C1444–C1474. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Plessner, H.L.; Voitenok, N.N.; Flynn, J.L. Tumor necrosis factor and tuberculosis. J. Investig. Dermatol. Symp. Proc. 2007, 12, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Kim, J.K.; Kim, I.S.; Jo, E.K. TNF in Human Tuberculosis: A Double-Edged Sword. Immune Netw. 2024, 24, e4. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Lussier, D.M.; Schreiber, R.D. Interferon γ and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb. Perspect. Biol. 2019, 11, a028480. [Google Scholar] [CrossRef] [PubMed]
- Shanmuganathan, G.; Orujyan, D.; Narinyan, W.; Poladian, N.; Dhama, S.; Parthasarathy, A.; Ha, A.; Tran, D.; Velpuri, P.; Nguyen, K.H.; et al. Role of Interferons in Mycobacterium tuberculosis Infection. Clin. Pract. 2022, 12, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.M.; Dalton, D.K.; Croft, M. A direct role for IFN-gamma in regulation of Th1 cell development. J. Immunol. 1996, 157, 1350–1358. [Google Scholar] [CrossRef]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.X.; Divangahi, M.; Remold, H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef]
- Chai, Q.; Wang, L.; Liu, C.H.; Ge, B. New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol. Immunol. 2020, 17, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, N.; Bergero, G.; Mazzocco Mariotta, Y.L.; Aoki, M.P. Purinergic modulation of the immune response to infections. Purinergic Signal. 2022, 18, 93–113. [Google Scholar] [CrossRef]
- Fremond, C.M.; Yeremeev, V.; Nicolle, D.M.; Jacobs, M.; Quesniaux, V.F.; Ryffel, B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J. Clin. Investig. 2004, 114, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Gern, B.H.; Delahaye, J.L.; Adams, K.N.; Plumlee, C.R.; Winkler, J.K.; Sherman, D.R.; Gerner, M.Y.; Urdahl, K.B. Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe 2018, 24, 439–446.e4. [Google Scholar] [CrossRef]
- Iantomasi, R.; Sali, M.; Cascioferro, A.; Palucci, I.; Zumbo, A.; Soldini, S.; Rocca, S.; Greco, E.; Maulucci, G.; De Spirito, M.; et al. PE_PGRS30 is required for the full virulence of Mycobacterium tuberculosis. Cell Microbiol. 2012, 14, 356–367. [Google Scholar] [CrossRef]
- van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef]
- Houben, D.; Demangel, C.; van Ingen, J.; Perez, J.; Baldeon, L.; Abdallah, A.M.; Caleechurn, L.; Bottai, D.; van Zon, M.; de Punder, K.; et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 2012, 14, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, A.A.; Alt, J.M.; Perera, T.V.; Dascher, C.C.; Behar, S.M. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect. Immun. 2002, 70, 4501–4509. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Ryan, L.; LaCourse, R.; North, R.J. Properties and protective value of the secondary versus primary T helper type 1 response to airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 2005, 201, 1915–1924. [Google Scholar] [CrossRef]
- Garcia-Romo, G.S.; Pedroza-Gonzalez, A.; Lambrecht, B.N.; Aguilar-Leon, D.; Estrada-Garcia, I.; Hernandez-Pando, R.; Flores-Romo, L. Mycobacterium tuberculosis manipulates pulmonary APCs subverting early immune responses. Immunobiology 2013, 218, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Grace, P.S.; Ernst, J.D. Antigen Export Reduces Antigen Presentation and Limits T Cell Control of M. tuberculosis. Cell Host Microbe 2016, 19, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.K.; Pennini, M.E.; Tobian, A.A.; Canaday, D.H.; Boom, W.H.; Harding, C.V. Prolonged toll-like receptor signaling by Mycobacterium tuberculosis and its 19-kilodalton lipoprotein inhibits gamma interferon-induced regulation of selected genes in macrophages. Infect. Immun. 2004, 72, 6603–6614. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Schuetz, A.; Ngwenyama, N.; Casazza, J.P.; Sanga, E.; Saathoff, E.; Boehme, C.; Geis, S.; Maboko, L.; Singh, M.; et al. Early depletion of Mycobacterium tuberculosis-specific T helper 1 cell responses after HIV-1 infection. J. Infect. Dis. 2008, 198, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Ernst, J.D. Cutting edge: Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J. Immunol. 2013, 191, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Difazio, R.; Flynn, J.L. IFN-gamma from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J. Immunol. 2013, 190, 270–277. [Google Scholar] [CrossRef]
- Lu, Y.J.; Barreira-Silva, P.; Boyce, S.; Powers, J.; Cavallo, K.; Behar, S.M. CD4 T cell help prevents CD8 T cell exhaustion and promotes control of Mycobacterium tuberculosis infection. Cell Rep. 2021, 36, 109696. [Google Scholar] [CrossRef]
- Mishra, A.; Singh, V.K.; Jagannath, C.; Subbian, S.; Restrepo, B.I.; Gauduin, M.C.; Khan, A. Human Macrophages Exhibit GM-CSF Dependent Restriction of Mycobacterium tuberculosis Infection via Regulating Their Self-Survival, Differentiation and Metabolism. Front. Immunol. 2022, 13, 859116. [Google Scholar] [CrossRef] [PubMed]
- Khader, S.A.; Partida-Sanchez, S.; Bell, G.; Jelley-Gibbs, D.M.; Swain, S.; Pearl, J.E.; Ghilardi, N.; Desauvage, F.J.; Lund, F.E.; Cooper, A.M. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J. Exp. Med. 2006, 203, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, S.C.; Lu, F.X.; Yu, J.; Perfetto, S.P.; Falloon, J.; Moser, S.; Evans, T.G.; Koup, R.; Miller, C.J.; Roederer, M. Vaccination in humans generates broad T cell cytokine responses. J. Immunol. 2004, 173, 5372–5380. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.O. The structure of mononuclear phagocytes differentiating in vivo. II. The effect of Mycobacterium tuberculosis. Am. J. Pathol. 1975, 80, 101–116. [Google Scholar] [PubMed]
- Algood, H.M.; Lin, P.L.; Flynn, J.L. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin. Infect. Dis. 2005, 41 (Suppl. S3), S189–S193. [Google Scholar] [CrossRef] [PubMed]
- Sandor, M.; Weinstock, J.V.; Wynn, T.A. Granulomas in schistosome and mycobacterial infections: A model of local immune responses. Trends Immunol. 2003, 24, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Knaak, T.; Satkamp, L.; Humbert, O.; Falkow, S.; Ramakrishnan, L. Complex pattern of Mycobacterium marinum gene expression during long-term granulomatous infection. Proc. Natl. Acad. Sci. USA 2002, 99, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Cardona, P.J.; Kim, M.J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Ulrichs, T.; Kosmiadi, G.A.; Jorg, S.; Pradl, L.; Titukhina, M.; Mishenko, V.; Gushina, N.; Kaufmann, S.H. Differential organization of the local immune response in patients with active cavitary tuberculosis or with nonprogressive tuberculoma. J. Infect. Dis. 2005, 192, 89–97. [Google Scholar] [CrossRef]
- Kaplan, G.; Post, F.A.; Moreira, A.L.; Wainwright, H.; Kreiswirth, B.N.; Tanverdi, M.; Mathema, B.; Ramaswamy, S.V.; Walther, G.; Steyn, L.M.; et al. Mycobacterium tuberculosis growth at the cavity surface: A microenvironment with failed immunity. Infect. Immun. 2003, 71, 7099–7108. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Virgilio, L.; Silva-Lucero, M.D.; Flores-Morelos, D.S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.M.; Zacapala-Gómez, A.E.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T. Autophagy as a defence against intracellular pathogens. Essays Biochem. 2013, 55, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kleinnijenhuis, J.; Oosting, M.; Plantinga, T.S.; van der Meer, J.W.; Joosten, L.A.; Crevel, R.V.; Netea, M.G. Autophagy modulates the Mycobacterium tuberculosis-induced cytokine response. Immunology 2011, 134, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Zihad, S.; Sifat, N.; Islam, M.A.; Monjur-Al-Hossain, A.S.M.; Sikdar, K.; Sarker, M.M.R.; Shilpi, J.A.; Uddin, S.J. Role of pattern recognition receptors in sensing Mycobacterium tuberculosis. Heliyon 2023, 9, e20636. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.K.; Silwal, P.; Yuk, J.M. AMPK-Targeted Effector Networks in Mycobacterial Infection. Front. Microbiol. 2019, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Silwal, P.; Kim, S.Y.; Yoshimori, T.; Jo, E.K. Autophagy-activating strategies to promote innate defense against mycobacteria. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy-Thandavan, S.; Jiang, M.; Schoenlein, P.; Dong, Z. Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Renal Physiol. 2009, 297, F244–F256. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Ganley, I.G. Autophagosome maturation and lysosomal fusion. Essays Biochem. 2013, 55, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Henault, J.; Kolbeck, R.; Sanjuan, M.A. Noncanonical autophagy: One small step for LC3, one giant leap for immunity. Curr. Opin. Immunol. 2014, 26, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Köster, S.; Upadhyay, S.; Chandra, P.; Papavinasasundaram, K.; Yang, G.; Hassan, A.; Grigsby, S.J.; Mittal, E.; Park, H.S.; Jones, V.; et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. USA 2017, 114, E8711–E8720. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.G.; Wang, X.; Yi, N.; Ma, J.; Turner, J.; Samten, B. Early Secreted Antigenic Target of 6-kDa of Mycobacterium tuberculosis Stimulates IL-6 Production by Macrophages through Activation of STAT3. Sci. Rep. 2017, 7, 40984. [Google Scholar] [CrossRef] [PubMed]
- Crotzer, V.L.; Blum, J.S. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Gordon, S.; Martinez, F.O. Foam Cell Macrophages in Tuberculosis. Front. Immunol. 2021, 12, 775326. [Google Scholar] [CrossRef]
- Almeida, P.E.; Roque, N.R.; Magalhães, K.G.; Mattos, K.A.; Teixeira, L.; Maya-Monteiro, C.; Almeida, C.J.; Castro-Faria-Neto, H.C.; Ryffel, B.; Quesniaux, V.F.; et al. Differential TLR2 downstream signaling regulates lipid metabolism and cytokine production triggered by Mycobacterium bovis BCG infection. Biochim. Biophys. Acta 2014, 1841, 97–107. [Google Scholar] [CrossRef]
- Knight, M.; Braverman, J.; Asfaha, K.; Gronert, K.; Stanley, S. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-γ/HIF-1α signaling and supports host defense. PLoS Pathog. 2018, 14, e1006874. [Google Scholar] [CrossRef] [PubMed]
- Sorgi, C.A.; Soares, E.M.; Rosada, R.S.; Bitencourt, C.S.; Zoccal, K.F.; Pereira, P.A.T.; Fontanari, C.; Brandão, I.; Masson, A.P.; Ramos, S.G.; et al. Eicosanoid pathway on host resistance and inflammation during Mycobacterium tuberculosis infection is comprised by LTB(4) reduction but not PGE(2) increment. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165574. [Google Scholar] [CrossRef] [PubMed]
- Nore, K.G.; Jørgensen, M.J.; Dyrhol-Riise, A.M.; Jenum, S.; Tonby, K. Elevated Levels of Anti-Inflammatory Eicosanoids and Monocyte Heterogeneity in Mycobacterium tuberculosis Infection and Disease. Front. Immunol. 2020, 11, 579849. [Google Scholar] [CrossRef] [PubMed]
- Fenton, S.E.; Saleiro, D.; Platanias, L.C. Type I and II Interferons in the Anti-Tumor Immune Response. Cancers 2021, 13, 1037. [Google Scholar] [CrossRef] [PubMed]
- Travar, M.; Petkovic, M.; Verhaz, A. Type I, II, and III Interferons: Regulating Immunity to Mycobacterium tuberculosis Infection. Arch. Immunol. Ther. Exp. (Warsz) 2016, 64, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Langer, J.A.; Zoon, K.C.; Samuel, C.E. Interferons and their actions. Annu. Rev. Biochem. 1987, 56, 727–777. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef]
- Moreira-Teixeira, L.; Mayer-Barber, K.; Sher, A.; O’Garra, A. Type I interferons in tuberculosis: Foe and occasionally friend. J. Exp. Med. 2018, 215, 1273–1285. [Google Scholar] [CrossRef]
- Ji, D.X.; Yamashiro, L.H.; Chen, K.J.; Mukaida, N.; Kramnik, I.; Darwin, K.H.; Vance, R.E. Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat. Microbiol. 2019, 4, 2128–2135. [Google Scholar] [CrossRef]
- Berry, M.P.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef]
- Madhvi, A.; Mishra, H.; Chegou, N.N.; Baker, B. Increased Interferon-Induced Protein with Tetracopeptides (IFITs) Reduces Mycobacterial Growth. Front. Cell Infect. Microbiol. 2022, 12, 828439. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zhang, K.; Singh, V.K.; Mishra, A.; Kachroo, P.; Bing, T.; Won, J.H.; Mani, A.; Papanna, R.; Mann, L.K.; et al. Human M1 macrophages express unique innate immune response genes after mycobacterial infection to defend against tuberculosis. Commun. Biol. 2022, 5, 480. [Google Scholar] [CrossRef] [PubMed]
- Cilloniz, C.; Pantin-Jackwood, M.J.; Ni, C.; Carter, V.S.; Korth, M.J.; Swayne, D.E.; Tumpey, T.M.; Katze, M.G. Molecular signatures associated with Mx1-mediated resistance to highly pathogenic influenza virus infection: Mechanisms of survival. J. Virol. 2012, 86, 2437–2446. [Google Scholar] [CrossRef] [PubMed]
- Leisching, G.; Wiid, I.; Baker, B. OAS1, 2, and 3: Significance During Active Tuberculosis? J. Infect. Dis. 2018, 217, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Cheng, Q.; Wen, Z.; Song, Y.; Zhu, Y.; Wang, L. IRF1 as a potential biomarker in Mycobacterium tuberculosis infection. J. Cell Mol. Med. 2021, 25, 7270–7279. [Google Scholar] [CrossRef] [PubMed]
- Zak, D.E.; Penn-Nicholson, A.; Scriba, T.J.; Thompson, E.; Suliman, S.; Amon, L.M.; Mahomed, H.; Erasmus, M.; Whatney, W.; Hussey, G.D.; et al. A blood RNA signature for tuberculosis disease risk: A prospective cohort study. Lancet 2016, 387, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Singhania, A.; Verma, R.; Graham, C.M.; Lee, J.; Tran, T.; Richardson, M.; Lecine, P.; Leissner, P.; Berry, M.P.R.; Wilkinson, R.J.; et al. A modular transcriptional signature identifies phenotypic heterogeneity of human tuberculosis infection. Nat. Commun. 2018, 9, 2308. [Google Scholar] [CrossRef]
- Esmail, H.; Lai, R.P.; Lesosky, M.; Wilkinson, K.A.; Graham, C.M.; Horswell, S.; Coussens, A.K.; Barry, C.E., 3rd; O’Garra, A.; Wilkinson, R.J. Complement pathway gene activation and rising circulating immune complexes characterize early disease in HIV-associated tuberculosis. Proc. Natl. Acad. Sci. USA 2018, 115, E964–E973. [Google Scholar] [CrossRef] [PubMed]
- Dorhoi, A.; Yeremeev, V.; Nouailles, G.; Weiner, J., 3rd; Jörg, S.; Heinemann, E.; Oberbeck-Müller, D.; Knaul, J.K.; Vogelzang, A.; Reece, S.T.; et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 2014, 44, 2380–2393. [Google Scholar] [CrossRef]
- Carmona, J.; Cruz, A.; Moreira-Teixeira, L.; Sousa, C.; Sousa, J.; Osorio, N.S.; Saraiva, A.L.; Svenson, S.; Kallenius, G.; Pedrosa, J.; et al. Mycobacterium tuberculosis Strains Are Differentially Recognized by TLRs with an Impact on the Immune Response. PLoS ONE 2013, 8, e67277. [Google Scholar] [CrossRef]
- Ordway, D.; Henao-Tamayo, M.; Harton, M.; Palanisamy, G.; Troudt, J.; Shanley, C.; Basaraba, R.J.; Orme, I.M. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J. Immunol. 2007, 179, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Manca, C.; Tsenova, L.; Freeman, S.; Barczak, A.K.; Tovey, M.; Murray, P.J.; Barry, C.; Kaplan, G. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 2005, 25, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Banks, D.A.; Ahlbrand, S.E.; Hughitt, V.K.; Shah, S.; Mayer-Barber, K.D.; Vogel, S.N.; El-Sayed, N.M.; Briken, V. Mycobacterium tuberculosis Inhibits Autocrine Type I IFN Signaling to Increase Intracellular Survival. J. Immunol. 2019, 202, 2348–2359. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.K.; Schlesinger, L.S. Characterization of mannose receptor-dependent phagocytosis mediated by Mycobacterium tuberculosis lipoarabinomannan. Infect. Immun. 1998, 66, 2769–2777. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Oddo, M.; Renno, T.; Attinger, A.; Bakker, T.; MacDonald, H.R.; Meylan, P.R. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J. Immunol. 1998, 160, 5448–5454. [Google Scholar] [CrossRef] [PubMed]
- Mohareer, K.; Medikonda, J.; Vadankula, G.R.; Banerjee, S. Mycobacterial Control of Host Mitochondria: Bioenergetic and Metabolic Changes Shaping Cell Fate and Infection Outcome. Front. Cell Infect. Microbiol. 2020, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.; Prabhu, R.; Gross, C.M.; Riesenberg, L.A.; Singh, V.; Aggarwal, S. Role of apoptosis and autophagy in tuberculosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L218–L229. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Booty, M.G.; Rosebrock, T.R.; Nunes-Alves, C.; Desjardins, D.M.; Keren, I.; Fortune, S.M.; Remold, H.G.; Behar, S.M. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 2012, 12, 289–300. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Costantini, P.; Jacotot, E.; Decaudin, D.; Kroemer, G. Mitochondrion as a novel target of anticancer chemotherapy. J. Natl. Cancer Inst. 2000, 92, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Bratton, S.B.; Langlais, C.; Walker, G.; Brown, D.G.; Sun, X.M.; Cohen, G.M. Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J. Biol. Chem. 2000, 275, 6067–6070. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Shroff, E.H.; Liu, J.; Chandel, N.S. Nitric oxide induces cell death by regulating anti-apoptotic BCL-2 family members. PLoS ONE 2009, 4, e7059. [Google Scholar] [CrossRef] [PubMed]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond-mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Herbst, S.; Schaible, U.E.; Schneider, B.E. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS ONE 2011, 6, e19105. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef]
- Kelly, D.M.; ten Bokum, A.M.; O’Leary, S.M.; O’Sullivan, M.P.; Keane, J. Bystander macrophage apoptosis after Mycobacterium tuberculosis H37Ra infection. Infect. Immun. 2008, 76, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Schaible, U.E.; Winau, F.; Sieling, P.A.; Fischer, K.; Collins, H.L.; Hagens, K.; Modlin, R.L.; Brinkmann, V.; Kaufmann, S.H. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat. Med. 2003, 9, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Keane, J.; Remold, H.G.; Kornfeld, H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol. 2000, 164, 2016–2020. [Google Scholar] [CrossRef] [PubMed]
- Keane, J.; Balcewicz-Sablinska, M.K.; Remold, H.G.; Chupp, G.L.; Meek, B.B.; Fenton, M.J.; Kornfeld, H. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect. Immun. 1997, 65, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Balcewicz-Sablinska, M.K.; Keane, J.; Kornfeld, H.; Remold, H.G. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J. Immunol. 1998, 161, 2636–2641. [Google Scholar] [CrossRef] [PubMed]
- Balcewicz-Sablinska, M.K.; Gan, H.; Remold, H.G. Interleukin 10 produced by macrophages inoculated with Mycobacterium avium attenuates mycobacteria-induced apoptosis by reduction of TNF-alpha activity. J. Infect. Dis. 1999, 180, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schreiber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.M.; Barbosa, A.M.; Barreira-Silva, P.; Silvestre, R.; Cunha, C.; Carvalho, A.; Rodrigues, F.; Correia-Neves, M.; Castro, A.G.; Torrado, E. Early IL-10 promotes vasculature-associated CD4+ T cells unable to control Mycobacterium tuberculosis infection. JCI Insight 2021, 6, e150060. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef]
- Wong, E.A.; Evans, S.; Kraus, C.R.; Engelman, K.D.; Maiello, P.; Flores, W.J.; Cadena, A.M.; Klein, E.; Thomas, K.; White, A.G.; et al. IL-10 Impairs Local Immune Response in Lung Granulomas and Lymph Nodes during Early Mycobacterium tuberculosis Infection. J. Immunol. 2020, 204, 644–659. [Google Scholar] [CrossRef]
- Wolf, A.J.; Desvignes, L.; Linas, B.; Banaiee, N.; Tamura, T.; Takatsu, K.; Ernst, J.D. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 2008, 205, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.; Raje, M.; Majumdar, S. Differential expression of NF-kappaB in mycobacteria infected THP-1 affects apoptosis. Biochim. Biophys. Acta 2007, 1770, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Sly, L.M.; Hingley-Wilson, S.M.; Reiner, N.E.; McMaster, W.R. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J. Immunol. 2003, 170, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Apoptosis and nuclear factor-kappa B: A tale of association and dissociation. Biochem. Pharmacol. 2000, 60, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Lu, Y.; Wang, X.; Zhang, S.; Wang, Y.; Wu, F.; Zhang, W.; Wang, X.; Zhang, L. Regulatory role and mechanism of the inhibition of the Mcl-1 pathway during apoptosis and polarization of H37Rv-infected macrophages. Medicine 2020, 99, e22438. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.I.; Choi, S.; Choi, H.G.; Kebede, S.G.; Dang, T.B.; Back, Y.W.; Park, H.S.; Kim, H.J. Recombinant Rv3261 protein of Mycobacterium tuberculosis induces apoptosis through a mitochondrion-dependent pathway in macrophages and inhibits intracellular bacterial growth. Cell Immunol. 2020, 354, 104145. [Google Scholar] [CrossRef]
- Medha; Priyanka; Bhatt, P.; Sharma, S.; Sharma, M. Role of C-terminal domain of Mycobacterium tuberculosis PE6 (Rv0335c) protein in host mitochondrial stress and macrophage apoptosis. Apoptosis 2023, 28, 136–165. [Google Scholar] [CrossRef]
- Lorenzo, H.K.; Susin, S.A. Mitochondrial effectors in caspase-independent cell death. FEBS Lett. 2004, 557, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Hmama, Z.; Peña-Díaz, S.; Joseph, S.; Av-Gay, Y. Immunoevasion and immunosuppression of the macrophage by Mycobacterium tuberculosis. Immunol. Rev. 2015, 264, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Jeon, B.Y.; Lee, H.M.; Jin, H.S.; Yuk, J.M.; Song, C.H.; Lee, S.H.; Lee, Z.W.; Cho, S.N.; Kim, J.M.; et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010, 6, e1001230. [Google Scholar] [CrossRef] [PubMed]
- Hussain Bhat, K.; Mukhopadhyay, S. Macrophage takeover and the host-bacilli interplay during tuberculosis. Future Microbiol. 2015, 10, 853–872. [Google Scholar] [CrossRef] [PubMed]
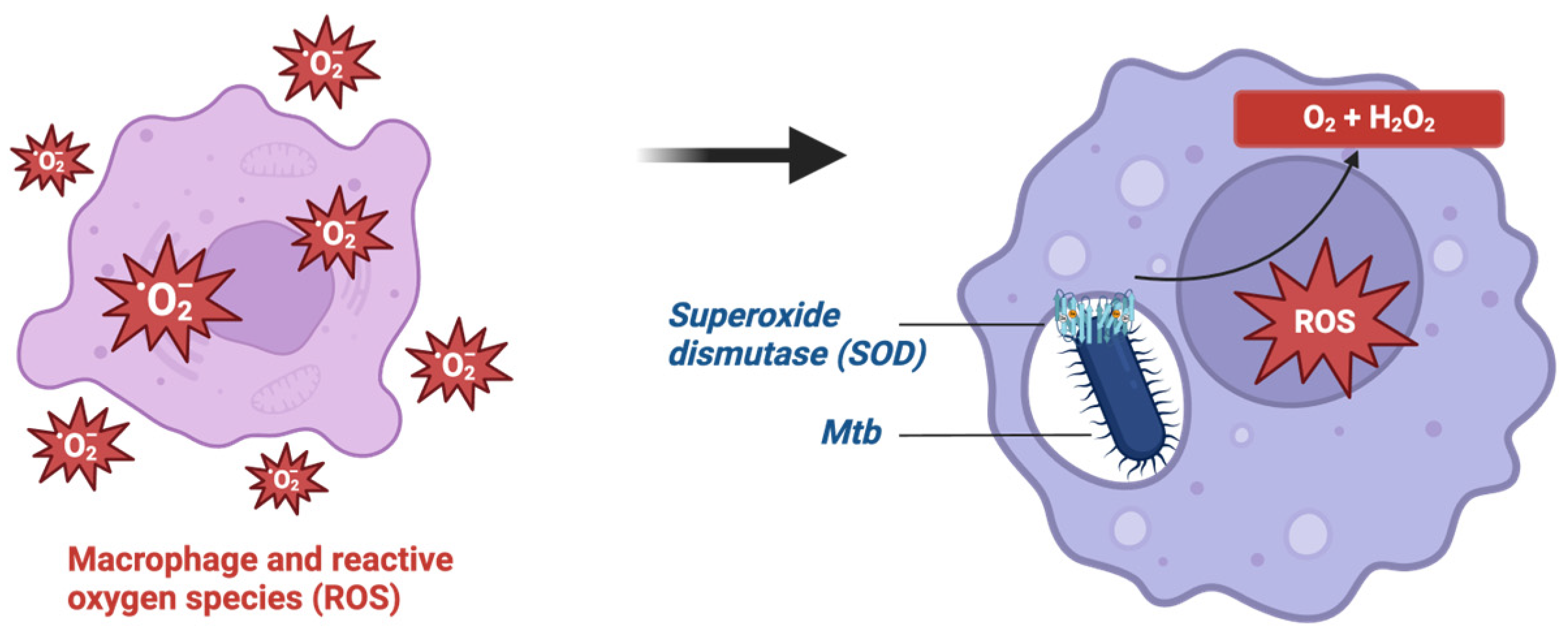
- Tyagi, P.; Dharmaraja, A.T.; Bhaskar, A.; Chakrapani, H.; Singh, A. Mycobacterium tuberculosis has diminished capacity to counteract redox stress induced by elevated levels of endogenous superoxide. Free Radic. Biol. Med. 2015, 84, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Nambi, S.; Long, J.E.; Mishra, B.B.; Baker, R.; Murphy, K.C.; Olive, A.J.; Nguyen, H.P.; Shaffer, S.A.; Sassetti, C.M. The Oxidative Stress Network of Mycobacterium tuberculosis Reveals Coordination between Radical Detoxification Systems. Cell Host Microbe 2015, 17, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Afriyie-Asante, A.; Dabla, A.; Dagenais, A.; Berton, S.; Smyth, R.; Sun, J. Mycobacterium tuberculosis Exploits Focal Adhesion Kinase to Induce Necrotic Cell Death and Inhibit Reactive Oxygen Species Production. Front. Immunol. 2021, 12, 742370. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, H.K.; Susin, S.A.; Penninger, J.; Kroemer, G. Apoptosis inducing factor (AIF): A phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999, 6, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.J.; Choi, J.A.; Choi, H.H.; Cho, S.N.; Kim, H.J.; Jo, E.K.; Park, J.K.; Song, C.H. Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of Mycobacterium tuberculosis. PLoS ONE 2011, 6, e28531. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Tang, J.; He, Z.G. Induction of Endoplasmic Reticulum Stress by CdhM Mediates Apoptosis of Macrophage During Mycobacterium tuberculosis Infection. Front. Cell Infect. Microbiol. 2022, 12, 877265. [Google Scholar] [CrossRef] [PubMed]
- Khalid, N.; Azimpouran, M. Necrosis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Liu, Z.G.; Jiao, D. Necroptosis, tumor necrosis and tumorigenesis. Cell Stress. 2019, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Amaral, E.P.; Costa, D.L.; Namasivayam, S.; Riteau, N.; Kamenyeva, O.; Mittereder, L.; Mayer-Barber, K.D.; Andrade, B.B.; Sher, A. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J. Exp. Med. 2019, 216, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gan, H.; Remold, H.G. A mechanism of virulence: Virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 2006, 176, 3707–3716. [Google Scholar] [CrossRef] [PubMed]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Lerner, T.R.; Borel, S.; Greenwood, D.J.; Repnik, U.; Russell, M.R.; Herbst, S.; Jones, M.L.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.G. Mycobacterium tuberculosis replicates within necrotic human macrophages. J. Cell Biol. 2017, 216, 583–594. [Google Scholar] [CrossRef]
- Andersson, A.M.; Andersson, B.; Lorell, C.; Raffetseder, J.; Larsson, M.; Blomgran, R. Autophagy induction targeting mTORC1 enhances Mycobacterium tuberculosis replication in HIV co-infected human macrophages. Sci. Rep. 2016, 6, 28171. [Google Scholar] [CrossRef]
- Singhal, A.; Jie, L.; Kumar, P.; Hong, G.S.; Leow, M.K.; Paleja, B.; Tsenova, L.; Kurepina, N.; Chen, J.; Zolezzi, F.; et al. Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 2014, 6, 263ra159. [Google Scholar] [CrossRef]
- Moreira, J.D.; Koch, B.E.V.; van Veen, S.; Walburg, K.V.; Vrieling, F.; Mara Pinto Dabés Guimarães, T.; Meijer, A.H.; Spaink, H.P.; Ottenhoff, T.H.M.; Haks, M.C.; et al. Functional Inhibition of Host Histone Deacetylases (HDACs) Enhances in vitro and in vivo Anti-mycobacterial Activity in Human Macrophages and in Zebrafish. Front. Immunol. 2020, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Adikesavalu, H.; Gopalaswamy, R.; Kumar, A.; Ranganathan, U.D.; Shanmugam, S. Autophagy Induction as a Host-Directed Therapeutic Strategy against Mycobacterium tuberculosis Infection. Medicina 2021, 57, 522. [Google Scholar] [CrossRef]
- Lam, K.K.; Zheng, X.; Forestieri, R.; Balgi, A.D.; Nodwell, M.; Vollett, S.; Anderson, H.J.; Andersen, R.J.; Av-Gay, Y.; Roberge, M. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012, 8, e1002691. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, K.M.; Ranganathan, U.D.; Tripathy, S.P.; Bethunaickan, R. Vitamin D—A host directed autophagy mediated therapy for tuberculosis. Mol. Immunol. 2020, 127, 238–244. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Falkenhorst, J.; Grunewald, S.; Mühlenberg, T.; Marino-Enriquez, A.; Reis, A.C.; Corless, C.; Heinrich, M.; Treckmann, J.; Podleska, L.E.; Schuler, M.; et al. Inhibitor of Apoptosis Proteins (IAPs) are commonly dysregulated in GIST and can be pharmacologically targeted to enhance the pro-apoptotic activity of imatinib. Oncotarget 2016, 7, 41390–41403. [Google Scholar] [CrossRef] [PubMed]
- Stutz, M.D.; Allison, C.C.; Ojaimi, S.; Preston, S.P.; Doerflinger, M.; Arandjelovic, P.; Whitehead, L.; Bader, S.M.; Batey, D.; Asselin-Labat, M.L.; et al. Macrophage and neutrophil death programs differentially confer resistance to tuberculosis. Immunity 2021, 54, 1758–1771.e7. [Google Scholar] [CrossRef]
- Lim, Y.J.; Lee, J.; Choi, J.A.; Cho, S.N.; Son, S.H.; Kwon, S.J.; Son, J.W.; Song, C.H. M1 macrophage dependent-p53 regulates the intracellular survival of mycobacteria. Apoptosis 2020, 25, 42–55. [Google Scholar] [CrossRef]
- Zhuang, L.; Yang, L.; Li, L.; Ye, Z.; Gong, W. Mycobacterium tuberculosis: Immune response, biomarkers, and therapeutic intervention. MedComm (2020) 2024, 5, e419. [Google Scholar] [CrossRef]
- Jeong, E.K.; Lee, H.J.; Jung, Y.J. Host-Directed Therapies for Tuberculosis. Pathogens 2022, 11, 1291. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, V.M.; Gröschel, M.I.; Martinson, N.; Zumla, A.; Maeurer, M.; van der Werf, T.S.; Vilaplana, C. Non-Steroidal Anti-inflammatory Drugs As Host-Directed Therapy for Tuberculosis: A Systematic Review. Front. Immunol. 2017, 8, 772. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.M. Host-Directed Therapies for Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 5, a021196. [Google Scholar] [CrossRef] [PubMed]
- Subbian, S.; Tsenova, L.; Holloway, J.; Peixoto, B.; O’Brien, P.; Dartois, V.; Khetani, V.; Zeldis, J.B.; Kaplan, G. Adjunctive Phosphodiesterase-4 Inhibitor Therapy Improves Antibiotic Response to Pulmonary Tuberculosis in a Rabbit Model. eBioMedicine 2016, 4, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Alsayed, S.S.R.; Gunosewoyo, H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. Int. J. Mol. Sci. 2023, 24, 5202. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.S.; Zhang, X.D.; Yan, J.W.; Huang, T.T.; Liu, Z.Z.; Li, Z.K.; Wang, L.; Li, F. Identification of Mycobacterium tuberculosis Resistance to Common Antibiotics: An Overview of Current Methods and Techniques. Infect. Drug Resist. 2024, 17, 1491–1506. [Google Scholar] [CrossRef]
- Negi, A.; Perveen, S.; Gupta, R.; Singh, P.P.; Sharma, R. Unraveling Dilemmas and Lacunae in the Escalating Drug Resistance of Mycobacterium tuberculosis to Bedaquiline, Delamanid, and Pretomanid. J. Med. Chem. 2024, 67, 2264–2286. [Google Scholar] [CrossRef] [PubMed]
- Mtimka, S.; Pillay, P.; Kwezi, L.; Pooe, O.J.; Tsekoa, T.L. An Exploratory Review of the Potential of Lytic Proteins and Bacteriophages for the Treatment of Tuberculosis. Microorganisms 2024, 12, 570. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.S.; Johnson, J.L.; Okwera, A.; Kanost, R.A.; Wu, M.; Peters, P.; Muhumuza, M.; Mayanja-Kizza, H.; Mugerwa, R.D.; Mugyenyi, P.; et al. Mechanisms of apoptosis of T-cells in human tuberculosis. J. Clin. Immunol. 2005, 25, 353–364. [Google Scholar] [CrossRef]
- Pagán, A.J.; Ramakrishnan, L. Immunity and Immunopathology in the Tuberculous Granuloma. Cold Spring Harb. Perspect. Med. 2014, 5, a018499. [Google Scholar] [CrossRef]
- Dallenga, T.; Repnik, U.; Corleis, B.; Eich, J.; Reimer, R.; Griffiths, G.W.; Schaible, U.E. M. tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe 2017, 22, 519–530.e3. [Google Scholar] [CrossRef] [PubMed]
- Toossi, Z. The inflammatory response in Mycobacterium tuberculosis infection. Arch. Immunol. Ther. Exp. 2000, 48, 513–519. [Google Scholar] [PubMed]
- Su, H.; Zhu, S.; Zhu, L.; Huang, W.; Wang, H.; Zhang, Z.; Xu, Y. Recombinant Lipoprotein Rv1016c Derived from Mycobacterium tuberculosis Is a TLR-2 Ligand that Induces Macrophages Apoptosis and Inhibits MHC II Antigen Processing. Front. Cell Infect. Microbiol. 2016, 6, 147. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Pathway | Mechanism |
|---|---|---|
| Rapamycin, histone deacetylase inhibitors | Autophagy | Inhibition of mTOR |
| Metformin | Autophagy | Activation of AMPK |
| Transcription factor EB | Autophagy | Regulates autophagy gene expression |
| Lysosomotropic agents | Autophagy | Enhance lysosomal acidity |
| Nitazoxanide | Autophagy | Activation of mTORC1 inhibition |
| Vitamin D | Autophagy | Synthesis of CAMP |
| BH3 mimetics | Apoptosis | BLC-2-mediated apoptosis |
| Nutlin-3 | Apoptosis | Activation of p53 |
| NSAIDs, corticosteroid, zileuton, desipramine | Necrosis | Eicosanoid modulation |
| Phosphodiesterase inhibitors | Necrosis | TNF reduction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, A.; Glassman, I.; Campbell, G.; Yeganyan, S.; Nguyen, J.; Shin, A.; Venketaraman, V. Host Cell Death and Modulation of Immune Response against Mycobacterium tuberculosis Infection. Int. J. Mol. Sci. 2024, 25, 6255. https://doi.org/10.3390/ijms25116255
Vu A, Glassman I, Campbell G, Yeganyan S, Nguyen J, Shin A, Venketaraman V. Host Cell Death and Modulation of Immune Response against Mycobacterium tuberculosis Infection. International Journal of Molecular Sciences. 2024; 25(11):6255. https://doi.org/10.3390/ijms25116255
Chicago/Turabian StyleVu, Annie, Ira Glassman, Giliene Campbell, Stephanie Yeganyan, Jessica Nguyen, Andrew Shin, and Vishwanath Venketaraman. 2024. "Host Cell Death and Modulation of Immune Response against Mycobacterium tuberculosis Infection" International Journal of Molecular Sciences 25, no. 11: 6255. https://doi.org/10.3390/ijms25116255