
Superelectrophilic Activation of Phosphacoumarins towards Weak Nucleophiles via Brønsted Acid Assisted Brønsted Acid Catalysis

 , , ,
, , ,  ,
,  ,
,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
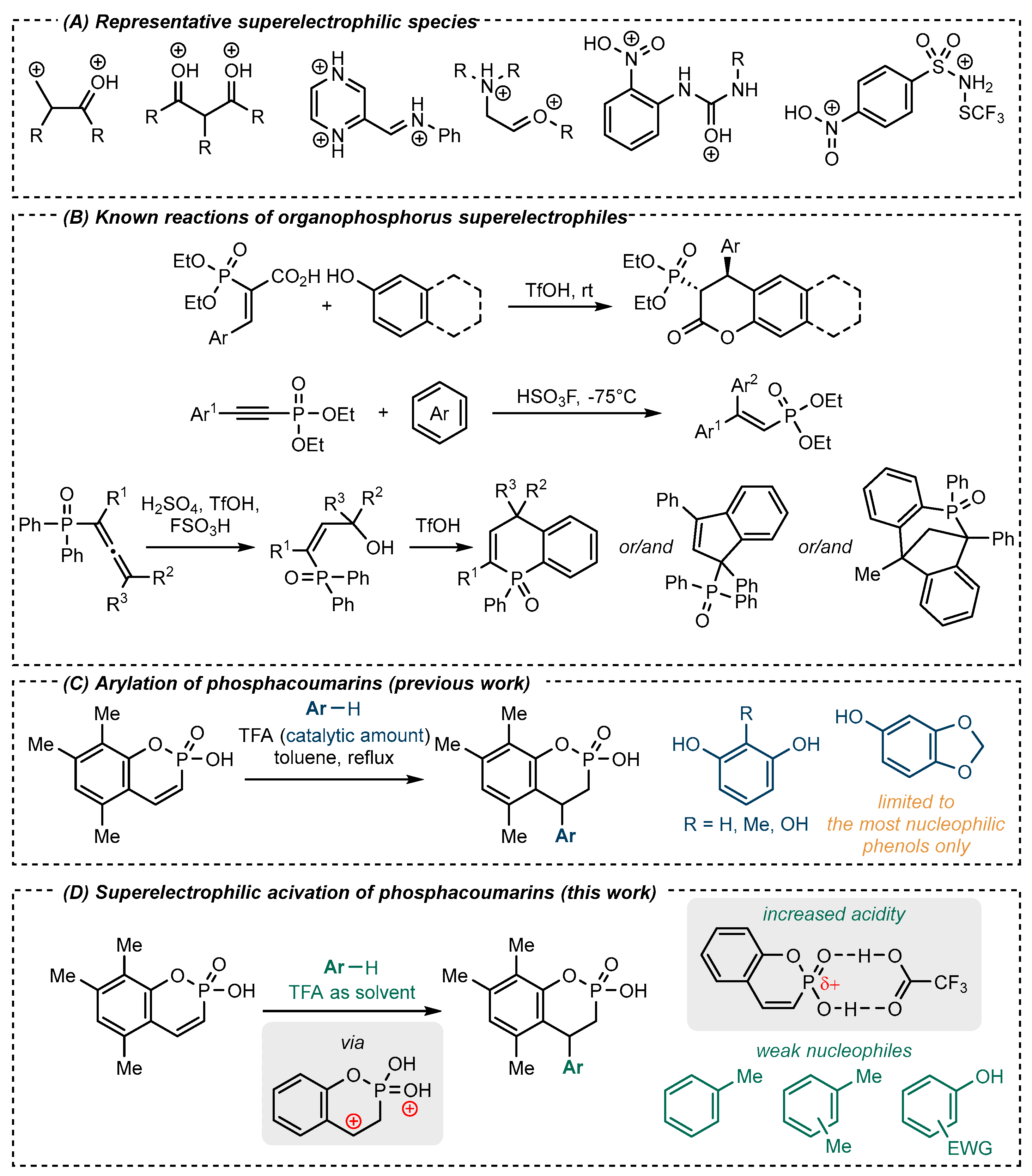
1. Introduction
2. Results and Discussion
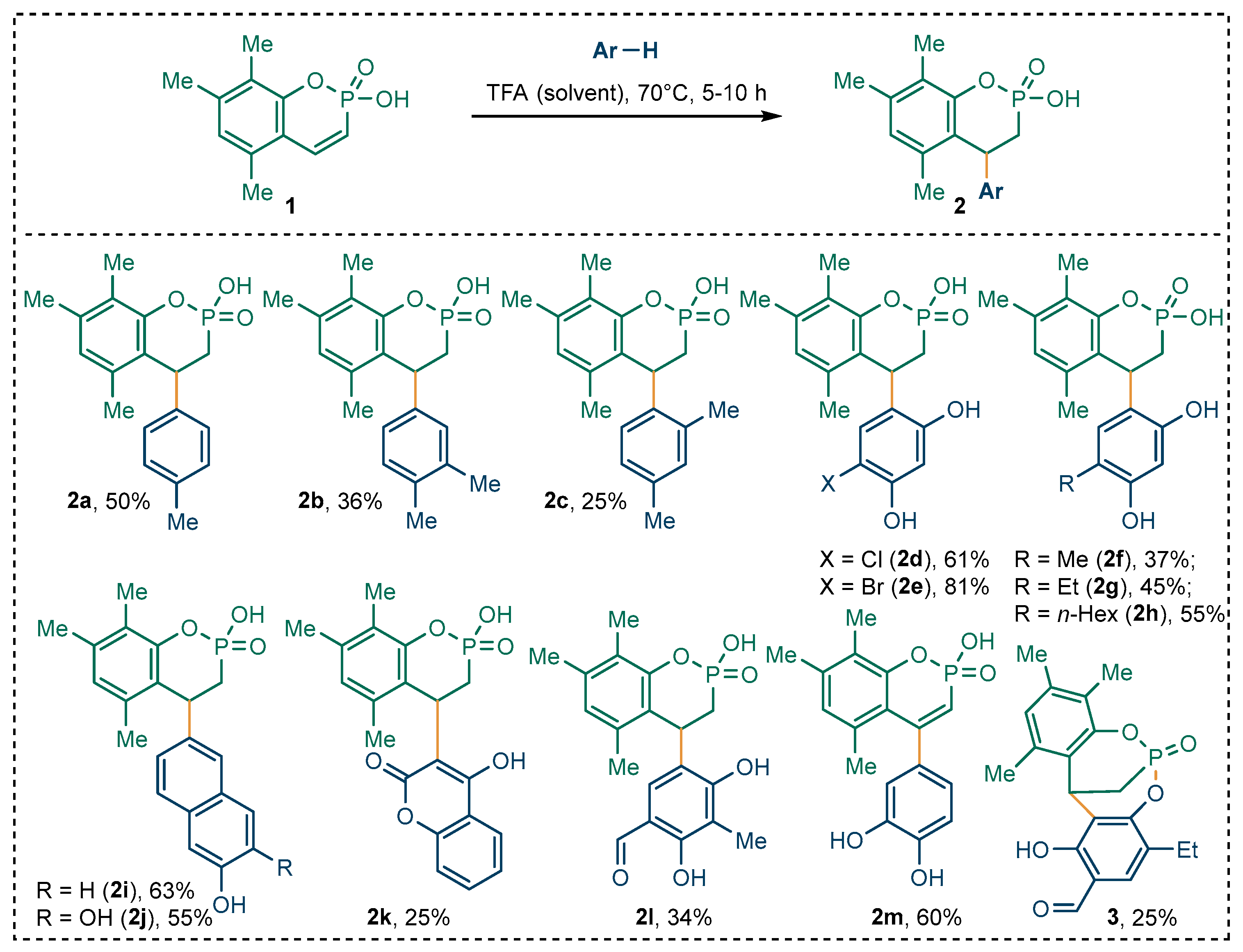
2.1. Synthesis of 4-(aryl)phosphaflavanoids in TFA Medium
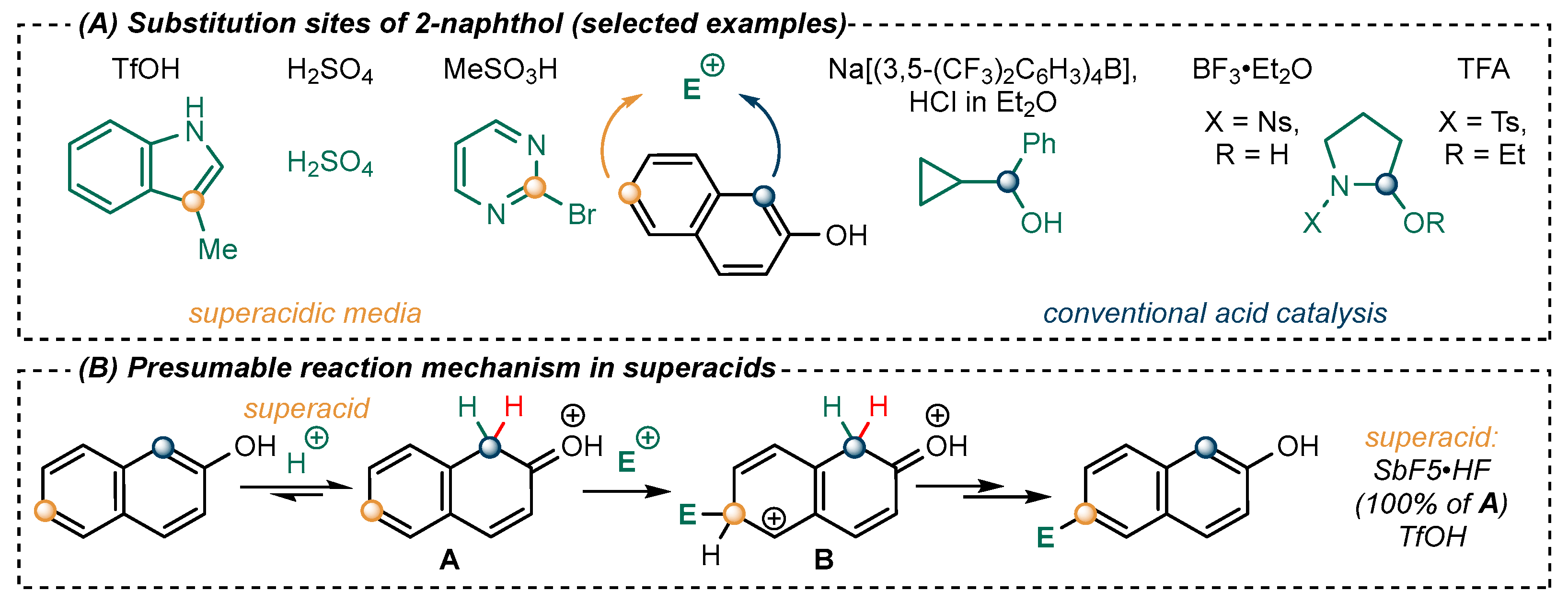
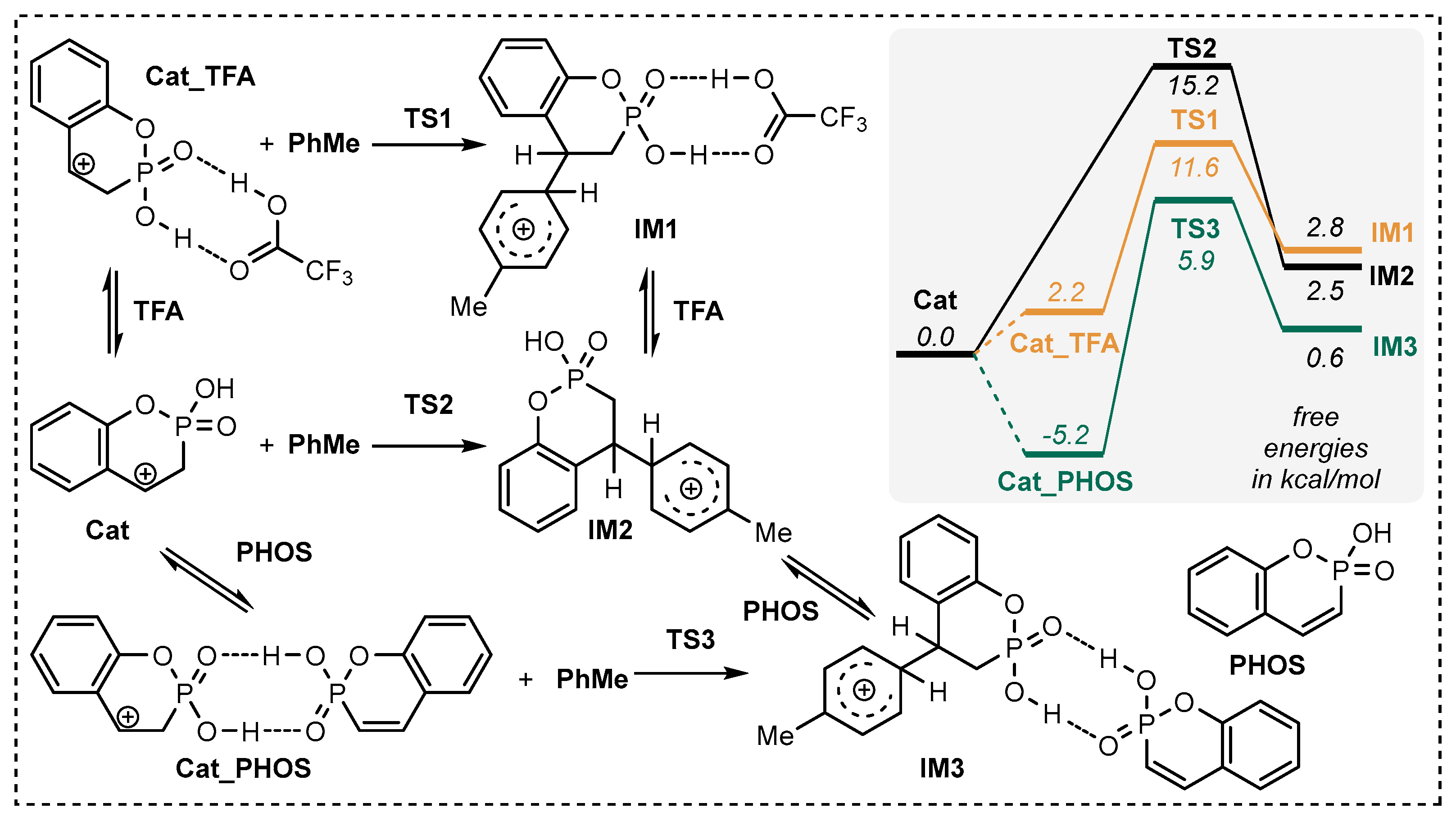
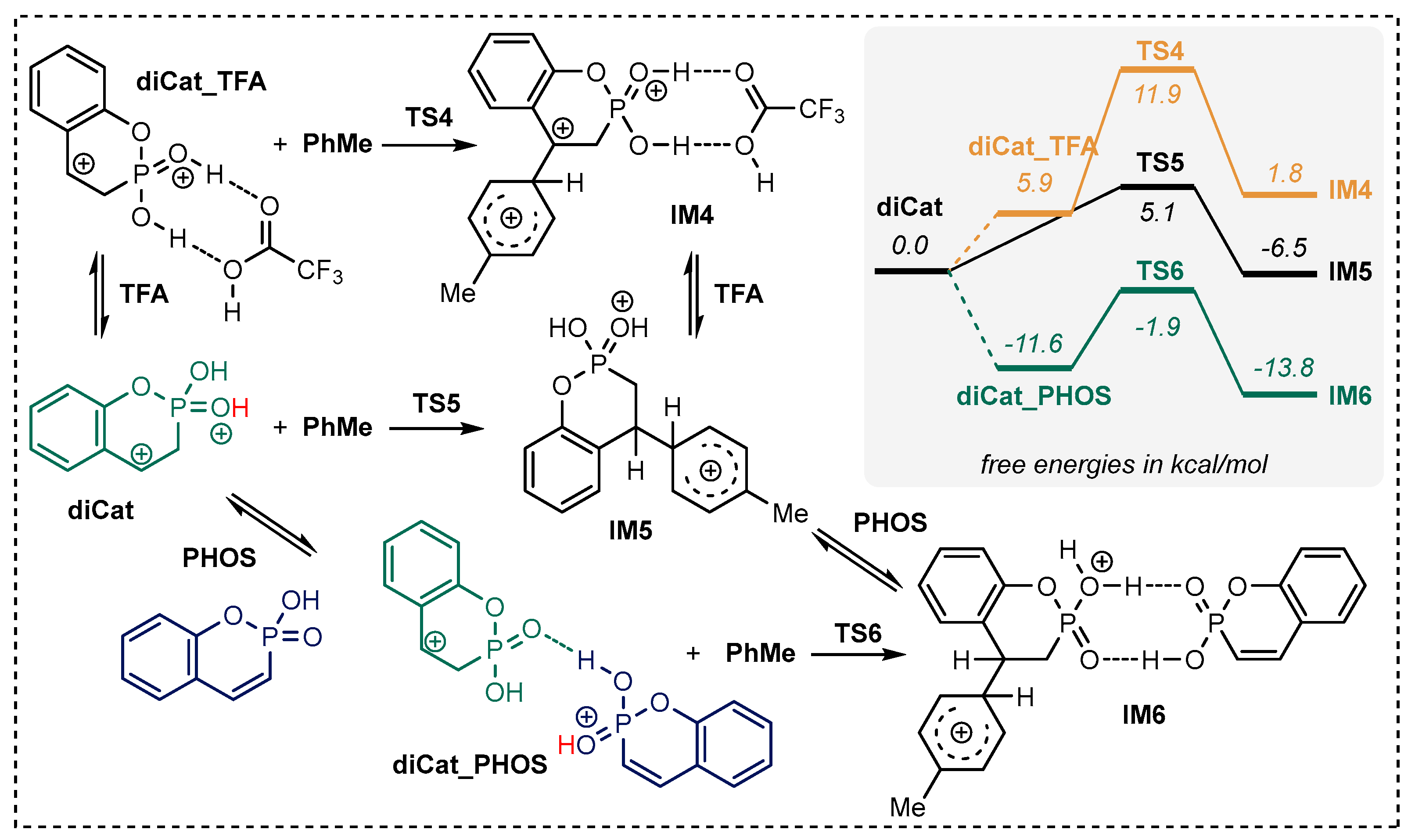
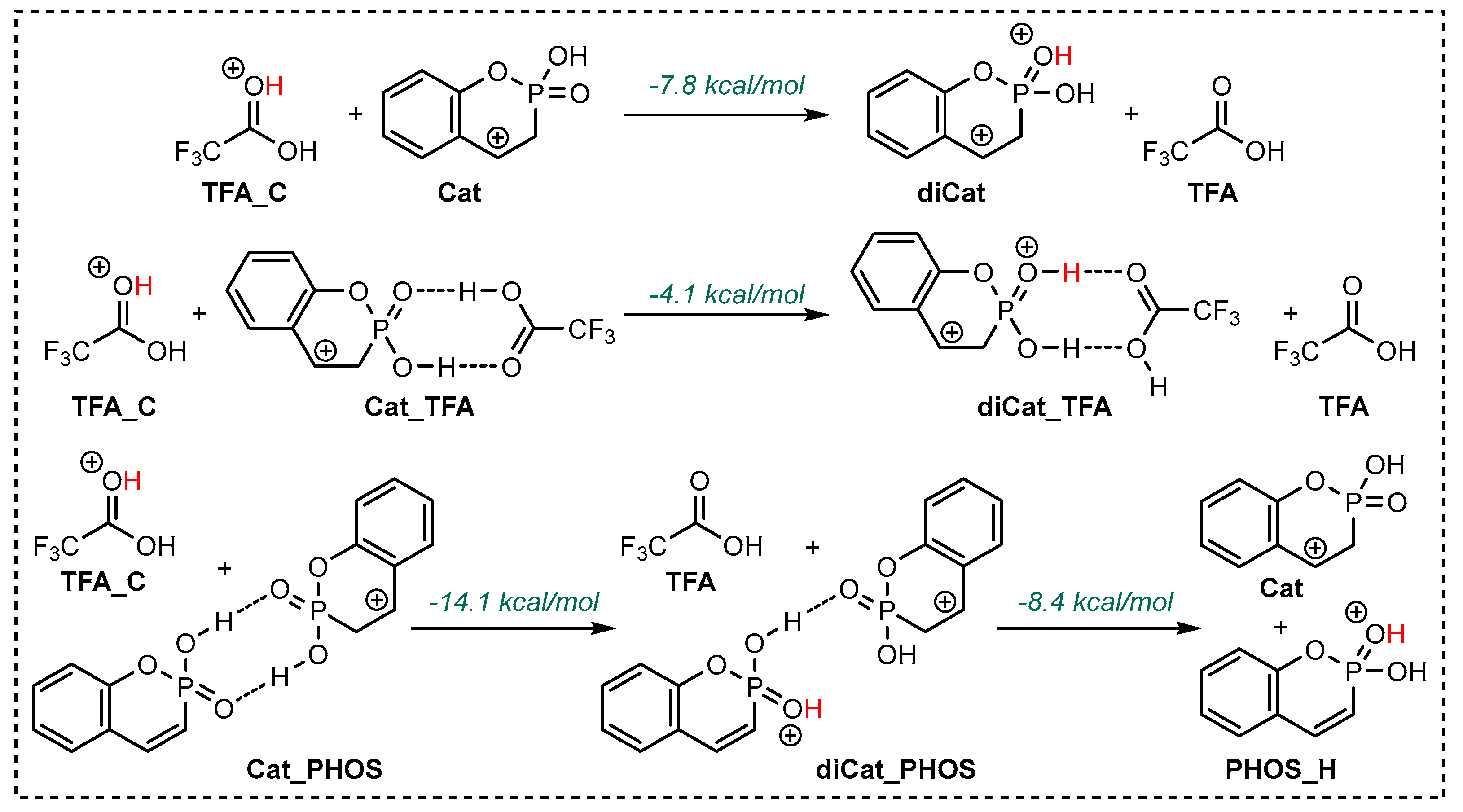
2.2. Exploration of the Reaction Mechanism by Quantum Chemistry Calculations
3. Materials and Methods
3.1. General Methods
3.2. X-Ray Analysis
3.3. Computational Details and Methods
3.4. Synthesis of Phosphacoumarin 1
3.5. General Procedure for Synthesis of Compounds 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Staskun, B. The Conversion of Benzoylacetanilides into 2- and 4-Hydroxyquinolines. J. Org. Chem. 1964, 29, 1153–1157. [Google Scholar] [CrossRef]
- Olah, G.A.; Germain, A.; Lin, H.C.; Forsyth, D.A. Electrophilic reactions at single bonds. XVIII. Indication of protosolvated de facto substituting agents in the reactions of alkanes with acetylium and nitronium ions in superacidic media. J. Am. Chem. Soc. 1975, 97, 2928–2929. [Google Scholar] [CrossRef]
- Olah, G.A. Superelectrophiles. Angew. Chem. Int. Ed. Engl. 1993, 32, 767–788. [Google Scholar] [CrossRef]
- Olah, G.A.; Klumpp, D.A. Superelectrophiles and Their Chemistry; Wiley: Hoboken, NJ, USA, 2007; ISBN 9780470049617. [Google Scholar]
- Vasilyev, A.V. Electrophilic activation of acetylene compounds in Brønsted superacids. Reactions of vinyl type cations. Russ. Chem. Rev. 2013, 82, 187–204. [Google Scholar] [CrossRef]
- Vasilyev, A.V. Superelectrophilic Activation of Alkynes, Alkenes, and Allenes. In Advances in Organic Synthesis; Bentham Science Publishers: Sharjah, United Arab Emirates, 2018; pp. 81–120. [Google Scholar]
- Klumpp, D.A.; Anokhin, M.V. Superelectrophiles: Recent Advances. Molecules 2020, 25, 3281. [Google Scholar] [CrossRef] [PubMed]
- Nenajdenko, V.G.; Shevchenko, N.E.; Balenkova, E.S.; Alabugin, I.V. 1,2-Dications in Organic Main Group Systems. Chem. Rev. 2003, 103, 229–282. [Google Scholar] [CrossRef]
- Olah, G.A.; Surya Prakash, G.K.; Molnár, Á.; Sommer, J. Superacid Chemistry; Wiley: Hoboken, NJ, USA, 2009; ISBN 9780471596684. [Google Scholar]
- Shchukin, A.O.; Vasilyev, A.V. Different reactivities of acetylene carbonyl compounds under the catalysis by Bronsted superacids and Lewis acids. Appl. Catal. A Gen. 2008, 336, 140–147. [Google Scholar] [CrossRef]
- Genaev, A.M.; Salnikov, G.E.; Koltunov, K.Y. DFT insights into superelectrophilic activation of α,β-unsaturated nitriles and ketones in superacids. Org. Biomol. Chem. 2022, 20, 6799–6808. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.C.G.; Zolotukhin, M.G.; Fomine, S.; Cedillo, G.; Morales, S.L.; Fröhlich, N.; Preis, E.; Scherf, U.; Salmón, M.; Chávez, M.I.; et al. Novel, Metal-Free, Superacid-Catalyzed “Click” Reactions of Isatins with Linear, Nonactivated, Multiring Aromatic Hydrocarbons. Macromolecules 2010, 43, 6968–6979. [Google Scholar] [CrossRef]
- Narayanan, A.; Nirmalchandar, A.; Paknia, F.; Rasul, G.; Mathew, T.; Surya Prakash, G.K. Superelectrophilic Activation of Phenylglyoxamides: Efficient Synthesis of Triarylacetamides and Fluorenecarboxamides by Superacid Catalysis. Top. Catal. 2018, 61, 652–663. [Google Scholar] [CrossRef]
- Kurouchi, H.; Sugimoto, H.; Otani, Y.; Ohwada, T. Cyclization of Arylacetoacetates to Indene and Dihydronaphthalene Derivatives in Strong Acids. Evidence for Involvement of Further Protonation of O,O-Diprotonated β-Ketoester, Leading to Enhancement of Cyclization. J. Am. Chem. Soc. 2010, 132, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Vuong, H.; Dash, B.P.; Nilsson Lill, S.O.; Klumpp, D.A. Diels–Alder Reactions with Ethylene and Superelectrophiles. Org. Lett. 2018, 20, 1849–1852. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.A.; Sanchez, G.V.; Aguirre, S.L.; Zhang, Y.; de Leon, S. Chemistry of Dicationic Electrophiles: Superacid-Catalyzed Reactions of Amino Acetals. J. Org. Chem. 2002, 67, 5028–5031. [Google Scholar] [CrossRef] [PubMed]
- Burilov, A.R.; Gazizov, A.S.; Pudovik, M.A.; Konovalov, A.I. Reaction of N-(2,2-Dimethoxyethyl)-N-methylamine and its N-functional derivatives with resorcinol and 2-methylresorcinol. Calix[4]resorcinols functionalized on the lower rim. Russ. J. Gen. Chem. 2007, 77, 98–102. [Google Scholar] [CrossRef]
- Raja, E.K.; Nilsson Lill, S.O.; Klumpp, D.A. Friedel–Crafts-type reactions with ureas and thioureas. Chem. Commun. 2012, 48, 8141–8143. [Google Scholar] [CrossRef] [PubMed]
- Bonazaba Milandou, L.J.C.; Carreyre, H.; Alazet, S.; Greco, G.; Martin-Mingot, A.; Nkounkou Loumpangou, C.; Ouamba, J.; Bouazza, F.; Billard, T.; Thibaudeau, S. Superacid-Catalyzed Trifluoromethylthiolation of Aromatic Amines. Angew. Chem. Int. Ed. 2017, 56, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, H.; Albrecht, Ł.; Wojciechowski, J.; Wolf, W.M. Trifluoromethanesulfonic acid mediated Friedel–Crafts reaction of (E)-3-aryl-2-(diethoxyphosphoryl)acrylic acids with electron-rich hydroxyarenes. A convenient approach to α-methylene-δ-valerolactones. Tetrahedron 2007, 63, 12583–12594. [Google Scholar] [CrossRef]
- Aristov, S.A.; Vasil’ev, A.V.; Fukin, G.K.; Rudenko, A.P. Reactions of acetylene ketones in superacids. Russ. J. Org. Chem. 2007, 43, 691–705. [Google Scholar] [CrossRef]
- Savechenkov, P.Y.; Rudenko, A.P.; Vasil’ev, A.V.; Fukin, G.K. Reactions of Vinyl Type Carbocations Generated in Fluorosulfonic Acid with Benzene Derivatives. New Synthesis of Alkyl 3,3-Diarylpropenoates. Russ. J. Org. Chem. 2005, 41, 1316–1328. [Google Scholar] [CrossRef]
- Lozovskiy, S.V.; Ivanov, A.Y.; Vasilyev, A.V. Different reactivity of phosphorylallenes under the action of Brønsted or Lewis acids: A crucial role of involvement of the P=O group in intra- or intermolecular interactions at the formation of cationic intermediates. Beilstein J. Org. Chem. 2019, 15, 1491–1504. [Google Scholar] [CrossRef]
- Lozovskiy, S.V.; Ivanov, A.Y.; Bogachenkov, A.S.; Vasilyev, A.V. 2,5-Dihydro-1,2-oxaphosphol-2-ium Ions, as Highly Reactive Phosphorus-Centered Electrophiles: Generation, NMR Study, and Reactions. ChemistrySelect 2017, 2, 4505–4510. [Google Scholar] [CrossRef]
- Lozovskiy, S.V.; Bogachenkov, A.S.; Dogadina, A.V.; Vasilyev, A.V. Acid-promoted transformations of aryl substituted diphenylphosphoryl allenes. Tetrahedron Lett. 2016, 57, 3167–3170. [Google Scholar] [CrossRef]
- Bogachenkov, A.S.; Dogadina, A.V.; Boyarskaya, I.A.; Boyarskiy, V.P.; Vasilyev, A.V. Synthesis of 1,4-dihydrophosphinoline 1-oxides by acid-promoted cyclization of 1-(diphenylphosphoryl)allenes. Org. Biomol. Chem. 2016, 14, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Bogachenkov, A.S.; Dogadina, A.V.; Boyarskiy, V.P.; Vasilyev, A.V. Acid-promoted transformations of 1-(diphenylphosphoryl)allenes: Synthesis of novel 1,4-dihydrophosphinoline 1-oxides. Org. Biomol. Chem. 2015, 13, 1333–1338. [Google Scholar] [CrossRef]
- Kazakova, A.N.; Vasilyev, A.V. Trifluoromethanesulfonic acid in organic synthesis. Russ. J. Org. Chem. 2017, 53, 485–509. [Google Scholar] [CrossRef]
- Olah, G.A.; Surya Prakash, G.K.; Wang, Q.; Li, X. Hydrogen Fluoride-Antimony(V) Fluoride. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Chichester, UK, 2001. [Google Scholar]
- Juhasz, M.; Hoffmann, S.; Stoyanov, E.; Kim, K.; Reed, C.A. The Strongest Isolable Acid. Angew. Chem. Int. Ed. 2004, 43, 5352–5355. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.A. Carborane acids. New “strong yet gentle” acids for organic and inorganic chemistry. Chem. Commun. 2005, 36, 1669–1677. [Google Scholar] [CrossRef]
- Avelar, A.; Tham, F.S.; Reed, C.A. Superacidity of Boron Acids H2(B12X12) (X=Cl, Br). Angew. Chem. Int. Ed. 2009, 48, 3491–3493. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H. From designer Lewis acid to designer Brønsted acid towards more reactive and selective acid catalysis. Proc. Jpn. Acad. Ser. B 2008, 84, 134–146. [Google Scholar] [CrossRef]
- Yamamoto, H.; Futatsugi, K. “Designer Acids”: Combined Acid Catalysis for Asymmetric Synthesis. Angew. Chem. Int. Ed. 2005, 44, 1924–1942. [Google Scholar] [CrossRef]
- Zalaltdinova, A.V.; Sadykova, Y.M.; Smailov, A.K.; Trofimova, L.M.; Burilov, A.R.; Pudovik, M.A. New intramolecular cyclization of 2 H -benzo[e]-1,2-oxaphosphorinine derivatives—A way to the synthesis of unsymmetrical cage phosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2022, 197, 549–550. [Google Scholar] [CrossRef]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef]
- Chen, S.; Wang, X.; Cheng, Y.; Gao, H.; Chen, X. A Review of Classification, Biosynthesis, Biological Activities and Potential Applications of Flavonoids. Molecules 2023, 28, 4982. [Google Scholar] [CrossRef]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Engel, R. Phosphonates as analogues of natural phosphates. Chem. Rev. 1977, 77, 349–367. [Google Scholar] [CrossRef]
- Amira, A.; Aouf, Z.; K’tir, H.; Chemam, Y.; Ghodbane, R.; Zerrouki, R.; Aouf, N. Recent Advances in the Synthesis of α-Aminophosphonates: A Review. ChemistrySelect 2021, 6, 6137–6149. [Google Scholar] [CrossRef]
- Sadykova, Y.M.; Sadikova, L.M.; Badrtdinova, A.R.; Dobrynin, A.B.; Burilov, A.R.; Pudovik, M.A. Condensation of 2-Ethoxyvinylphosphonic Acid Dichloroanhydride with 2,3,5-Trimethylphenol. Novel Method for Preparation of Phosphacoumarins. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 2267–2272. [Google Scholar] [CrossRef]
- Chang, Z.; Huang, J.; Wang, S.; Chen, G.; Zhao, H.; Wang, R.; Zhao, D. Copper catalyzed late-stage C(sp3)-H functionalization of nitrogen heterocycles. Nat. Commun. 2021, 12, 4342. [Google Scholar] [CrossRef]
- Yadav, N.; Khan, J.; Tyagi, A.; Singh, S.; Hazra, C.K. Rapid Access to Arylated and Allylated Cyclopropanes via Bronsted Acid-Catalyzed Dehydrative Coupling of Cyclopropylcarbinols. J. Org. Chem. 2022, 87, 6886–6901. [Google Scholar] [CrossRef]
- Gazizov, A.S.; Smolobochkin, A.V.; Anikina, E.A.; Strelnik, A.G.; Burilov, A.R.; Pudovik, M.A. Acid-Mediated C–N Bond Cleavage in 1-Sulfonylpyrrolidines: An Efficient Route towards Dibenzoxanthenes, Diarylmethanes, and Resorcinarenes. Synlett 2018, 29, 467–472. [Google Scholar] [CrossRef]
- Sabat, N.; Zhou, W.; Gandon, V.; Guinchard, X.; Vincent, G. Unbiased C3-Electrophilic Indoles: Triflic Acid Mediated C3-Regioselective Hydroarylation of N-H Indoles. Angew. Chem. Int. Ed. 2022, 61, e202204400. [Google Scholar] [CrossRef]
- Umrigar, V.M.; Chakraborty, M.; Parikh, P.A. Microwave Assisted Sulfonation of 2-Naphthol by Sulfuric Acid: Cleaner Production of Schaeffer’s Acid. Ind. Eng. Chem. Res. 2007, 46, 6217–6220. [Google Scholar] [CrossRef]
- Shcherbakov, S.S.; Magometov, A.Y.; Shcherbakova, V.Y.; Aksenov, A.V.; Domenyuk, D.A.; Zelensky, V.A.; Rubin, M. Electrophilic alkylation of arenes with 5-bromopyrimidine en route to 4-aryl-5-alkynylpyrimidines. RSC Adv. 2020, 10, 10315–10321. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Mateescu, G.D.; Mo, Y.K. Stable carbocations. CXL. Naphthalenium ions. J. Am. Chem. Soc. 1973, 95, 1865–1874. [Google Scholar] [CrossRef]
- Hartmann, H.; Yu, X. On the Protonation and Deuteration of Hydroxy-Substituted Naphthalenes—A 1H NMR Study**. ChemistrySelect 2022, 7, e202202836. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Milne, J.B. Trifluoroacetic Acid. In The Chemistry of Nonaqueous Solvents; Elsevier: Amsterdam, The Netherlands, 1978; pp. 1–52. [Google Scholar]
- Sheldrick, G.M. SADABS; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXTL v.6.12, Structure Determination Software Suite; Bruker AXS: Madison, WI, USA, 2000. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
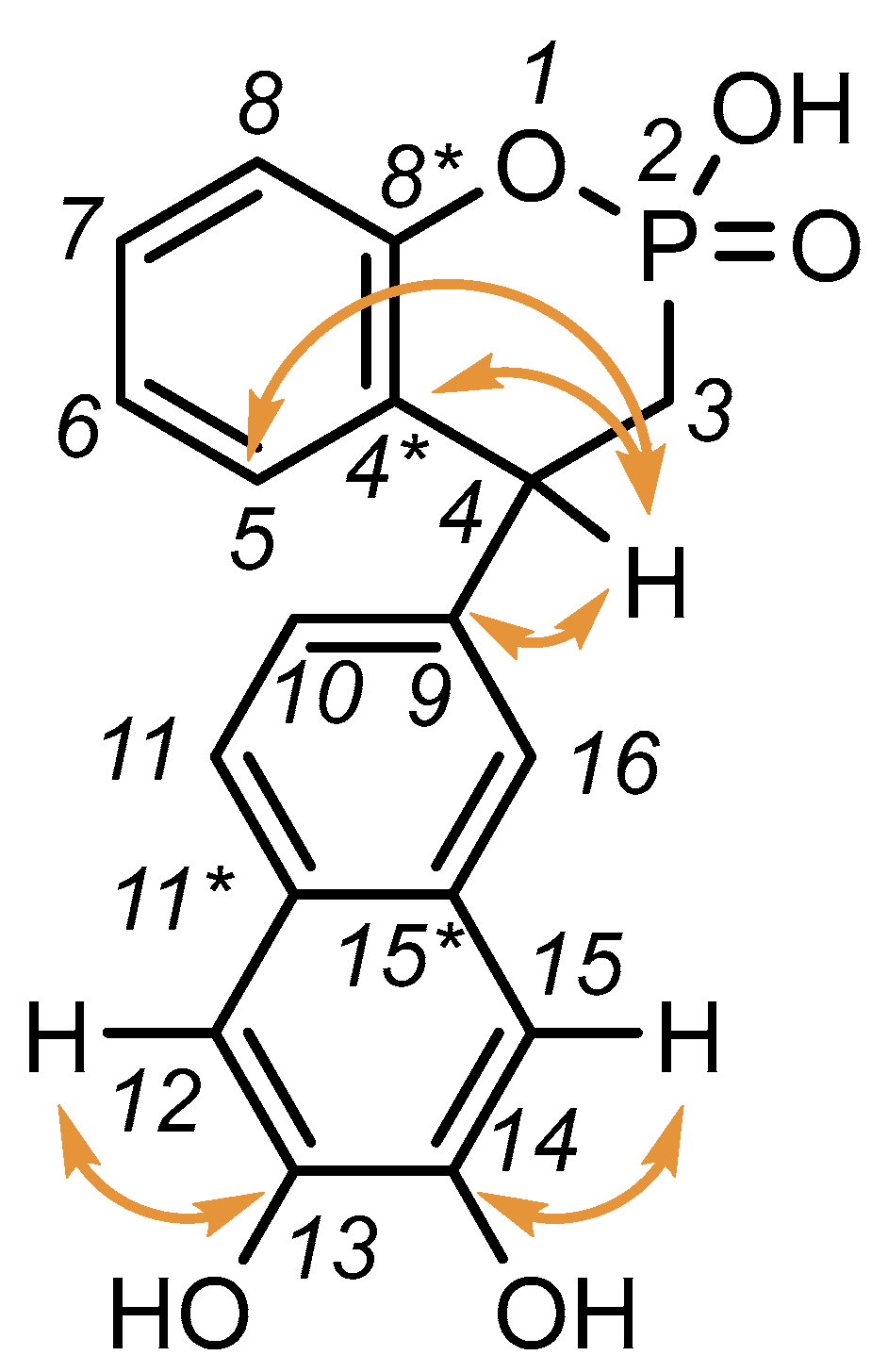
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zalaltdinova, A.V.; Sadykova, Y.M.; Gazizov, A.S.; Smailov, A.K.; Syakaev, V.V.; Gerasimova, D.P.; Chugunova, E.A.; Akylbekov, N.I.; Zhapparbergenov, R.U.; Appazov, N.O.; et al. Superelectrophilic Activation of Phosphacoumarins towards Weak Nucleophiles via Brønsted Acid Assisted Brønsted Acid Catalysis. Int. J. Mol. Sci. 2024, 25, 6327. https://doi.org/10.3390/ijms25126327
Zalaltdinova AV, Sadykova YM, Gazizov AS, Smailov AK, Syakaev VV, Gerasimova DP, Chugunova EA, Akylbekov NI, Zhapparbergenov RU, Appazov NO, et al. Superelectrophilic Activation of Phosphacoumarins towards Weak Nucleophiles via Brønsted Acid Assisted Brønsted Acid Catalysis. International Journal of Molecular Sciences. 2024; 25(12):6327. https://doi.org/10.3390/ijms25126327
Chicago/Turabian StyleZalaltdinova, Alena V., Yulia M. Sadykova, Almir S. Gazizov, Atabek K. Smailov, Victor V. Syakaev, Daria P. Gerasimova, Elena A. Chugunova, Nurgali I. Akylbekov, Rakhmetulla U. Zhapparbergenov, Nurbol O. Appazov, and et al. 2024. "Superelectrophilic Activation of Phosphacoumarins towards Weak Nucleophiles via Brønsted Acid Assisted Brønsted Acid Catalysis" International Journal of Molecular Sciences 25, no. 12: 6327. https://doi.org/10.3390/ijms25126327
APA StyleZalaltdinova, A. V., Sadykova, Y. M., Gazizov, A. S., Smailov, A. K., Syakaev, V. V., Gerasimova, D. P., Chugunova, E. A., Akylbekov, N. I., Zhapparbergenov, R. U., Appazov, N. O., Burilov, A. R., Pudovik, M. A., Alabugin, I. V., & Sinyashin, O. G. (2024). Superelectrophilic Activation of Phosphacoumarins towards Weak Nucleophiles via Brønsted Acid Assisted Brønsted Acid Catalysis. International Journal of Molecular Sciences, 25(12), 6327. https://doi.org/10.3390/ijms25126327