

Fibrostenosing Crohn’s Disease: Pathogenetic Mechanisms and New Therapeutic Horizons
 , , , , , , and
, , , , , , and
Abstract
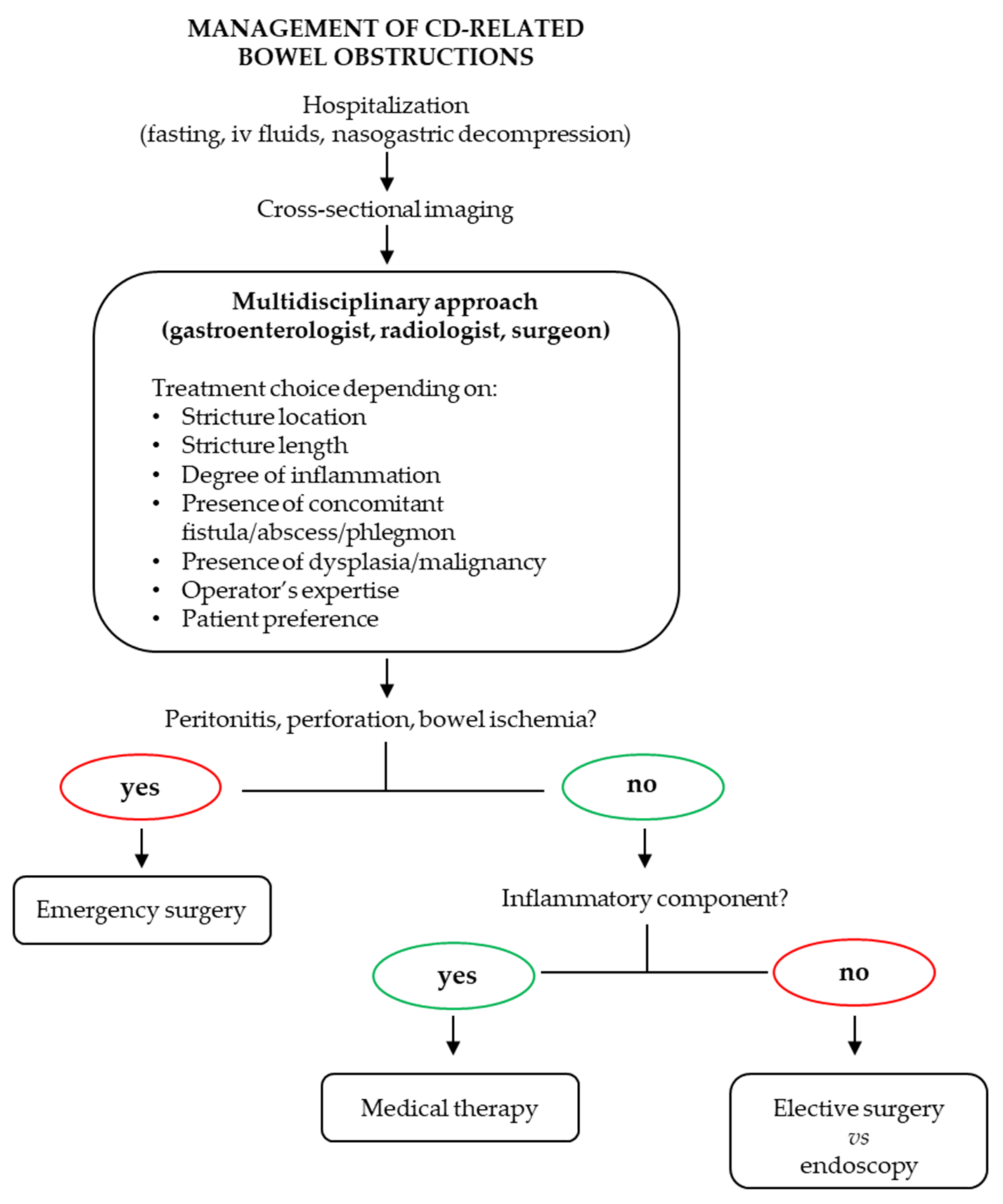
:1. Introduction
2. Pathogenetic Mechanisms of Intestinal Fibrosis in CD
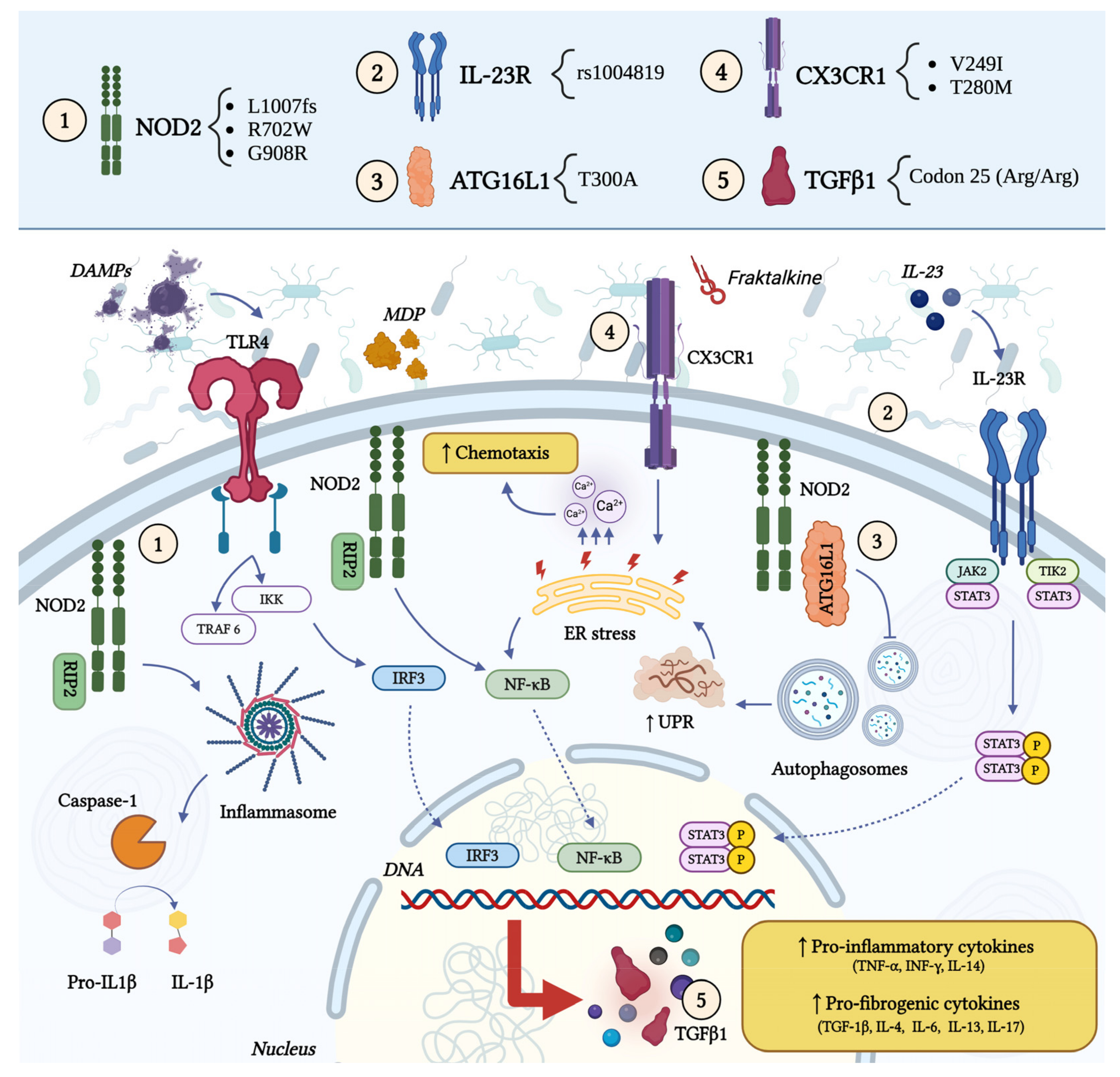
2.1. Genetic Factors
2.2. Epigenetic Factors
2.3. Cytokines and Other Soluble Factors
2.4. Creeping Fat
2.5. Extracellular Matrix
2.6. Gut Microbiota
3. Anti-Fibrotic Therapies
3.1. Approved Systemic Medical Therapies
3.2. Intralesional Medical Therapies
3.3. Novel Potential Medical Therapies
3.4. Ongoing Clinical Trials
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gasche, C.; Scholmerich, J.; Brynskov, J.; D’Haens, G.; Hanauer, S.B.; Irvine, E.J.; Jewell, D.P.; Rachmilewitz, D.; Sachar, D.B.; Sandborn, W.J.; et al. A Simple Classification of Crohn’s Disease: Report of the Working Party for the World Congresses of Gastroenterology, Vienna 1998. Inflamm. Bowel Dis. 2000, 6, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, M.S.; Satsangi, J.; Ahmad, T.; Arnott, I.D.R.; Bernstein, C.N.; Brant, S.R.; Caprilli, R.; Colombel, J.-F.; Gasche, C.; Geboes, K.; et al. Toward an Integrated Clinical, Molecular and Serological Classification of Inflammatory Bowel Disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can. J. Gastroenterol. 2005, 19 (Suppl. A), 5A–36A. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.; Collard, A.; Oger, A.F.; Degroote, E.; Aboul Nasr El Yafi, F.A.; Belaiche, J. Behaviour of Crohn’s Disease According to the Vienna Classification: Changing Pattern over the Course of the Disease. Gut 2001, 49, 777–782. [Google Scholar] [CrossRef]
- Rieder, F.; Zimmermann, E.M.; Remzi, F.H.; Sandborn, W.J. Crohn’s Disease Complicated by Strictures: A Systematic Review. Gut 2013, 62, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- El Ouali, S.; Click, B.; Holubar, S.D.; Rieder, F. Natural History, Diagnosis and Treatment Approach to Fibrostenosing Crohn’s Disease. United Eur. Gastroenterol. J. 2020, 8, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Thia, K.T.; Sandborn, W.J.; Harmsen, W.S.; Zinsmeister, A.R.; Loftus, E.V. Risk Factors Associated with Progression to Intestinal Complications of Crohn’s Disease in a Population-Based Cohort. Gastroenterology 2010, 139, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Latella, G.; Magro, F.; Yuksel, E.S.; Higgins, P.D.R.; Di Sabatino, A.; de Bruyn, J.R.; Rimola, J.; Brito, J.; Bettenworth, D.; et al. European Crohn’s and Colitis Organisation Topical Review on Prediction, Diagnosis and Management of Fibrostenosing Crohn’s Disease. J. Crohn’s Colitis 2016, 10, 873–885. [Google Scholar] [CrossRef]
- Adamina, M.; Bonovas, S.; Raine, T.; Spinelli, A.; Warusavitarne, J.; Armuzzi, A.; Bachmann, O.; Bager, P.; Biancone, L.; Bokemeyer, B.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Surgical Treatment. J. Crohn’s Colitis 2020, 14, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Bettenworth, D.; Ma, C.; Parker, C.E.; Williamson, L.A.; Nelson, S.A.; van Assche, G.; Di Sabatino, A.; Bouhnik, Y.; Stidham, R.W.; et al. An Expert Consensus to Standardise Definitions, Diagnosis and Treatment Targets for Anti-Fibrotic Stricture Therapies in Crohn’s Disease. Aliment. Pharmacol. Ther. 2018, 48, 347–357. [Google Scholar] [CrossRef]
- Diebold, R.J.; Eis, M.J.; Yin, M.; Ormsby, I.; Boivin, G.P.; Darrow, B.J.; Saffitz, J.E.; Doetschman, T. Early-Onset Multifocal Inflammation in the Transforming Growth Factor Beta 1-Null Mouse Is Lymphocyte Mediated. Proc. Natl. Acad. Sci. USA 1995, 92, 12215–12219. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Abrogation of TGFbeta Signaling in T Cells Leads to Spontaneous T Cell Differentiation and Autoimmune Disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Ward, J.M.; Yaswen, L.; Mackall, C.L.; Bauer, S.R.; Huh, C.G.; Gress, R.E.; Karlsson, S. Transforming Growth Factor-Beta 1 Null Mice. An Animal Model for Inflammatory Disorders. Am. J. Pathol. 1995, 146, 264–275. [Google Scholar]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-Wide Association Defines More than 30 Distinct Susceptibility Loci for Crohn’s Disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Latella, G.; Di Gregorio, J.; Flati, V.; Rieder, F.; Lawrance, I.C. Mechanisms of Initiation and Progression of Intestinal Fibrosis in IBD. Scand. J. Gastroenterol. 2015, 50, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Cleynen, I. Genetic Influences on the Development of Fibrosis in Crohn’s Disease. Front. Med. 2016, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Cuthbert, A.; Croucher, P.J.; Mirza, M.M.; Mascheretti, S.; Fisher, S.; Frenzel, H.; King, K.; Hasselmeyer, A.; MacPherson, A.J.; et al. Association between Insertion Mutation in NOD2 Gene and Crohn’s Disease in German and British Populations. Lancet 2001, 357, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 Is a General Sensor of Peptidoglycan through Muramyl Dipeptide (MDP) Detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, Y.; Zhou, C.; Wu, H.; Zhao, J.; Wu, L.; Zhao, M.; Zhang, F.; Liu, H. The Role of Autophagy and Related MicroRNAs in Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2018, 2018, 7565076. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Koseki, T.; Lin, J.; del Peso, L.; Lucas, P.C.; Chen, F.F.; Ogura, Y.; Núñez, G. An Induced Proximity Model for NF-κB Activation in the Nod1/RICK and RIP Signaling Pathways. J. Biol. Chem. 2000, 275, 27823–27831. [Google Scholar] [CrossRef]
- McDermott, M.F.; Tschopp, J. From Inflammasomes to Fevers, Crystals and Hypertension: How Basic Research Explains Inflammatory Diseases. Trends Mol. Med. 2007, 13, 381–388. [Google Scholar] [CrossRef]
- Bonen, D.K.; Ogura, Y.; Nicolae, D.L.; Inohara, N.; Saab, L.; Tanabe, T.; Chen, F.F.; Foster, S.J.; Duerr, R.H.; Brant, S.R.; et al. Crohn’s Disease-Associated NOD2 Variants Share a Signaling Defect in Response to Lipopolysaccharide and Peptidoglycan. Gastroenterology 2003, 124, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A Frameshift Mutation in NOD2 Associated with Susceptibility to Crohn’s Disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 Leucine-Rich Repeat Variants with Susceptibility to Crohn’s Disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Shaw, M.H.; Warner, N.; Park, J.-H.; Chen, F.; Ogura, Y.; Núñez, G. Cutting Edge: Crohn’s Disease-Associated Nod2 Mutation Limits Production of Proinflammatory Cytokines to Protect the Host from Enterococcus Faecalis-Induced Lethality. J. Immunol. 2011, 187, 2849–2852. [Google Scholar] [CrossRef]
- Salem, M.; Seidelin, J.B.; Eickhardt, S.; Alhede, M.; Rogler, G.; Nielsen, O.H. Species-Specific Engagement of Human Nucleotide Oligomerization Domain 2 (NOD)2 and Toll-like Receptor (TLR) Signalling upon Intracellular Bacterial Infection: Role of Crohn’s Associated NOD2 Gene Variants. Clin. Exp. Immunol. 2015, 179, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.; Rangwalla, S.C.; Dwamena, B.A.; Higgins, P.D.R. The Prognostic Power of the NOD2 Genotype for Complicated Crohn’s Disease: A Meta-Analysis. Am. J. Gastroenterol. 2011, 106, 699–712. [Google Scholar] [CrossRef]
- Dang, J.T.; Dang, T.T.; Wine, E.; Dicken, B.; Madsen, K.; Laffin, M. The Genetics of Postoperative Recurrence in Crohn Disease: A Systematic Review, Meta-Analysis, and Framework for Future Work. Crohn’s Colitis 360 2021, 3, otaa094. [Google Scholar] [CrossRef]
- Li, C.; Kuemmerle, J.F. Mechanisms That Mediate the Development of Fibrosis in Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2014, 20, 1250–1258. [Google Scholar] [CrossRef]
- Naser, S.A.; Arce, M.; Khaja, A.; Fernandez, M.; Naser, N.; Elwasila, S.; Thanigachalam, S. Role of ATG16L, NOD2 and IL23R in Crohn’s Disease Pathogenesis. World J. Gastroenterol. 2012, 18, 412–424. [Google Scholar] [CrossRef]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A Genome-Wide Association Study Identifies IL23R as an Inflammatory Bowel Disease Gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef]
- Glas, J.; Seiderer, J.; Wetzke, M.; Konrad, A.; Török, H.-P.; Schmechel, S.; Tonenchi, L.; Grassl, C.; Dambacher, J.; Pfennig, S.; et al. Rs1004819 Is the Main Disease-Associated IL23R Variant in German Crohn’s Disease Patients: Combined Analysis of IL23R, CARD15, and OCTN1/2 Variants. PLoS ONE 2007, 2, e819. [Google Scholar] [CrossRef]
- Borecki, K.; Zawada, I.; Salkić, N.N.; Karakiewicz, B.; Adler, G. Relationship between the IL23R SNPs and Crohn’s Disease Susceptibility and Phenotype in the Polish and Bosnian Populations: A Case-Control Study. Int. J. Environ. Res. Public Health 2019, 16, 1551. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A Genome-Wide Association Scan of Nonsynonymous SNPs Identifies a Susceptibility Variant for Crohn Disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Prescott, N.J.; Fisher, S.A.; Franke, A.; Hampe, J.; Onnie, C.M.; Soars, D.; Bagnall, R.; Mirza, M.M.; Sanderson, J.; Forbes, A.; et al. A Nonsynonymous SNP in ATG16L1 Predisposes to Ileal Crohn’s Disease and Is Independent of CARD15 and IBD5. Gastroenterology 2007, 132, 1665–1671. [Google Scholar] [CrossRef]
- Cummings, J.R.F.; Cooney, R.; Pathan, S.; Anderson, C.A.; Barrett, J.C.; Beckly, J.; Geremia, A.; Hancock, L.; Guo, C.; Ahmad, T.; et al. Confirmation of the Role of ATG16l1 as a Crohn’s Disease Susceptibility Gene. Inflamm. Bowel Dis. 2007, 13, 941–946. [Google Scholar] [CrossRef]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-Wide Association Study Identifies New Susceptibility Loci for Crohn Disease and Implicates Autophagy in Disease Pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Levin, A.D.; Koelink, P.J.; Bloemendaal, F.M.; Vos, A.C.W.; D’Haens, G.R.; van den Brink, G.R.; Wildenberg, M.E. Autophagy Contributes to the Induction of Anti-TNF Induced Macrophages. J. Crohn’s Colitis 2016, 10, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Fowler, E.V.; Doecke, J.; Simms, L.A.; Zhao, Z.Z.; Webb, P.M.; Hayward, N.K.; Whiteman, D.C.; Florin, T.H.; Montgomery, G.W.; Cavanaugh, J.A.; et al. ATG16L1 T300A Shows Strong Associations with Disease Subgroups in a Large Australian IBD Population: Further Support for Significant Disease Heterogeneity. Am. J. Gastroenterol. 2008, 103, 2519–2526. [Google Scholar] [CrossRef]
- Cleynen, I.; González, J.R.; Figueroa, C.; Franke, A.; McGovern, D.; Bortlík, M.; Crusius, B.J.A.; Vecchi, M.; Artieda, M.; Szczypiorska, M.; et al. Genetic Factors Conferring an Increased Susceptibility to Develop Crohn’s Disease Also Influence Disease Phenotype: Results from the IBDchip European Project. Gut 2013, 62, 1556–1565. [Google Scholar] [CrossRef]
- Brand, S.; Hofbauer, K.; Dambacher, J.; Schnitzler, F.; Staudinger, T.; Pfennig, S.; Seiderer, J.; Tillack, C.; Konrad, A.; Göke, B.; et al. Increased Expression of the Chemokine Fractalkine in Crohn’s Disease and Association of the Fractalkine Receptor T280M Polymorphism with a Fibrostenosing Disease Phenotype. Am. J. Gastroenterol. 2006, 101, 99–106. [Google Scholar] [CrossRef]
- Sabate, J.-M.; Ameziane, N.; Lamoril, J.; Jouet, P.; Farmachidi, J.-P.; Soulé, J.-C.; Harnois, F.; Sobhani, I.; Jian, R.; Deybach, J.-C.; et al. The V249I Polymorphism of the CX3CR1 Gene Is Associated with Fibrostenotic Disease Behavior in Patients with Crohn’s Disease. Eur. J. Gastroenterol. Hepatol. 2008, 20, 748–755. [Google Scholar] [CrossRef]
- Daoudi, M.; Lavergne, E.; Garin, A.; Tarantino, N.; Debré, P.; Pincet, F.; Combadière, C.; Deterre, P. Enhanced Adhesive Capacities of the Naturally Occurring Ile249-Met280 Variant of the Chemokine Receptor CX3CR1. J. Biol. Chem. 2004, 279, 19649–19657. [Google Scholar] [CrossRef]
- Schulte, C.M.; Goebell, H.; Röher, H.D.; Schulte, K.M. C-509T Polymorphism in the TGFB1 Gene Promoter: Impact on Crohn’s Disease Susceptibility and Clinical Course? Immunogenetics 2001, 53, 178–182. [Google Scholar] [CrossRef]
- García-González, M.A.; Crusius, J.B.; Strunk, M.H.; Bouma, G.; Pérez-Centeno, C.M.; Pals, G.; Meuwissen, S.G.; Peña, A.S. TGFB1 Gene Polymorphisms and Inflammatory Bowel Disease. Immunogenetics 2000, 51, 869–872. [Google Scholar] [CrossRef]
- Hume, G.E.; Fowler, E.V.; Lincoln, D.; Eri, R.; Templeton, D.; Florin, T.H.; Cavanaugh, J.A.; Radford-Smith, G.L. Angiotensinogen and Transforming Growth Factor Beta1: Novel Genes in the Pathogenesis of Crohn’s Disease. J. Med. Genet. 2006, 43, e51. [Google Scholar] [CrossRef]
- Sadler, T.; Bhasin, J.M.; Xu, Y.; Barnholz-Sloan, J.; Chen, Y.; Ting, A.H.; Stylianou, E. Genome-Wide Analysis of DNA Methylation and Gene Expression Defines Molecular Characteristics of Crohn’s Disease-Associated Fibrosis. Clin. Epigenetics 2016, 8, 30. [Google Scholar] [CrossRef]
- Behm-Ansmant, I.; Rehwinkel, J.; Izaurralde, E. MicroRNAs Silence Gene Expression by Repressing Protein Expression and/or by Promoting mRNA Decay. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 523–530. [Google Scholar] [CrossRef]
- Van Wynsberghe, P.M.; Chan, S.-P.; Slack, F.J.; Pasquinelli, A.E. Analysis of microRNA Expression and Function. Methods Cell Biol. 2011, 106, 219–252. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lindsay, M.A. microRNAs and the Immune Response. Trends Immunol. 2008, 29, 343–351. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and Pathological Roles for microRNAs in the Immune System. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef]
- Xue, T.; Qiu, X.; Liu, H.; Gan, C.; Tan, Z.; Xie, Y.; Wang, Y.; Ye, T. Epigenetic Regulation in Fibrosis Progress. Pharmacol. Res. 2021, 173, 105910. [Google Scholar] [CrossRef]
- Nijhuis, A.; Biancheri, P.; Lewis, A.; Bishop, C.L.; Giuffrida, P.; Chan, C.; Feakins, R.; Poulsom, R.; Di Sabatino, A.; Corazza, G.R.; et al. In Crohn’s Disease Fibrosis-Reduced Expression of the miR-29 Family Enhances Collagen Expression in Intestinal Fibroblasts. Clin. Sci. (Lond.) 2014, 127, 341–350. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after Myocardial Infarction Reveals a Role of miR-29 in Cardiac Fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef]
- Qin, W.; Chung, A.C.K.; Huang, X.R.; Meng, X.-M.; Hui, D.S.C.; Yu, C.-M.; Sung, J.J.Y.; Lan, H.Y. TGF-β/Smad3 Signaling Promotes Renal Fibrosis by Inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef]
- Roderburg, C.; Urban, G.-W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA Profiling Reveals a Role for miR-29 in Human and Murine Liver Fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef]
- Sengupta, S.; den Boon, J.A.; Chen, I.-H.; Newton, M.A.; Stanhope, S.A.; Cheng, Y.-J.; Chen, C.-J.; Hildesheim, A.; Sugden, B.; Ahlquist, P. MicroRNA 29c Is Down-Regulated in Nasopharyngeal Carcinomas, up-Regulating mRNAs Encoding Extracellular Matrix Proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 5874–5878. [Google Scholar] [CrossRef]
- Brain, O.; Owens, B.M.J.; Pichulik, T.; Allan, P.; Khatamzas, E.; Leslie, A.; Steevels, T.; Sharma, S.; Mayer, A.; Catuneanu, A.M.; et al. The Intracellular Sensor NOD2 Induces microRNA-29 Expression in Human Dendritic Cells to Limit IL-23 Release. Immunity 2013, 39, 521–536. [Google Scholar] [CrossRef]
- Lewis, A.; Mehta, S.; Hanna, L.N.; Rogalski, L.A.; Jeffery, R.; Nijhuis, A.; Kumagai, T.; Biancheri, P.; Bundy, J.G.; Bishop, C.L.; et al. Low Serum Levels of MicroRNA-19 Are Associated with a Stricturing Crohn’s Disease Phenotype. Inflamm. Bowel Dis. 2015, 21, 1926–1934. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, H.; Cheng, F.; Zhang, Z.; Long, S. miR-21 Negatively Regulates the PTEN-PI3K-Akt-mTOR Signaling Pathway in Crohn’s Disease by Altering Immune Tolerance and Epithelial-Mesenchymal Transition. Discov. Med. 2022, 34, 45–58. [Google Scholar] [PubMed]
- Wang, Z.; Zhou, H.; Cheng, F.; Zhang, Z.; Long, S. MiR-21 Regulates Epithelial-Mesenchymal Transition in Intestinal Fibrosis of Crohn’s Disease by Targeting PTEN/mTOR. Dig. Liver Dis. 2022, 54, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Sadler, T.; Scarpa, M.; Rieder, F.; West, G.; Stylianou, E. Cytokine-Induced Chromatin Modifications of the Type I Collagen Alpha 2 Gene during Intestinal Endothelial-to-Mesenchymal Transition. Inflamm. Bowel Dis. 2013, 19, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Somineni, H.K.; Venkateswaran, S.; Kilaru, V.; Marigorta, U.M.; Mo, A.; Okou, D.T.; Kellermayer, R.; Mondal, K.; Cobb, D.; Walters, T.D.; et al. Blood-Derived DNA Methylation Signatures of Crohn’s Disease and Severity of Intestinal Inflammation. Gastroenterology 2019, 156, 2254–2265.e3. [Google Scholar] [CrossRef]
- Ahmad, S.; Sands, M.; Greenberg, E.; Tangen, L.; Huang, J.; Irudayaraj, J.M.K. Mucosal DNA Methylome Alteration in Crohn’s Disease: Surgical and Non-Surgical Groups. Front. Genet. 2023, 14, 1244513. [Google Scholar] [CrossRef]
- Rieder, F.; Fiocchi, C. Intestinal Fibrosis in IBD--a Dynamic, Multifactorial Process. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 228–235. [Google Scholar] [CrossRef]
- Li, J.; Qiu, S.-J.; She, W.-M.; Wang, F.-P.; Gao, H.; Li, L.; Tu, C.-T.; Wang, J.-Y.; Shen, X.-Z.; Jiang, W. Significance of the Balance between Regulatory T (Treg) and T Helper 17 (Th17) Cells during Hepatitis B Virus Related Liver Fibrosis. PLoS ONE 2012, 7, e39307. [Google Scholar] [CrossRef]
- Vaday, G.G.; Hershkoviz, R.; Rahat, M.A.; Lahat, N.; Cahalon, L.; Lider, O. Fibronectin-Bound TNF-Alpha Stimulates Monocyte Matrix Metalloproteinase-9 Expression and Regulates Chemotaxis. J. Leukoc. Biol. 2000, 68, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Harpel, J.G.; Gleizes, P.E.; Mazzieri, R.; Nunes, I.; Rifkin, D.B. Latent Transforming Growth Factor-Beta: Structural Features and Mechanisms of Activation. Kidney Int. 1997, 51, 1376–1382. [Google Scholar] [CrossRef]
- Santacroce, G.; Lenti, M.V.; Di Sabatino, A. Therapeutic Targeting of Intestinal Fibrosis in Crohn’s Disease. Cells 2022, 11, 429. [Google Scholar] [CrossRef]
- Katsumoto, T.R.; Violette, S.M.; Sheppard, D. Blocking TGFβ via Inhibition of the Avβ6 Integrin: A Possible Therapy for Systemic Sclerosis Interstitial Lung Disease. Int. J. Rheumatol. 2011, 2011, 208219. [Google Scholar] [CrossRef] [PubMed]
- Lyons, R.M.; Keski-Oja, J.; Moses, H.L. Proteolytic Activation of Latent Transforming Growth Factor-Beta from Fibroblast-Conditioned Medium. J. Cell Biol. 1988, 106, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.S.; Datta, P.K. Targeting the Transforming Growth Factor-Beta Signaling Pathway in Human Cancer. Expert. Opin. Investig. Drugs 2010, 19, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Feng, X.-H. TGF-β Signaling in Cell Fate Control and Cancer. Curr. Opin. Cell Biol. 2019, 61, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ Signalling in Context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G. The Role of Matrix Stiffness in Regulating Cell Behavior. Hepatology 2008, 47, 1394–1400. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of Canonical Wnt Signalling Is Required for TGF-β-Mediated Fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef]
- Hanyu, A.; Ishidou, Y.; Ebisawa, T.; Shimanuki, T.; Imamura, T.; Miyazono, K. The N Domain of Smad7 Is Essential for Specific Inhibition of Transforming Growth Factor-Beta Signaling. J. Cell Biol. 2001, 155, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Soroosh, A.; Albeiroti, S.; West, G.A.; Willard, B.; Fiocchi, C.; de la Motte, C.A. Crohn’s Disease Fibroblasts Overproduce the Novel Protein KIAA1199 to Create Proinflammatory Hyaluronan Fragments. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 358–368.e4. [Google Scholar] [CrossRef]
- Zhou, L.; Todorovic, V. Interleukin-36: Structure, Signaling and Function. Adv. Exp. Med. Biol. 2021, 21, 191–210. [Google Scholar] [CrossRef]
- Melton, E.; Qiu, H. Interleukin-36 Cytokine/Receptor Signaling: A New Target for Tissue Fibrosis. Int. J. Mol. Sci. 2020, 21, 6458. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Nishida, A. Pro- and Anti-Inflammatory Roles of Interleukin (IL)-33, IL-36, and IL-38 in Inflammatory Bowel Disease. J. Gastroenterol. 2023, 58, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, W.; Wang, Z.; Lu, Y.; Zhang, J. New Insights on IL-36 in Intestinal Inflammation and Colorectal Cancer (Review). Exp. Ther. Med. 2023, 25, 275. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, K.; Kersten, C.; Schmied, A.; Vieth, M.; Primbs, T.; Carlé, B.; Knieling, F.; Claussen, J.; Klimowicz, A.C.; Zheng, J.; et al. Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice with Chronic Intestinal Inflammation. Gastroenterology 2019, 156, 1082–1097.e11. [Google Scholar] [CrossRef] [PubMed]
- Koop, K.; Enderle, K.; Hillmann, M.; Ruspeckhofer, L.; Vieth, M.; Sturm, G.; Trajanoski, Z.; Kühl, A.A.; Atreya, R.; Leppkes, M.; et al. Interleukin 36 Receptor-Inducible Matrix Metalloproteinase 13 Mediates Intestinal Fibrosis. Front. Immunol. 2023, 14, 1163198. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Long, S.H.; He, Y.; Chen, M.H.; Cao, K.; Chen, Y.J.; Chen, B.L.; Mao, R.; Zhang, S.H.; Zhu, Z.H.; Zeng, Z.R.; et al. Activation of PI3K/Akt/mTOR Signaling Pathway Triggered by PTEN Downregulation in the Pathogenesis of Crohn’s Disease. J. Dig. Dis. 2013, 14, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K.; Qi, H.; Liu, L.F.; Zheng, X.F.S. Targeting Mammalian Target of Rapamycin (mTOR) for Health and Diseases. Drug Discov. Today 2007, 12, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Crohn, B.B.; Ginzburg, L.; Oppenheimer, G.D. Regional Ileitis; a Pathologic and Clinical Entity. Am. J. Med. 1952, 13, 583–590. [Google Scholar] [CrossRef]
- Desreumaux, P.; Ernst, O.; Geboes, K.; Gambiez, L.; Berrebi, D.; Müller-Alouf, H.; Hafraoui, S.; Emilie, D.; Ectors, N.; Peuchmaur, M.; et al. Inflammatory Alterations in Mesenteric Adipose Tissue in Crohn’s Disease. Gastroenterology 1999, 117, 73–81. [Google Scholar] [CrossRef]
- Ha, C.W.Y.; Martin, A.; Sepich-Poore, G.D.; Shi, B.; Wang, Y.; Gouin, K.; Humphrey, G.; Sanders, K.; Ratnayake, Y.; Chan, K.S.L.; et al. Translocation of Viable Gut Microbiota to Mesenteric Adipose Drives Formation of Creeping Fat in Humans. Cell 2020, 183, 666–683.e17. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, C.; Hegazy, A.N.; Siegmund, B. The Role of Adipose Tissue in Inflammatory Bowel Diseases. Curr. Opin. Gastroenterol. 2018, 34, 183–186. [Google Scholar] [CrossRef]
- Kredel, L.I.; Jödicke, L.J.; Scheffold, A.; Gröne, J.; Glauben, R.; Erben, U.; Kühl, A.A.; Siegmund, B. T-Cell Composition in Ileal and Colonic Creeping Fat—Separating Ileal from Colonic Crohn’s Disease. J. Crohn’s Colitis 2019, 13, 79–91. [Google Scholar] [CrossRef]
- Fiocchi, C.; Lund, P.K. Themes in Fibrosis and Gastrointestinal Inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G677–G683. [Google Scholar] [CrossRef] [PubMed]
- Speca, S.; Giusti, I.; Rieder, F.; Latella, G. Cellular and Molecular Mechanisms of Intestinal Fibrosis. World J. Gastroenterol. 2012, 18, 3635–3661. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Huber, N.; Lang, S.; Fürst, A.; Jehle, E.; Rogler, G. Hallmarks of Epithelial to Mesenchymal Transition Are Detectable in Crohn’s Disease Associated Intestinal Fibrosis. Clin. Transl. Med. 2015, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Frei, S.; Pesch, T.; Kellermeier, S.; Arikkat, J.; Frei, P.; Fried, M.; Weber, A.; Jehle, E.; Rühl, A.; et al. Interleukin-13 and Transforming Growth Factor β Synergise in the Pathogenesis of Human Intestinal Fistulae. Gut 2013, 62, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Flier, S.N.; Tanjore, H.; Kokkotou, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of Epithelial to Mesenchymal Transition as a Novel Source of Fibroblasts in Intestinal Fibrosis. J. Biol. Chem. 2010, 285, 20202–20212. [Google Scholar] [CrossRef] [PubMed]
- Uehara, H.; Nakagawa, T.; Katsuno, T.; Sato, T.; Isono, A.; Noguchi, Y.; Saito, Y. Emergence of Fibrocytes Showing Morphological Changes in the Inflamed Colonic Mucosa. Dig. Dis. Sci. 2010, 55, 253–260. [Google Scholar] [CrossRef]
- Rieder, F.; Fiocchi, C.; Rogler, G. Mechanisms, Management, and Treatment of Fibrosis in Patients with Inflammatory Bowel Diseases. Gastroenterology 2017, 152, 340–350.e6. [Google Scholar] [CrossRef]
- Pucilowska, J.B.; McNaughton, K.K.; Mohapatra, N.K.; Hoyt, E.C.; Zimmermann, E.M.; Sartor, R.B.; Lund, P.K. IGF-I and Procollagen Alpha1(I) Are Coexpressed in a Subset of Mesenchymal Cells in Active Crohn’s Disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G1307–G1322. [Google Scholar] [CrossRef] [PubMed]
- Dammeier, J.; Brauchle, M.; Falk, W.; Grotendorst, G.R.; Werner, S. Connective Tissue Growth Factor: A Novel Regulator of Mucosal Repair and Fibrosis in Inflammatory Bowel Disease? Int. J. Biochem. Cell Biol. 1998, 30, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Geboes, K.P.; Cabooter, L.; Geboes, K. Contribution of Morphology for the Comprehension of Mechanisms of Fibrosis in Inflammatory Enterocolitis. Acta Gastroenterol. Belg. 2000, 63, 371–376. [Google Scholar] [PubMed]
- Stallmach, A.; Schuppan, D.; Riese, H.H.; Matthes, H.; Riecken, E.O. Increased Collagen Type III Synthesis by Fibroblasts Isolated from Strictures of Patients with Crohn’s Disease. Gastroenterology 1992, 102, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, N.A.; Jass, J.R.; Duval, I.; Moskowitz, R.L.; Nicholls, R.J.; Morson, B.C. Restorative Proctocolectomy with Ileal Reservoir: Pathological and Histochemical Study of Mucosal Biopsy Specimens. J. Clin. Pathol. 1987, 40, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Koukoulis, G.; Ke, Y.; Henley, J.D.; Cummings, O.W. Obliterative Muscularization of the Small Bowel Submucosa in Crohn Disease: A Possible Mechanism of Small Bowel Obstruction. Arch. Pathol. Lab. Med. 2001, 125, 1331–1334. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.; Masamunt, M.C.; Lawrance, I.C.; Sans, M. Mesenchymal Cell Proliferation and Programmed Cell Death: Key Players in Fibrogenesis and New Targets for Therapeutic Intervention. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G703–G708. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.-J.; Han, L.-H.; Cong, R.-S.; Liang, J. Caspase Family Proteases and Apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Ravi, A.; Garg, P.; Sitaraman, S.V. Matrix Metalloproteinases in Inflammatory Bowel Disease: Boon or a Bane? Inflamm. Bowel Dis. 2007, 13, 97–107. [Google Scholar] [CrossRef]
- Pender, S.L.F. Do Metalloproteinases Contribute to Tissue Destruction or Remodeling in the Inflamed Gut? Inflamm. Bowel Dis. 2008, 14 (Suppl. S2), S136–S137. [Google Scholar] [CrossRef]
- Rieder, F.; Kugathasan, S. Circulating Antibodies against Bacterial Wall Products: Are There Arguments for Early Immunosuppression? Dig. Dis. 2012, 30 (Suppl. S3), 55–66. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, F.; D’Amico, F.; Bencardino, S.; Faggiani, I.; Fanizza, J.; Zilli, A.; Parigi, T.L.; Allocca, M.; Danese, S.; Furfaro, F. Gut Microbiota Metabolites: Unveiling Their Role in Inflammatory Bowel Diseases and Fibrosis. Pharmaceuticals 2024, 17, 347. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the Pathogenesis of Inflammatory Bowel Disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, C.P.; Neut, C.; Desreumaux, P.; Colombel, J.F. Dysbiosis in Inflammatory Bowel Disease. Gut 2004, 53, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kaakoush, N.O.; Mitchell, H.M. The Role of Bacteria and Pattern-Recognition Receptors in Crohn’s Disease. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 152–168. [Google Scholar] [CrossRef] [PubMed]
- Grassl, G.A.; Valdez, Y.; Bergstrom, K.S.B.; Vallance, B.A.; Finlay, B.B. Chronic Enteric Salmonella Infection in Mice Leads to Severe and Persistent Intestinal Fibrosis. Gastroenterology 2008, 134, 768–780. [Google Scholar] [CrossRef]
- McKaig, B.C.; Hughes, K.; Tighe, P.J.; Mahida, Y.R. Differential Expression of TGF-Beta Isoforms by Normal and Inflammatory Bowel Disease Intestinal Myofibroblasts. Am. J. Physiol. Cell Physiol. 2002, 282, C172–C182. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.B.; Harrop, J.; Reddy, M.; Young, P.; Terrett, J.; Emery, J.; Moore, G.; Truneh, A. Characterization of a Novel TNF-like Ligand and Recently Described TNF Ligand and TNF Receptor Superfamily Genes and Their Constitutive and Inducible Expression in Hematopoietic and Non-Hematopoietic Cells. Gene 1997, 204, 35–46. [Google Scholar] [CrossRef]
- Jacob, N.; Jacobs, J.P.; Kumagai, K.; Ha, C.W.Y.; Kanazawa, Y.; Lagishetty, V.; Altmayer, K.; Hamill, A.M.; Von Arx, A.; Sartor, R.B.; et al. Inflammation-Independent TL1A-Mediated Intestinal Fibrosis Is Dependent on the Gut Microbiome. Mucosal Immunol. 2018, 11, 1466–1476. [Google Scholar] [CrossRef]
- Maeda, S.; Hsu, L.-C.; Liu, H.; Bankston, L.A.; Iimura, M.; Kagnoff, M.F.; Eckmann, L.; Karin, M. Nod2 Mutation in Crohn’s Disease Potentiates NF-kappaB Activity and IL-1beta Processing. Science 2005, 307, 734–738. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nuñez, G.; Flavell, R.A. Nod2-Dependent Regulation of Innate and Adaptive Immunity in the Intestinal Tract. Science 2005, 307, 731–734. [Google Scholar] [CrossRef]
- Zhao, S.; Dejanovic, D.; Yao, P.; Bhilocha, S.; Sadler, T.; Schirbel, A.; West, G.; Doyon, G.; Lopez, R.; Mao, R.; et al. Selective Deletion of MyD88 Signaling in α-SMA Positive Cells Ameliorates Experimental Intestinal Fibrosis via Post-Transcriptional Regulation. Mucosal Immunol. 2020, 13, 665–678. [Google Scholar] [CrossRef]
- Winstanley, C.; Morgan, J.A.W. The Bacterial Flagellin Gene as a Biomarker for Detection, Population Genetics and Epidemiological Analysis. Microbiology 1997, 143 Pt 10, 3071–3084. [Google Scholar] [CrossRef]
- Lodes, M.J.; Cong, Y.; Elson, C.O.; Mohamath, R.; Landers, C.J.; Targan, S.R.; Fort, M.; Hershberg, R.M. Bacterial Flagellin Is a Dominant Antigen in Crohn Disease. J. Clin. Investig. 2004, 113, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Solitano, V.; Dal Buono, A.; Gabbiadini, R.; Wozny, M.; Repici, A.; Spinelli, A.; Vetrano, S.; Armuzzi, A. Fibro-Stenosing Crohn’s Disease: What Is New and What Is Next? J. Clin. Med. 2023, 12, 3052. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.O.; Bettenworth, D.; Bokemeyer, A.; Srivastava, A.; Rosty, C.; de Hertogh, G.; Robert, M.E.; Valasek, M.A.; Mao, R.; Kurada, S.; et al. Histopathology Scoring Systems of Stenosis Associated with Small Bowel Crohn’s Disease: A Systematic Review. Gastroenterology 2020, 158, 137–150.e1. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.O.; Bettenworth, D.; Bokemeyer, A.; Srivastava, A.; Rosty, C.; de Hertogh, G.; Robert, M.E.; Valasek, M.A.; Mao, R.; Li, J.; et al. International Consensus to Standardise Histopathological Scoring for Small Bowel Strictures in Crohn’s Disease. Gut 2022, 71, 479–486. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (CAPACITY): Two Randomised Trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Richeldi, L.; Kolb, M.; Jouneau, S.; Wuyts, W.A.; Schinzel, B.; Stowasser, S.; Quaresma, M.; Raghu, G. Efficacy and Safety of Nintedanib in Patients with Advanced Idiopathic Pulmonary Fibrosis. BMC Pulm. Med. 2020, 20, 3. [Google Scholar] [CrossRef]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered Fibroblast Growth Factor 19 Reduces Liver Injury and Resolves Sclerosing Cholangitis in Mdr2-Deficient Mice. Hepatology 2016, 63, 914–929. [Google Scholar] [CrossRef]
- So, K.; McGrouther, D.A.; Bush, J.A.; Durani, P.; Taylor, L.; Skotny, G.; Mason, T.; Metcalfe, A.; O’Kane, S.; Ferguson, M.W.J. Avotermin for Scar Improvement Following Scar Revision Surgery: A Randomized, Double-Blind, within-Patient, Placebo-Controlled, Phase II Clinical Trial. Plast. Reconstr. Surg. 2011, 128, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-N.; Mao, R.; Qian, C.; Bettenworth, D.; Wang, J.; Li, J.; Bruining, D.H.; Jairath, V.; Feagan, B.G.; Chen, M.-H.; et al. Development of Antifibrotic Therapy for Stricturing Crohn’s Disease: Lessons from Randomized Trials in Other Fibrotic Diseases. Physiol. Rev. 2022, 102, 605–652. [Google Scholar] [CrossRef]
- Torres, J.; Bonovas, S.; Doherty, G.; Kucharzik, T.; Gisbert, J.P.; Raine, T.; Adamina, M.; Armuzzi, A.; Bachmann, O.; Bager, P.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohn’s Colitis 2020, 14, 4–22. [Google Scholar] [CrossRef]
- Lu, C.; Baraty, B.; Lee Robertson, H.; Filyk, A.; Shen, H.; Fung, T.; Novak, K.; Ma, C.; Panaccione, R.; Achkar, J.-P.; et al. Systematic Review: Medical Therapy for Fibrostenosing Crohn’s Disease. Aliment. Pharmacol. Ther. 2020, 51, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Toy, L.; Scherl, E.; Kornbluth, A.; Marion, J.; Greenstein, A.; Agus, S.; Gerson, C.; Fox, N. Complete Bowel Obstruction Following Initial Response to Infliximab Therapy for Crohn’s Disease: A Series of a Newly Described Complication. Gastroenterology 2000, 118, A569. [Google Scholar] [CrossRef]
- Vasilopoulos, S.; Kugathasan, S.; Saeian, K.; Emmons, J.; Hogan, W.; Otterson, M.; Telford, G.; Binion, D. Intestinal Strictures Complicating Initially Successful Infliximab Treatment for Luminal Crohn’s Disease. Am. J. Gastroenterol. 2000, 95, 2503. [Google Scholar] [CrossRef]
- Chiba, M.; Tanaka, Y.; Ono, I. Early Intestinal Obstruction after Infliximab Therapy in Crohn’s Disease. Autops. Case Rep. 2019, 9, e2018068. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Ciccocioppo, R.; Benazzato, L.; Sturniolo, G.C.; Corazza, G.R. Infliximab Downregulates Basic Fibroblast Growth Factor and Vascular Endothelial Growth Factor in Crohn’s Disease Patients. Aliment. Pharmacol. Ther. 2004, 19, 1019–1024. [Google Scholar] [CrossRef]
- Pallotta, N.; Barberani, F.; Hassan, N.-A.; Guagnozzi, D.; Vincoli, G.; Corazziari, E. Effect of Infliximab on Small Bowel Stenoses in Patients with Crohn’s Disease. World J. Gastroenterol. 2008, 14, 1885–1890. [Google Scholar] [CrossRef]
- Pelletier, A.-L.; Kalisazan, B.; Wienckiewicz, J.; Bouarioua, N.; Soulé, J.-C. Infliximab Treatment for Symptomatic Crohn’s Disease Strictures. Aliment. Pharmacol. Ther. 2009, 29, 279–285. [Google Scholar] [CrossRef]
- Allocca, M.; Bonifacio, C.; Fiorino, G.; Spinelli, A.; Furfaro, F.; Balzarini, L.; Bonovas, S.; Danese, S. Efficacy of Tumour Necrosis Factor Antagonists in Stricturing Crohn’s Disease: A Tertiary Center Real-Life Experience. Dig. Liver Dis. 2017, 49, 872–877. [Google Scholar] [CrossRef]
- Bouhnik, Y.; Carbonnel, F.; Laharie, D.; Stefanescu, C.; Hébuterne, X.; Abitbol, V.; Nachury, M.; Brixi, H.; Bourreille, A.; Picon, L.; et al. Efficacy of Adalimumab in Patients with Crohn’s Disease and Symptomatic Small Bowel Stricture: A Multicentre, Prospective, Observational Cohort (CREOLE) Study. Gut 2018, 67, 53–60. [Google Scholar] [CrossRef]
- Schulberg, J.D.; Wright, E.K.; Holt, B.A.; Hamilton, A.L.; Sutherland, T.R.; Ross, A.L.; Vogrin, S.; Miller, A.M.; Connell, W.C.; Lust, M.; et al. Intensive Drug Therapy versus Standard Drug Therapy for Symptomatic Intestinal Crohn’s Disease Strictures (STRIDENT): An Open-Label, Single-Centre, Randomised Controlled Trial. Lancet Gastroenterol. Hepatol. 2022, 7, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Vuyyuru, S.K.; Kante, B.; Kumar, P.; Sahu, P.; Kedia, S.; Ranjan, M.K.; Sharma, R.; Panwar, R.; Makharia, G.; Ahuja, V. Real World Analysis on the Efficacy and Safety of Anti-Tumor Necrosis Factor Therapy in Patients with Stricturing Crohn’s Disease. Sci. Rep. 2021, 11, 11704. [Google Scholar] [CrossRef]
- Rodríguez-Lago, I.; Hoyo, J.D.; Pérez-Girbés, A.; Garrido-Marín, A.; Casanova, M.J.; Chaparro, M.; Fernández-Clotet, A.; Castro-Poceiro, J.; García, M.J.; Sánchez, S.; et al. Early Treatment with Anti-Tumor Necrosis Factor Agents Improves Long-Term Effectiveness in Symptomatic Stricturing Crohn’s Disease. United Eur. Gastroenterol. J. 2020, 8, 1056–1066. [Google Scholar] [CrossRef]
- Ma, C.; Fedorak, R.N.; Kaplan, G.G.; Dieleman, L.A.; Devlin, S.M.; Stern, N.; Kroeker, K.I.; Seow, C.H.; Leung, Y.; Novak, K.L.; et al. Clinical, Endoscopic and Radiographic Outcomes with Ustekinumab in Medically-Refractory Crohn’s Disease: Real World Experience from a Multicentre Cohort. Aliment. Pharmacol. Ther. 2017, 45, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Elmoursi, A.; Barrett, T.A.; Perry, C. Double Biologic Therapy for Refractory Stricturing Crohn’s Disease: A Successful Case of Deep Remission with Ustekinumab and Vedolizumab. Inflamm. Bowel Dis. 2020, 26, e62–e63. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Murate, K.; Nakamura, M.; Furukawa, K.; Kakushima, N.; Yamamura, T.; Maeda, K.; Sawada, T.; Ishikawa, E.; Ishikawa, T.; et al. The Effects of Ustekinumab on Small Intestinal Lesions and Stenotic Lesions. Nagoya J. Med. Sci. 2022, 84, 825–838. [Google Scholar] [CrossRef]
- Bevan, R.; Rees, C.J.; Rutter, M.D.; Macafee, D.A.L. Review of the Use of Intralesional Steroid Injections in the Management of Ileocolonic Crohn’s Strictures. Frontline Gastroenterol. 2013, 4, 238–243. [Google Scholar] [CrossRef]
- Ramboer, C.; Verhamme, M.; Dhondt, E.; Huys, S.; Van Eygen, K.; Vermeire, L. Endoscopic Treatment of Stenosis in Recurrent Crohn’s Disease with Balloon Dilation Combined with Local Corticosteroid Injection. Gastrointest. Endosc. 1995, 42, 252–255. [Google Scholar] [CrossRef]
- Brooker, J.C.; Beckett, C.G.; Saunders, B.P.; Benson, M.J. Long-Acting Steroid Injection after Endoscopic Dilation of Anastomotic Crohn’s Strictures May Improve the Outcome: A Retrospective Case Series. Endoscopy 2003, 35, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Lavy, A. Triamcinolone Improves Outcome in Crohn’s Disease Strictures. Dis. Colon. Rectum. 1997, 40, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.V.; Draganov, P.; Valentine, J. Efficacy and Safety of Endoscopic Balloon Dilation of Symptomatic Upper and Lower Gastrointestinal Crohn’s Disease Strictures. J. Clin. Gastroenterol. 2005, 39, 284–290. [Google Scholar] [CrossRef] [PubMed]
- East, J.E.; Brooker, J.C.; Rutter, M.D.; Saunders, B.P. A Pilot Study of Intrastricture Steroid versus Placebo Injection after Balloon Dilatation of Crohn’s Strictures. Clin. Gastroenterol. Hepatol. 2007, 5, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, G.; Oliva, S.; Passariello, M.; Pallotta, N.; Civitelli, F.; Frediani, S.; Gualdi, G.; Gandullia, P.; Mallardo, S.; Cucchiara, S. Intralesional Steroid Injection after Endoscopic Balloon Dilation in Pediatric Crohn’s Disease with Stricture: A Prospective, Randomized, Double-Blind, Controlled Trial. Gastrointest. Endosc. 2010, 72, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Feleshtynskiy, Y.; Mylianovska, A.; Pirogovsky, V.; Dyadyk, O. Evaluation of the Endoscopic Treatment with Topical Prednisolone Administration for Intestinal Strictures in Crohn’s Disease. Pol. Przegl. Chir. 2021, 94, 28–33. [Google Scholar] [CrossRef]
- Teich, N.; Wallstabe, I.; Schiefke, I. Topic Infliximab Injection for Refractory Rectal Stenosis in Crohn’s Disease: Long-Term Follow-up in Two Patients. Int. J. Colorectal Dis. 2017, 32, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Swaminath, A.; Lichtiger, S. Dilation of Colonic Strictures by Intralesional Injection of Infliximab in Patients with Crohn’s Colitis. Inflamm. Bowel Dis. 2008, 14, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Hendel, J.; Karstensen, J.G.; Vilmann, P. Serial Intralesional Injections of Infliximab in Small Bowel Crohn’s Strictures Are Feasible and Might Lower Inflammation. United Eur. Gastroenterol. J. 2014, 2, 406–412. [Google Scholar] [CrossRef]
- Imai, J.; Yahata, T.; Ichikawa, H.; Ibrahim, A.A.; Yazawa, M.; Sumiyoshi, H.; Inagaki, Y.; Matsushima, M.; Suzuki, T.; Mine, T.; et al. Inhibition of Plasminogen Activator Inhibitor-1 Attenuates against Intestinal Fibrosis in Mice. Intest. Res. 2020, 18, 219–228. [Google Scholar] [CrossRef]
- Lewis, A.; Sánchez, S.; Berti, G.; Pan-Castillo, B.; Nijhuis, A.; Mehta, S.; Eleia, L.; Gordon, H.; Gadhok, R.; Kimberley, C.; et al. Small-Molecule Wnt Inhibitors Are a Potential Novel Therapy for Intestinal Fibrosis in Crohns Disease. Clin. Sci. 2022, 136, 1405–1423. [Google Scholar] [CrossRef]
- Hiraishi, K.; Kurahara, L.-H.; Sumiyoshi, M.; Hu, Y.-P.; Koga, K.; Onitsuka, M.; Kojima, D.; Yue, L.; Takedatsu, H.; Jian, Y.-W.; et al. Daikenchuto (Da-Jian-Zhong-Tang) Ameliorates Intestinal Fibrosis by Activating Myofibroblast Transient Receptor Potential Ankyrin 1 Channel. World J. Gastroenterol. 2018, 24, 4036–4053. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, C.-Z.; Zhu, R.; Fan, H.; Liu, X.-X.; Duan, X.-Y.; Tang, Q.; Shou, Z.-X.; Zuo, D.-M. miR-200b-Containing Microvesicles Attenuate Experimental Colitis Associated Intestinal Fibrosis by Inhibiting Epithelial-Mesenchymal Transition. J. Gastroenterol. Hepatol. 2017, 32, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Truffi, M.; Sorrentino, L.; Monieri, M.; Fociani, P.; Mazzucchelli, S.; Bonzini, M.; Zerbi, P.; Sampietro, G.M.; Di Sabatino, A.; Corsi, F. Inhibition of Fibroblast Activation Protein Restores a Balanced Extracellular Matrix and Reduces Fibrosis in Crohn’s Disease Strictures Ex Vivo. Inflamm. Bowel Dis. 2018, 24, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Koo, J.B.; Kim, H.Y.; Seo, J.W.; Lee, E.J.; Kim, W.R.; Cho, J.Y.; Hahm, K.B.; Hong, S.P.; Kim, D.H.; et al. Umbilical Cord/Placenta-Derived Mesenchymal Stem Cells Inhibit Fibrogenic Activation in Human Intestinal Myofibroblasts via Inhibition of Myocardin-Related Transcription Factor A. Stem Cell Res. Ther. 2019, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Vieujean, S.; Loly, J.-P.; Boutaffala, L.; Meunier, P.; Reenaers, C.; Briquet, A.; Lechanteur, C.; Baudoux, E.; Beguin, Y.; Louis, E. Mesenchymal Stem Cell Injection in Crohn’s Disease Strictures: A Phase I-II Clinical Study. J. Crohn’s Colitis 2022, 16, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, X.; Hao, Y.; Ding, J.; Shen, J.; Xue, Z.; Qi, W.; Li, Z.; Song, Y.; Zhang, T.; et al. Protective Effects of a Novel Probiotic Strain, Lactococcus Lactis ML2018, in Colitis: In Vivo and in Vitro Evidence. Food Funct. 2019, 10, 1132–1145. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Choi, J.; Kwon, J.Y.; Jung, K.-A.; Yang, C.W.; Park, S.-H.; Cho, M.-L. A Probiotic Complex, Rosavin, Zinc, and Prebiotics Ameliorate Intestinal Inflammation in an Acute Colitis Mouse Model. J. Transl. Med. 2018, 16, 37. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zheng, C.; Wang, S.; Yang, R.; Liu, Z.; Chen, T. Treatment with a Probiotic Combination Reduces Abdominal Adhesion in Rats by Decreasing Intestinal Inflammation and Restoring Microbial Composition. Oncol. Rep. 2020, 43, 986–998. [Google Scholar] [CrossRef]
- Lombardi, F.; Augello, F.R.; Palumbo, P.; Mollsi, E.; Giuliani, M.; Cimini, A.M.; Cifone, M.G.; Cinque, B. Soluble Fraction from Lysate of a High Concentration Multi-Strain Probiotic Formulation Inhibits TGF-Β1-Induced Intestinal Fibrosis on CCD-18Co Cells. Nutrients 2021, 13, 882. [Google Scholar] [CrossRef]
- Kashima, S.; Fujiya, M.; Konishi, H.; Ueno, N.; Inaba, Y.; Moriichi, K.; Tanabe, H.; Ikuta, K.; Ohtake, T.; Kohgo, Y. Polyphosphate, an Active Molecule Derived from Probiotic Lactobacillus Brevis, Improves the Fibrosis in Murine Colitis. Transl. Res. 2015, 166, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F. Efficacy and Safety of Rapamycin in the Treatment of Crohn’s Disease-Related Stricture; National Library of Medicine: Bethesda, MD, USA, 2020. [Google Scholar]
- Varea, S. A Randomized, Double-Blinded, Placebo-Controlled Study on the Effects of Adalimumab Intralesional Intestinal Strictures of Crohn’s Disease Patients; National Library of Medicine: Bethesda, MD, USA, 2018. [Google Scholar]
- National Library of Medicine. Boehringer Ingelheim Multi-Center, Double-Blind, Randomized, Placebo-Controlled, Phase IIa Trial to Evaluate Spesolimab (BI 655130) Efficacy in Patients with Fibrostenotic Crohn’s Disease; National Library of Medicine: Bethesda, MD, USA, 2023. [Google Scholar]
- National Library of Medicine. Nantes University Hospital SyMptomAtic Stricturing Small Bowel CRohn’s Disease—Medical Treatment Versus Surgery, a Prospective, Multi-Centre, Randomized, Non-Inferiority Trial; National Library of Medicine: Bethesda, MD, USA, 2022. [Google Scholar]
- National Library of Medicine. Second Affiliated Hospital, School of Medicine, Zhejiang University Efficacy of Ustekinumab-Based Integrated Medicine Therapy in Patients with Symptomatic Stricturing Crohn’s Disease: A Multicentre, Prospective, Observational Cohort Study; National Library of Medicine: Bethesda, MD, USA, 2023. [Google Scholar]
- Weiming, Z. Surgical Intervention Versus Biologics Treatment for Symptomatic Stricturing Crohn’s Disease (SIBTC): An Open-Label, Single-Center, Randomized Controlled Trial; National Library of Medicine: Bethesda, MD, USA, 2022. [Google Scholar]
- National Library of Medicine. Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz Clinical Trial Phase IIa to Evaluate the Safety and Effectiveness of Treatment with Fat-Derived Mesenchymal Allogenic Mesenchymal Troncal Cells in Patients with Single Inflammatory Stenosis in the Context of Crohn’s Disease; National Library of Medicine: Bethesda, MD, USA, 2023. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Cytokines Soluble Factors | Mechanism | Activity | |
| TGF-β |  | SMAD pathway → ECM deposition. | PROFIBROTIC |
| IL-6 |  | Cell migration-inducing hyaluronan-binding protein → hyaluronan degradation. | |
| IL-36 |  | Expression of the metalloproteinase MMP13. | |
| IL-4 IL-5 IL 13 | 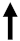 | Th2 pathway → ECM deposition. | |
| IL-17 |  | Th17 pathway → ECM deposition. | |
| mTOR complex | 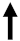 | Synthesis of collagen, fibroblasts, and myofibroblast.Production of profibrogenic cytokines (IL-4, IL-6, IL-13, IL-17, and TGF-β1). | |
 | |||
| IFN-γ | 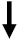 | Th1 pathway → ECM deposition. | ANTIFIBROTIC |
| Cytokines/Soluble Factors | ECM | Microbiota | |||
|---|---|---|---|---|---|
| Therapeutic Agent | Target Pathway | Therapeutic Agent | Target Pathway | Therapeutic Agent | Target Pathway |
| TM5275 [160] | TGF-β signaling; inhibition of PAI-1 | Anti-FAP antibodies [164] | Inhibition of FAP; decrease in type-I collagen; reduced TIMP-1 levels | Polyphosphate [171] | Reduced levels of TGF-β1, TNFα and IL-1β |
| 3235-0367 Wnt-C59 ICG-001 [161] | TGF-β signaling; inhibition of Wnt signaling | Perinatal stem cells (umbilical/placenta) [165] | Inhibition of RhoA, MRTF-A, and SRF expression;reduced expression of procollagen1A1, fibronectin, and α-SMA | 12 probiotics, prebiotics, rosavin, and zinc [168] | Decrease in α-SMA and type I collagen |
| Daikenchuto [162] | TGF-β signaling; activation of TRPA1; reduced production of type I collagen and α-SMA | Allogenic bone marrow stem cells [166] | NK | Lactococcus lactis ML2018 [167] | |
| Rapamycin [172] | Inhibition of mTOR complex | Adipose-derived allogenic mesenchymal stem cells [173] | NK | Lactobacillus plantarum, L. acidophilus, L. rhamnosus, and Bifidobacterium animalis [169] | TGF-β1/Smad signalling |
| Spesolimab (monoclonal antibody) [174] | Inhibition of IL-36 receptor | Microvesicles containing mi-RNA-200b [163] | Inhibition of ZEB1 and ZEB2; inhibition of EMT | Multi-Strain Probiotic Formulation (Vivomixx®) [170] | TGF-β1/Smad signalling |
| Anti-IL-36R antibodies [84] | Inhibition of IL-36 receptor | ||||
| Title | Study ID | Study Type | Phase | Drug | Arms | Randomization | Placebo | Primary Outcome | Status |
|---|---|---|---|---|---|---|---|---|---|
| A Randomized, Double-blinded, Placebo-controlled Study on the Effects of Adalimumab Intralesional Intestinal Strictures of Crohn’s Disease Patients [173] | NCT01986127 | Interventional | III | Adalimumab (intralesional administration during endoscopy) | Two arms: dilatation and adalimumab vs. dilatation and placebo | Yes | Yes | Successful dilatation at week 8 | Concluded |
| SyMptomAtic Stricturing Small Bowel CRohn’s Disease—Medical Treatment Versus Surgery, a Prospective, Multi-centre, Randomized, Non-inferiority Trial [175] | NCT05584228 | Interventional | NA | Azathioprine per os and infliximab sc | Two arms: medical therapy vs. surgery | Yes | Yes | Clinical: IBD-related quality of life at 12 months | Not yet recruiting |
| Efficacy of Ustekinumab-based Integrated Medicine Therapy in Patients With Symptomatic Stricturing Crohn’s Disease: a Multicentre, Prospective, Observational Cohort Study [176] | NCT05387031 | Observational | NA | Ustekinumab | Single arm | NA | NA | Treatment success at week 52 | Recruiting |
| Surgical Intervention Versus Biologics Treatment for Symptomatic Stricturing Crohn’s Disease (SIBTC): an Open-label, Single-center, Randomized Controlled Trial [177] | NCT05421455 | Interventional | NA | Biologics | Two arms: (up to two) biologics vs. surgery | Yes | No | Clinical remission rate at 1 year | Recruiting |
| Efficacy and Safety of Rapamycin in the Treatment of Crohn’s Disease-related Stricture [172] | NCT02675153 | Interventional | NA | Rapamycin 2 mg/day for six months | Single arm | No | No (open label) | Response rate at 24 weeks (ability to tolerate normal diet; need for endoscopy or surgery; adverse events) | Unknown |
| Multi-center, Double-blind, Randomized, Placebo-controlled, Phase IIa Trial to Evaluate Spesolimab (BI 655130) Efficacy in Patients With Fibrostenotic Crohn’s Disease [174] | NCT05013385 | Interventional | IIa | Spesolimab | Two arms: spesolimab vs. placebo | Yes | Yes |
| Concluded |
| Clinical Trial Phase IIa to Evaluate the Safety and Effectiveness of Treatment With Fat-derived Mesenchymal Allogenic Mesenchymal Troncal Cells in Patients With Single Inflammatory Stenosis in the Context of Crohn’s Disease [178] | NCT05521672 | Interventional | IIa | Adipose-derived allogenic mesenchymal stem cells (perilesional injection during laparoscopic procedure) | Single arm | No | No |
| Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mignini, I.; Blasi, V.; Termite, F.; Esposto, G.; Borriello, R.; Laterza, L.; Scaldaferri, F.; Ainora, M.E.; Gasbarrini, A.; Zocco, M.A. Fibrostenosing Crohn’s Disease: Pathogenetic Mechanisms and New Therapeutic Horizons. Int. J. Mol. Sci. 2024, 25, 6326. https://doi.org/10.3390/ijms25126326
Mignini I, Blasi V, Termite F, Esposto G, Borriello R, Laterza L, Scaldaferri F, Ainora ME, Gasbarrini A, Zocco MA. Fibrostenosing Crohn’s Disease: Pathogenetic Mechanisms and New Therapeutic Horizons. International Journal of Molecular Sciences. 2024; 25(12):6326. https://doi.org/10.3390/ijms25126326
Chicago/Turabian StyleMignini, Irene, Valentina Blasi, Fabrizio Termite, Giorgio Esposto, Raffaele Borriello, Lucrezia Laterza, Franco Scaldaferri, Maria Elena Ainora, Antonio Gasbarrini, and Maria Assunta Zocco. 2024. "Fibrostenosing Crohn’s Disease: Pathogenetic Mechanisms and New Therapeutic Horizons" International Journal of Molecular Sciences 25, no. 12: 6326. https://doi.org/10.3390/ijms25126326