
Gene Expression Analysis of Yeast Strains with a Nonsense Mutation in the eRF3-Coding Gene Highlights Possible Mechanisms of Adaptation

 , , , , , , and
, , , , , , and 
Abstract
1. Introduction
2. Results
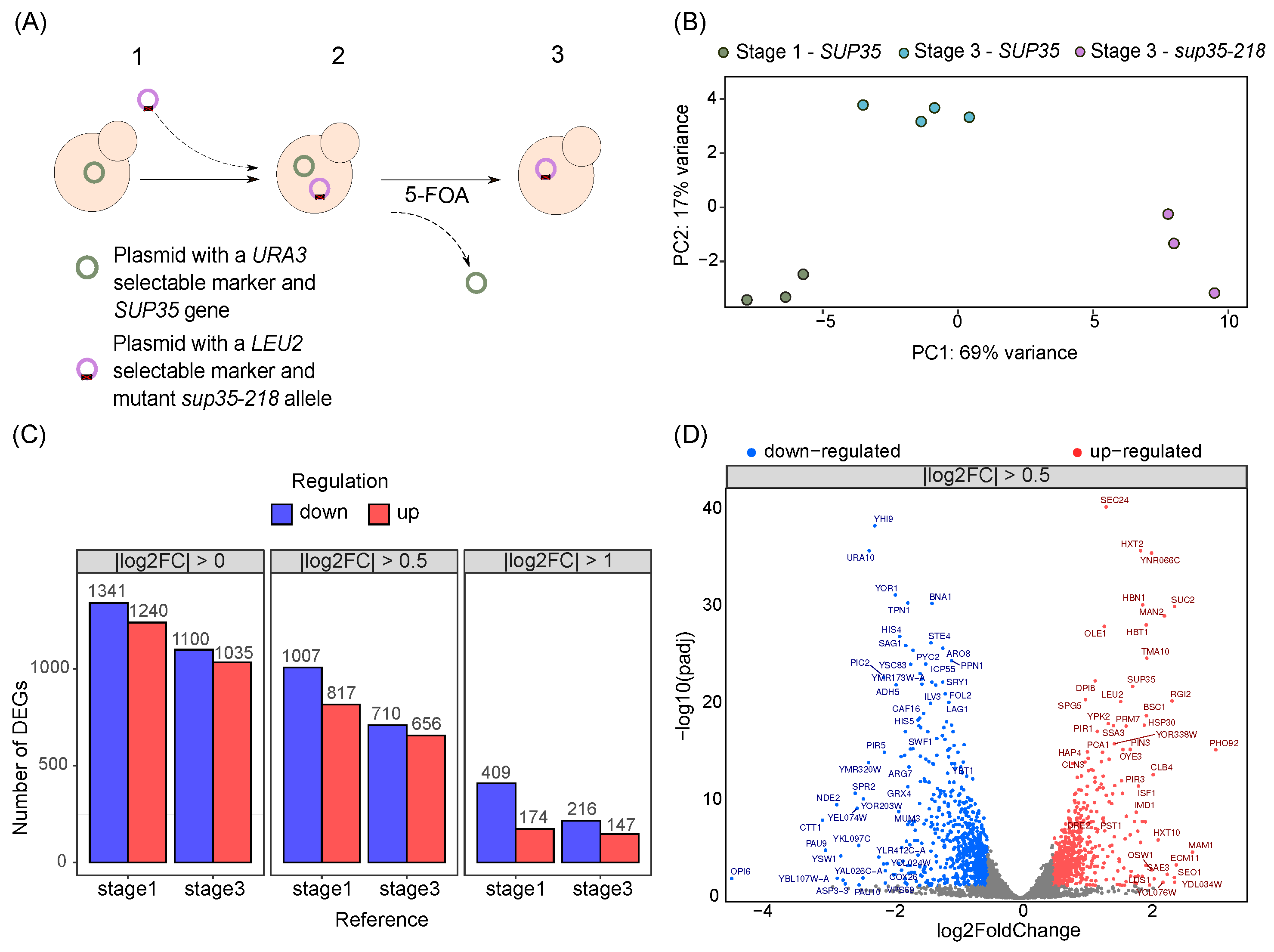
2.1. Global Transcriptional Changes in Yeast Strains with Nonsense Mutations in the Release Factor Genes
2.2. Identification of Biological Processes Involved in Adaptation to the Presence of sup35-218
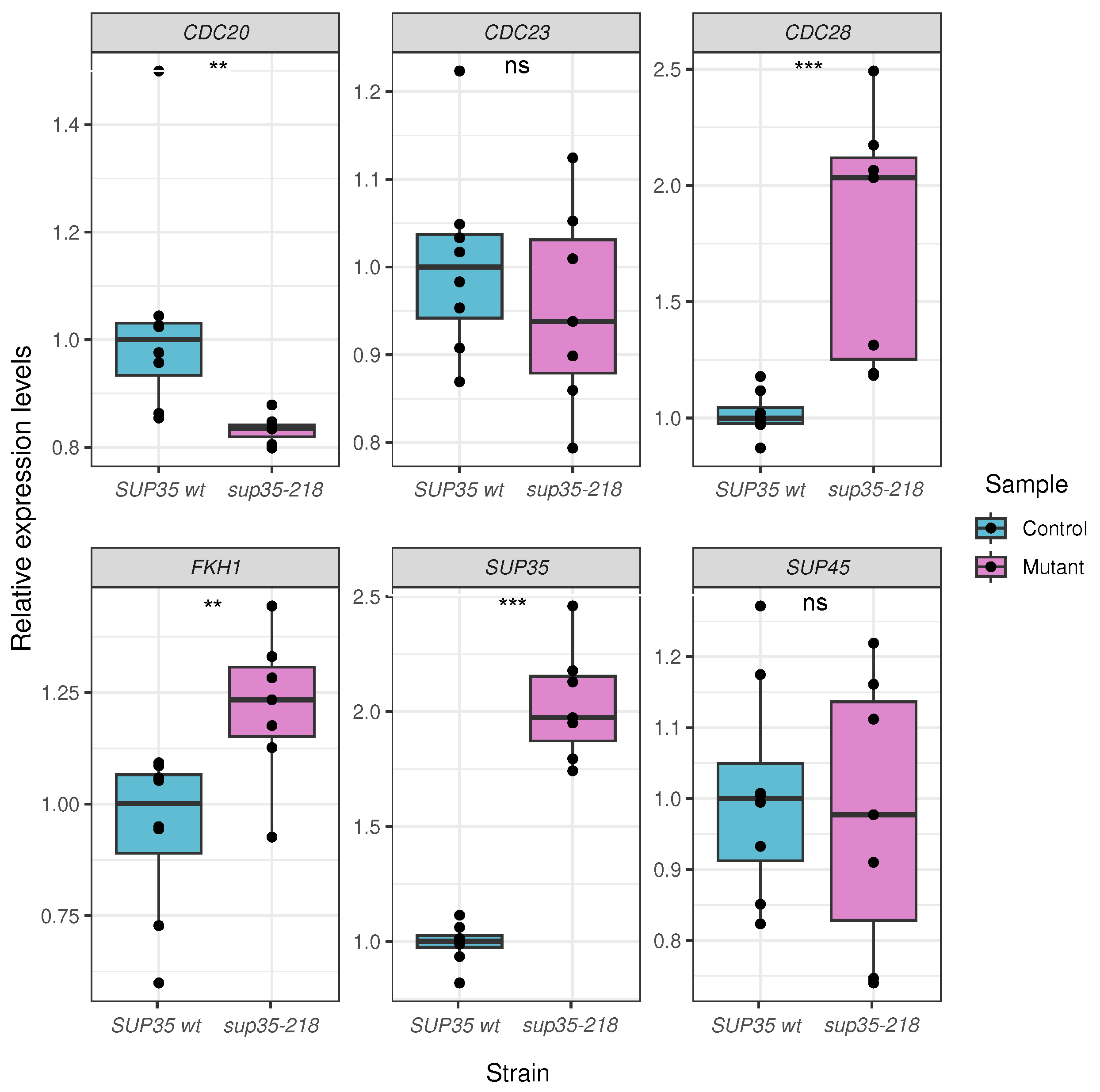
2.3. Validation of High-Throughput Sequencing Results Using qPCR
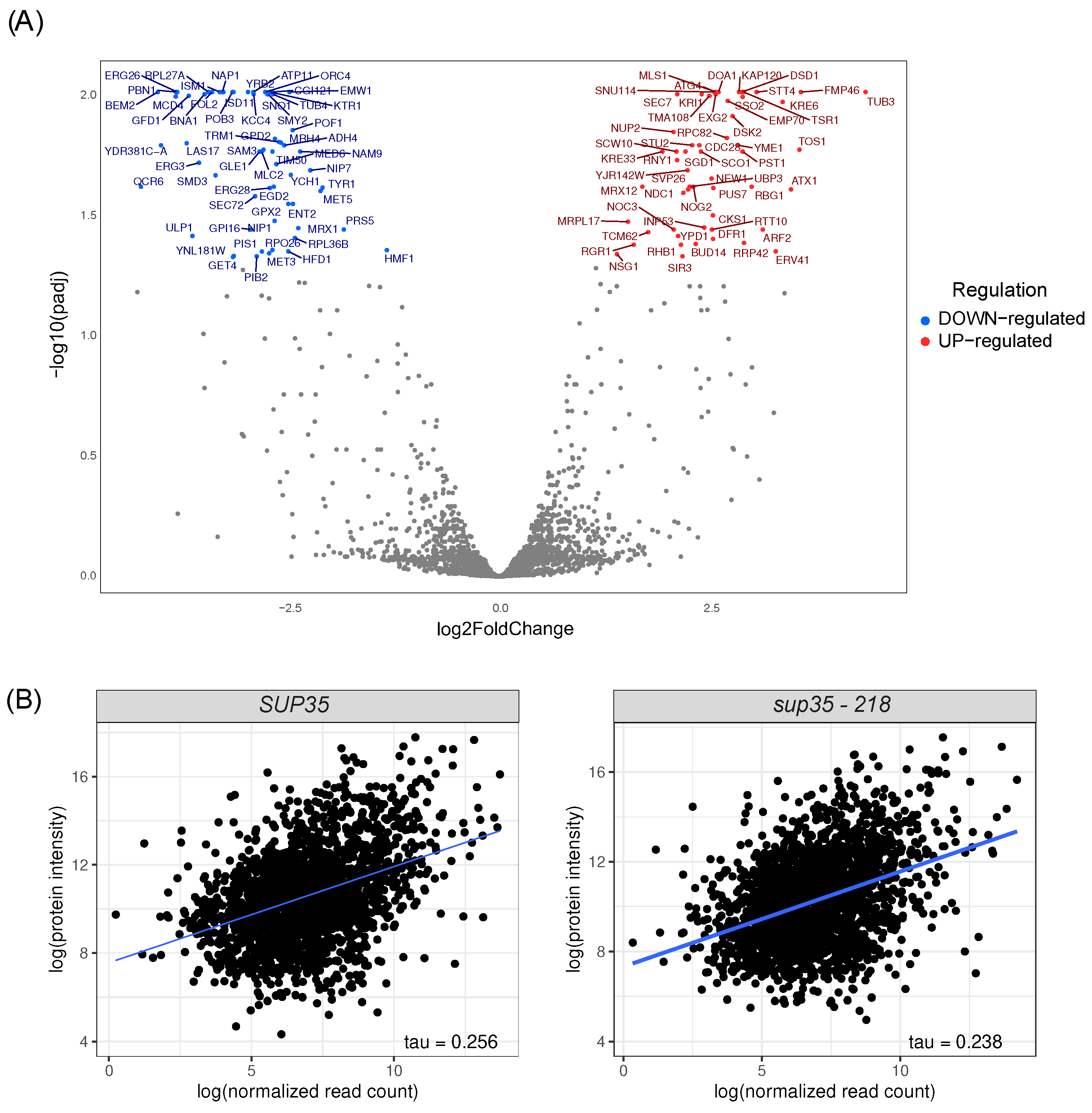
2.4. Proteome Analysis of Cells Harboring sup35-218 Allele
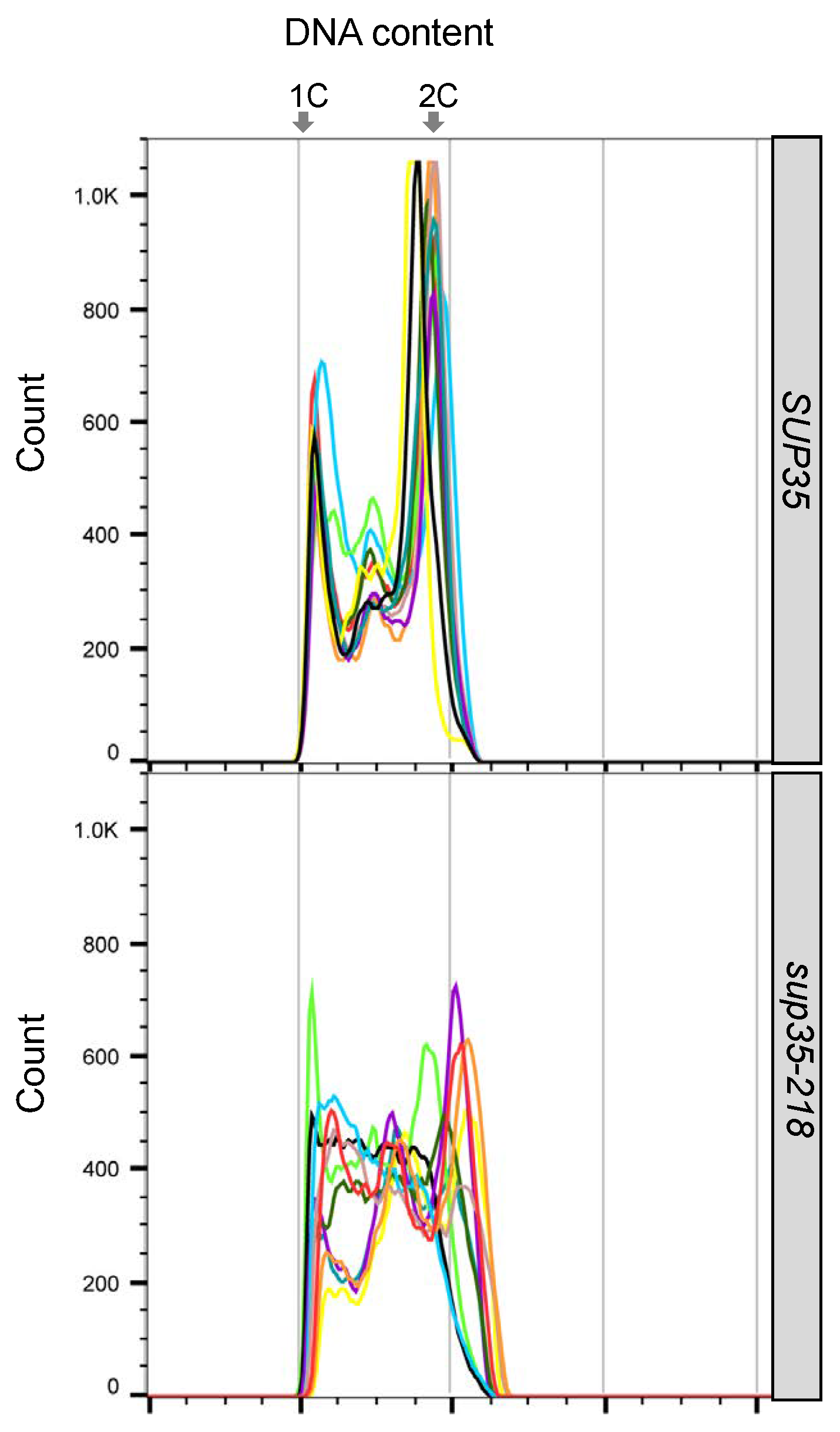
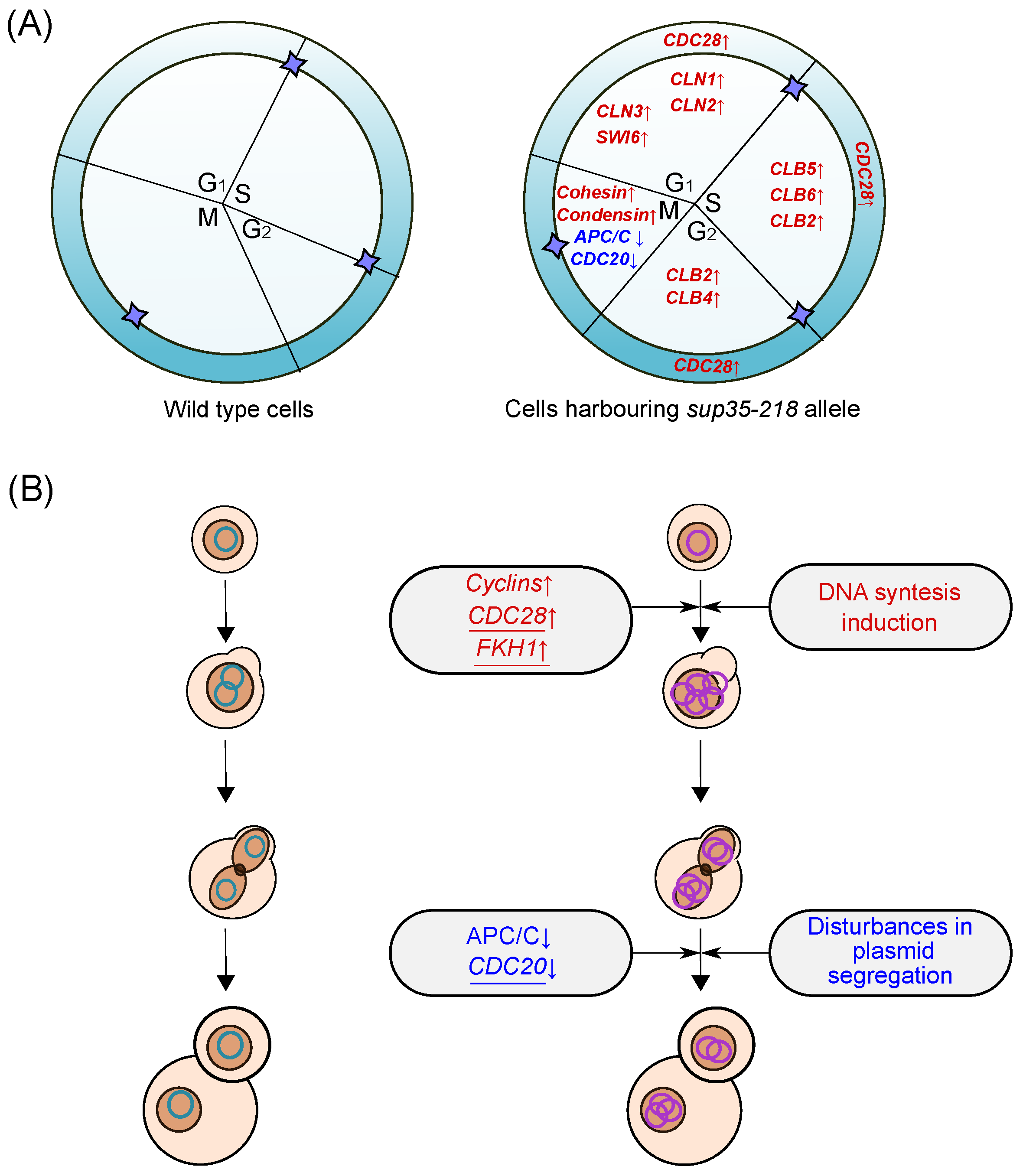
2.5. Effects of the sup35-218 Mutation on Cell Cycle Progression
3. Discussion
4. Materials and Methods
4.1. Yeast Strains and Media
4.2. Cell Cycle Analysis by Flow Cytometry
4.3. RNA-Seq Library Preparation and Sequencing
4.4. RNA-Seq Data Analysis
4.5. RNA Extraction and cDNA Generation for qPCR
4.6. qPCR
4.7. Protein Extraction
4.8. Proteome Analysis
4.9. Code Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| eRF3 | eukaryotic release factor 3 |
| APC | anaphase-promoting complex |
| PTC | premature termination codon |
| DEGs | differentially expressed genes |
| GO | Gene Ontology |
| TF | transcription factor |
References
- Hellen, C.U.T. Translation termination and ribosome recycling in eukaryotes. Cold Spring Harb. Perspect. Biol. 2018, 10, a032656. [Google Scholar] [CrossRef] [PubMed]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Martins-Dias, P.; Romão, L. Nonsense suppression therapies in human genetic diseases. Cell. Mol. Life Sci. 2021, 78, 4677–4701. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Tang, B.; He, X.; Zou, P.; Zeng, Z.; Xiao, R. Nonsense suppression therapy: An emerging treatment for hereditary skin diseases. Acta Derm.Venereol. 2022, 102, adv00658. [Google Scholar] [CrossRef]
- Frolova, L.; Le Goff, X.; Rasmussen, H.H.; Cheperegin, S.; Drugeon, G.; Kress, M.; Arman, I.; Haenni, A.L.; Celis, J.E.; Philippe, M. A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature 1994, 372, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Stansfield, I.; Jones, K.M.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Poznyakovski, A.I.; Paushkin, S.V.; Nierras, C.R.; Cox, B.S.; Ter-Avanesyan, M.D.; Tuite, M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373. [Google Scholar] [CrossRef] [PubMed]
- Zhouravleva, G.; Frolova, L.; Le Goff, X.; Le Guellec, R.; Inge-Vechtomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995, 14, 4065–4072. [Google Scholar] [CrossRef]
- Moskalenko, S.E.; Chabelskaya, S.V.; Inge-Vechtomov, S.G.; Philippe, M.; Zhouravleva, G.A. Viable nonsense mutants for the essential gene SUP45 of Saccharomyces cerevisiae. BMC Mol. Biol. 2003, 4, 2. [Google Scholar] [CrossRef]
- Chabelskaya, S.; Kiktev, D.; Inge-Vechtomov, S.; Philippe, M.; Zhouravleva, G. Nonsense mutations in the essential gene SUP35 of Saccharomyces cerevisiae are non-lethal. Mol. Genet. Genom. 2004, 272, 297–307. [Google Scholar] [CrossRef]
- Maksiutenko, E.M.; Barbitoff, Y.A.; Matveenko, A.G.; Moskalenko, S.E.; Zhouravleva, G.A. Gene amplification as a mechanism of yeast adaptation to nonsense mutations in release factor genes. Genes 2021, 12, 2019. [Google Scholar] [CrossRef]
- Bonetti, B.; Fu, L.; Moon, J.; Bedwell, D.M. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J. Mol. Biol. 1995, 251, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Bertram, G.; Innes, S.; Minella, O.; Richardson, J.P.; Stansfield, I. Endless possibilities: Translation termination and stop codon recognition. Microbiology 2001, 147, 255–269. [Google Scholar] [CrossRef]
- Hamza, A.; Baetz, K. Iron-responsive transcription factor Aft1 interacts with kinetochore protein Iml3 and promotes pericentromeric cohesin. J. Biol. Chem. 2012, 287, 4139–4147. [Google Scholar] [CrossRef]
- Pramila, T.; Miles, S.; GuhaThakurta, D.; Jemiolo, D.; Breeden, L.L. Conserved homeodomain proteins interact with MADS box protein Mcm1 to restrict ECB—dependent transcription to the M/G1 phase of the cell cycle. Genes Dev. 2002, 16, 3034–3045. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.M.; Primig, M.; Washburn, B.K.; Winzeler, E.A.; Bellis, M.; Sarrauste de Menthiere, C.; Davis, R.W.; Esposito, R.E. The Ume6 regulon coordinates metabolic and meiotic gene expression in yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 13431–13436. [Google Scholar] [CrossRef]
- Raithatha, S.A.; Vaza, S.; Islam, M.T.; Greenwood, B.; Stuart, D.T. Ume6 acts as a stable platform to coordinate repression and activation of early meiosis-specific genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 2021, 41, e0037820. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hou, R.; Liu, C.; Yang, T.; Qiao, C.; Wei, J. Integration of transcriptome and proteome profiles in placenta accreta reveals trophoblast over-migration as the underlying pathogenesis. Clin. Proteom. 2021, 18, 31. [Google Scholar] [CrossRef]
- Causton, H.C.; Ren, B.; Koh, S.S.; Harbison, C.T.; Kanin, E.; Jennings, E.G.; Lee, T.I.; True, H.L.; Lander, E.S.; Young, R.A. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 2001, 12, 323–337. [Google Scholar] [CrossRef]
- Taymaz-Nikerel, H.; Cankorur-Cetinkaya, A.; Kirdar, B. Genome-Wide transcriptional response of Saccharomyces cerevisiae to stress-induced perturbations. Front. Bioeng. Biotechnol. 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Rupes, I. Checking cell size in yeast. Trends Genet. 2002, 18, 479–485. [Google Scholar] [CrossRef]
- Jiménez, J.; Bru, S.; Ribeiro, M.; Clotet, J. Live fast, die soon: Cell cycle progression and lifespan in yeast cells. Microb. Cell 2015, 2, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Enserink, J.M.; Chymkowitch, P. Cell cycle-dependent transcription: The cyclin dependent kinase Cdk1 is a direct regulator of basal transcription machineries. Int. J. Mol. Sci. 2022, 23, 1293. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, M.D.; Hodge, A.E. Regulation of Cdc28 cyclin-dependent protein kinase activity during the cell cycle of the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1998, 62, 1191–1243. [Google Scholar] [CrossRef] [PubMed]
- Hoggard, T.; Hollatz, A.J.; Cherney, R.E.; Seman, M.R.; Fox, C.A. The Fkh1 Forkhead associated domain promotes ORC binding to a subset of DNA replication origins in budding yeast. Nucleic Acids Res. 2021, 49, 10207–10220. [Google Scholar] [CrossRef] [PubMed]
- Postnikoff, S.D.L.; Malo, M.E.; Wong, B.; Harkness, T.A.A. The yeast forkhead transcription factors FKH1 and FKH2 regulate lifespan and stress response together with the anaphase-promoting complex. PLoS Genet. 2012, 8, e1002583. [Google Scholar] [CrossRef] [PubMed]
- Adler, S.O.; Spiesser, T.W.; Uschner, F.; Münzner, U.; Hahn, J.; Krantz, M.; Klipp, E. A yeast cell cycle model integrating stress, signaling, and physiology. FEMS Yeast Res. 2022, 22, foac026. [Google Scholar] [CrossRef] [PubMed]
- Shirayama, M.; Tóth, A.; Gálová, M.; Nasmyth, K. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature 1999, 402, 203–207. [Google Scholar] [CrossRef]
- Schwab, M.; Lutum, A.S.; Seufert, W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 1997, 90, 683–693. [Google Scholar] [CrossRef]
- Zachariae, W.; Shevchenko, A.; Andrews, P.D.; Ciosk, R.; Galova, M.; Stark, M.J.; Mann, M.; Nasmyth, K. Mass spectrometric analysis of the anaphase-promoting complex from yeast: Identification of a subunit related to cullins. Science 1998, 279, 1216–1219. [Google Scholar] [CrossRef]
- Newlon, C.S.; Fangman, W.L. Mitochondrial DNA synthesis in cell cycle mutants of Saccharomyces cerevisiae. Cell 1975, 5, 423–428. [Google Scholar] [CrossRef]
- Lamb, J.R.; Michaud, W.A.; Sikorski, R.S.; Hieter, P.A. Cdc16p, Cdc23p and Cdc27p form a complex essential for mitosis. EMBO J. 1994, 13, 4321–4328. [Google Scholar] [CrossRef] [PubMed]
- Gregan, J.; Lindner, K.; Brimage, L.; Franklin, R.; Namdar, M.; Hart, E.A.; Aves, S.J.; Kearsey, S.E. Fission yeast Cdc23/Mcm10 functions after pre-replicative complex formation to promote Cdc45 chromatin binding. Mol. Biol. Cell 2003, 14, 3876–3887. [Google Scholar] [CrossRef] [PubMed]
- Hart, E.A.; Bryant, J.A.; Moore, K.; Aves, S.J. Fission yeast Cdc23 interactions with DNA replication initiation proteins. Curr. Genet. 2002, 41, 342–348. [Google Scholar] [CrossRef]
- Lee, J.K.; Seo, Y.S.; Hurwitz, J. The Cdc23 (Mcm10) protein is required for the phosphorylation of minichromosome maintenance complex by the Dfp1-Hsk1 kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 2334–2339. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, J.; Yue, W.; Li, A.; Meng, T.G.; Lei, W.L.; Fan, L.H.; Ouyang, Y.C.; Schatten, H.; Wang, Z.B.; et al. Cell division cycle 23 is required for mouse oocyte meiotic maturation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 8990–9002. [Google Scholar] [CrossRef]
- Wang, R.; Burton, J.L.; Solomon, M.J. Transcriptional and post-transcriptional regulation of Cdc20 during the spindle assembly checkpoint in S. cerevisiae. Cell. Signal. 2017, 33, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; York, J.P.; Zhang, P. Loss of Cdc20 causes a securin-dependent metaphase arrest in two-cell mouse embryos. Mol. Cell. Biol. 2007, 27, 3481–3488. [Google Scholar] [CrossRef]
- Chica, N.; Portantier, M.; Nyquist-Andersen, M.; Espada-Burriel, S.; Lopez-Aviles, S. Uncoupling of mitosis and cytokinesis upon a prolonged arrest in metaphase is influenced by protein phosphatases and mitotic transcription in fission yeast. Front. Cell Dev. Biol. 2022, 10, 876810. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Shimatake, H.; Kikuchi, A. A yeast gene required for the G1-to-S transition encodes a protein containing an A-kinase target site and GTPase domain. EMBO J. 1988, 7, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Borkhsenius, A.S.; Inge-Vechtomov, S.G. The role of SUP35 and SUP45 genes in controlling Saccharomycetes cell cycle. Dokl. Akad. Nauk. 1997, 353, 553–556. [Google Scholar] [PubMed]
- Valouev, I.A.; Kushnirov, V.V.; Ter-Avanesyan, M.D. Yeast polypeptide chain release factors eRF1 and eRF3 are involved in cytoskeleton organization and cell cycle regulation. Cell Motil. Cytoskelet. 2002, 52, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Amoussouvi, A.; Teufel, L.; Reis, M.; Seeger, M.; Schlichting, J.K.; Schreiber, G.; Herrmann, A.; Klipp, E. Transcriptional timing and noise of yeast cell cycle regulators—A single cell and single molecule approach. NPJ Syst. Biol. Appl. 2018, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Matveenko, A.G.; Matiiv, A.B.; Maksiutenko, E.M.; Moskalenko, S.E.; Drozdova, P.B.; Polev, D.E.; Beliavskaia, A.Y.; Danilov, L.G.; Predeus, A.V.; et al. Chromosome-level genome assembly and structural variant analysis of two laboratory yeast strains from the Peterhof Genetic Collection lineage. G3 2021, 11, jkab029. [Google Scholar] [CrossRef] [PubMed]
- Volkov, K.V.; Aksenova, A.Y.; Soom, M.J.; Osipov, K.V.; Svitin, A.V.; Kurischko, C.; Shkundina, I.S.; Ter-Avanesyan, M.D.; Inge-Vechtomov, S.G.; Mironova, L.N. Novel non-Mendelian determinant involved in the control of translation accuracy in Saccharomyces cerevisiae. Genetics 2002, 160, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.; Michaelis, S.; Mitchell, A. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1994. [Google Scholar]
- Gietz, R.D.; Woods, R.A. Transformation of yeast by lithium acetate single-stranded carrier DNA polyethylene glycol method. Methods Enzymol. 2002, 350, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Riera, A.; Evrin, C.; Sun, J.; Li, H.; Speck, C.; Weinreich, M. Cdc6 ATPase activity disengages Cdc6 from the pre-replicative complex to promote DNA replication. eLife 2015, 4, e05795. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kushnirov, V.V.; Alexandrov, I.M.; Mitkevich, O.V.; Shkundina, I.S.; Ter-Avanesyan, M.D. Purification and analysis of prion and amyloid aggregates. Methods 2006, 39, 50–55. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulation | Gene | Protein | Description |
|---|---|---|---|
| UP | YPR190C | Rpc82 | RNA polymerase III subunit C82 |
| UP | YLR045C | Stu2 | required for elongation of mitotic spindle during anaphase |
| UP | YBR160W | Cdc28 | master regulator of mitotic and meiotic cell cycles |
| UP | YDR055W | Pst1 | cell wall protein, up-regulated by activation of the cell integrity pathway |
| DOWN | YGR267C | Fol2 | catalyzes first step in folic acid biosynthesis |
| DOWN | YGL001C | Erg26 | take part in ergosterol biosynthesis |
| DOWN | YJR025C | Bna1 | required for the de novo biosynthesis of NAD from tryptophan to kynurenine |
| DOWN | YER044C | Erg28 | Endoplasmic reticulum membrane protein |
| DOWN | YER057C | Hmf1 | takes part in maintenance of the mitochondrial genome |
| Primer Name | Target Gene | Sequence 5’→3’ |
|---|---|---|
| ACT1-F | ACT1 | TAACGGTTCTGGTATGTGTAAAGC |
| ACT1-R | ACT1 | GCTTCATCACCAACGTAGGAGTC |
| SUP35-F | SUP35 | ACAACAAGGTAACAACAGATACC |
| SUP35-R | SUP35 | GGATTGAATTGCTGCTGATAAC |
| SUP45-F | SUP45 | CGATCCAAGACTAGCATGTAAG |
| SUP45-R | SUP45 | CTTGAACATACTTGACATTGGC |
| FKH1-RT-F | FKH1 | CGGATCATGGGAATTACAG |
| FKH1-RT_R | FKH1 | CACACCACCTATGTCTATG |
| CDC20-RT-F | CDC20 | GTCGATCCTGAAACCTTAC |
| CDC20-RT-R | CDC20 | CATATCTAACCCACACGC |
| CDC23-RT-F2 | CDC23 | AGATGTCACTAATCCATACCT |
| CDC23-RT-R2 | CDC23 | CATTTCGTCCGTAAACTTCC |
| CDC28-RT-F | CDC28 | AGAGTAGTCGCATTGAAGAAA |
| CDC28-RT-R | CDC28 | GCATCAGAGTGAACAATATCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maksiutenko, E.M.; Barbitoff, Y.A.; Danilov, L.G.; Matveenko, A.G.; Zemlyanko, O.M.; Efremova, E.P.; Moskalenko, S.E.; Zhouravleva, G.A. Gene Expression Analysis of Yeast Strains with a Nonsense Mutation in the eRF3-Coding Gene Highlights Possible Mechanisms of Adaptation. Int. J. Mol. Sci. 2024, 25, 6308. https://doi.org/10.3390/ijms25126308
Maksiutenko EM, Barbitoff YA, Danilov LG, Matveenko AG, Zemlyanko OM, Efremova EP, Moskalenko SE, Zhouravleva GA. Gene Expression Analysis of Yeast Strains with a Nonsense Mutation in the eRF3-Coding Gene Highlights Possible Mechanisms of Adaptation. International Journal of Molecular Sciences. 2024; 25(12):6308. https://doi.org/10.3390/ijms25126308
Chicago/Turabian StyleMaksiutenko, Evgeniia M., Yury A. Barbitoff, Lavrentii G. Danilov, Andrew G. Matveenko, Olga M. Zemlyanko, Elena P. Efremova, Svetlana E. Moskalenko, and Galina A. Zhouravleva. 2024. "Gene Expression Analysis of Yeast Strains with a Nonsense Mutation in the eRF3-Coding Gene Highlights Possible Mechanisms of Adaptation" International Journal of Molecular Sciences 25, no. 12: 6308. https://doi.org/10.3390/ijms25126308
APA StyleMaksiutenko, E. M., Barbitoff, Y. A., Danilov, L. G., Matveenko, A. G., Zemlyanko, O. M., Efremova, E. P., Moskalenko, S. E., & Zhouravleva, G. A. (2024). Gene Expression Analysis of Yeast Strains with a Nonsense Mutation in the eRF3-Coding Gene Highlights Possible Mechanisms of Adaptation. International Journal of Molecular Sciences, 25(12), 6308. https://doi.org/10.3390/ijms25126308