
Homozygosity for a Rare Plec Variant Suggests a Contributory Role in Congenital Insensitivity to Pain
 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
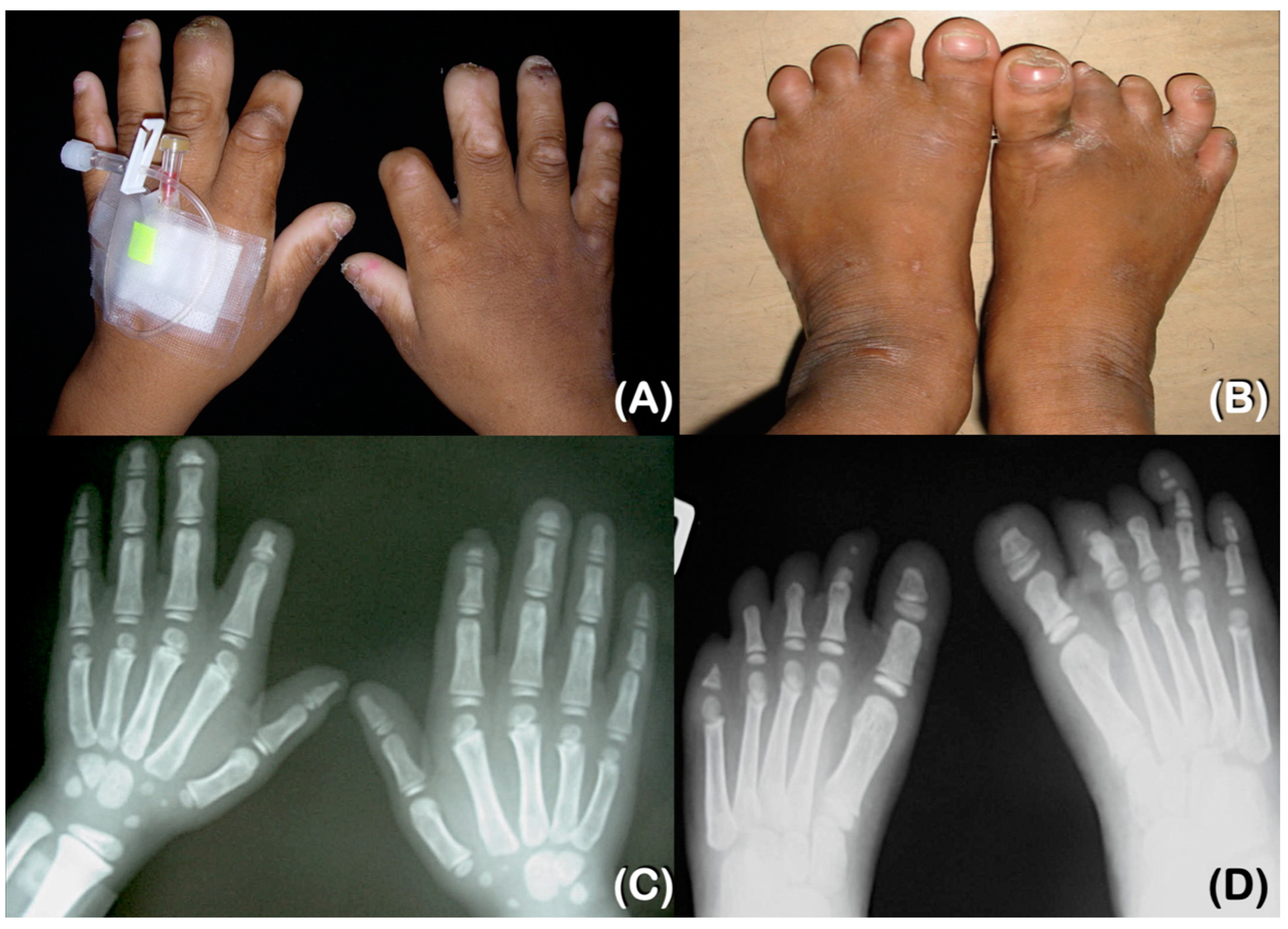
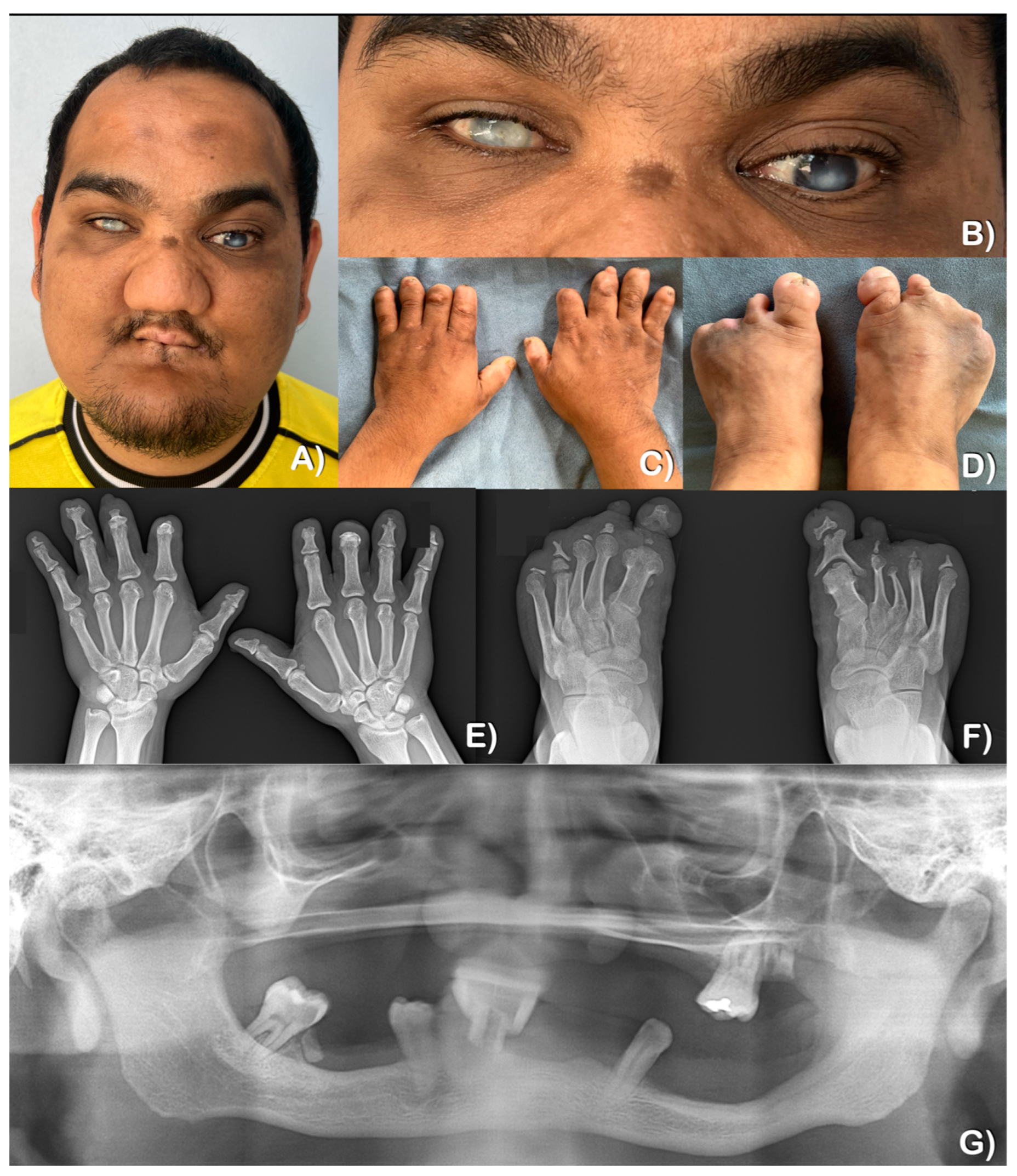
2.1. Patient Reports
2.1.1. Patient 1
2.1.2. Patient 2
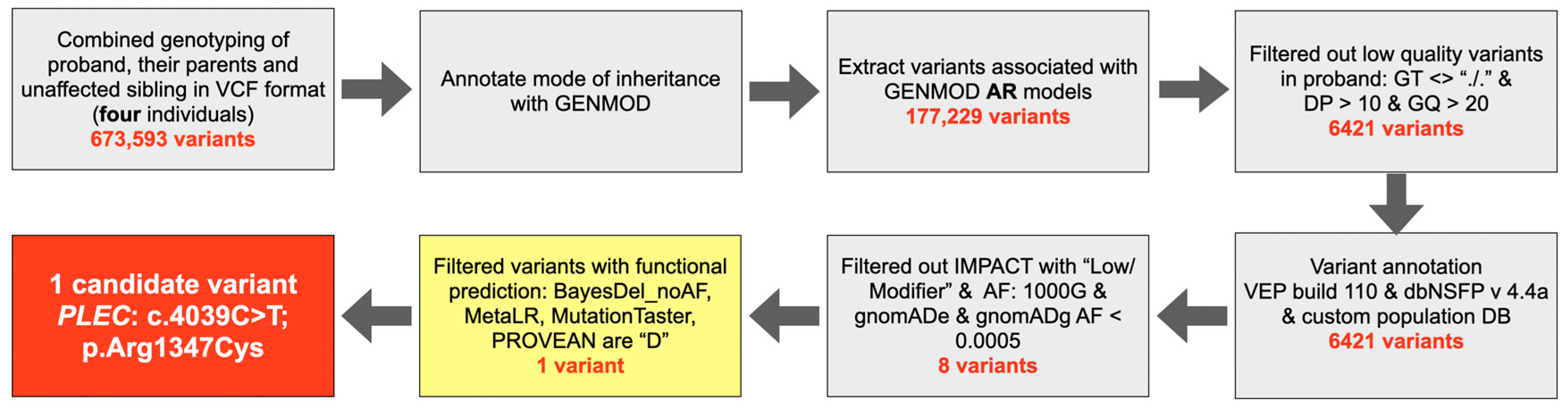
2.2. Whole Exome Sequencing and Bioinformatic Analysis
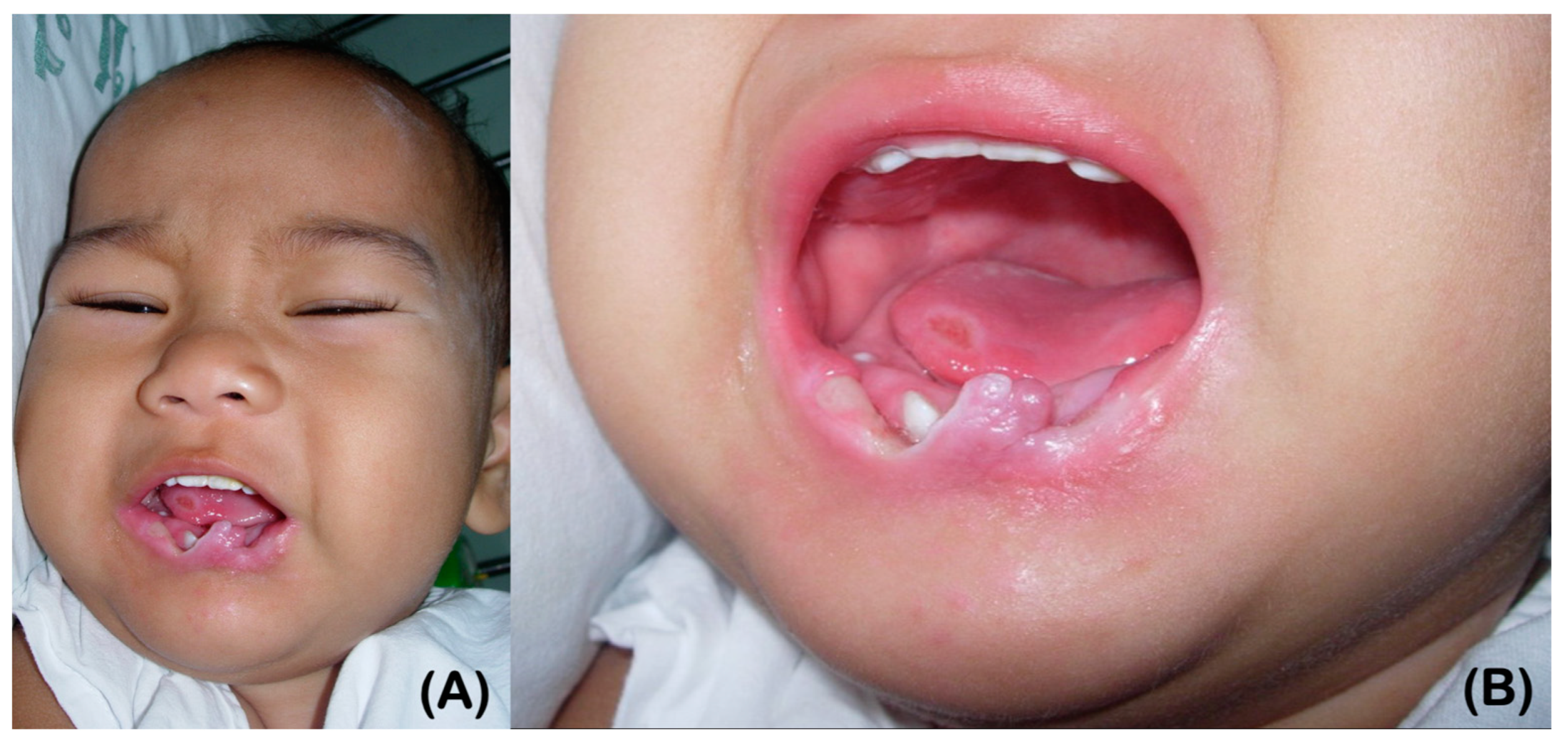
2.3. Histopathological Study of Skin Biopsy
2.4. Immunohistochemistry of the Skin
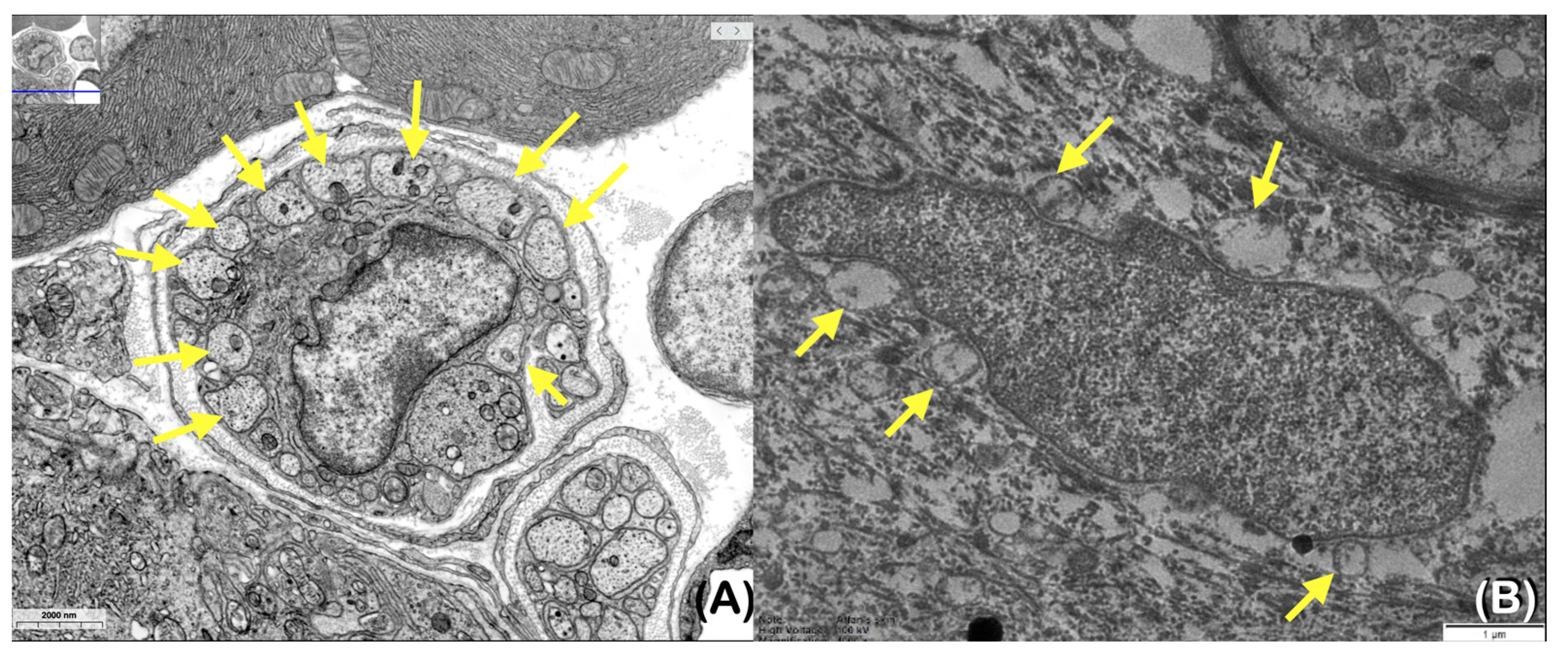
2.5. Transmission Electron Microscopic Study of Skin
2.6. Plec Expression during Early Development
2.6.1. Plectin Expression in the Spinal Dorsal Horn
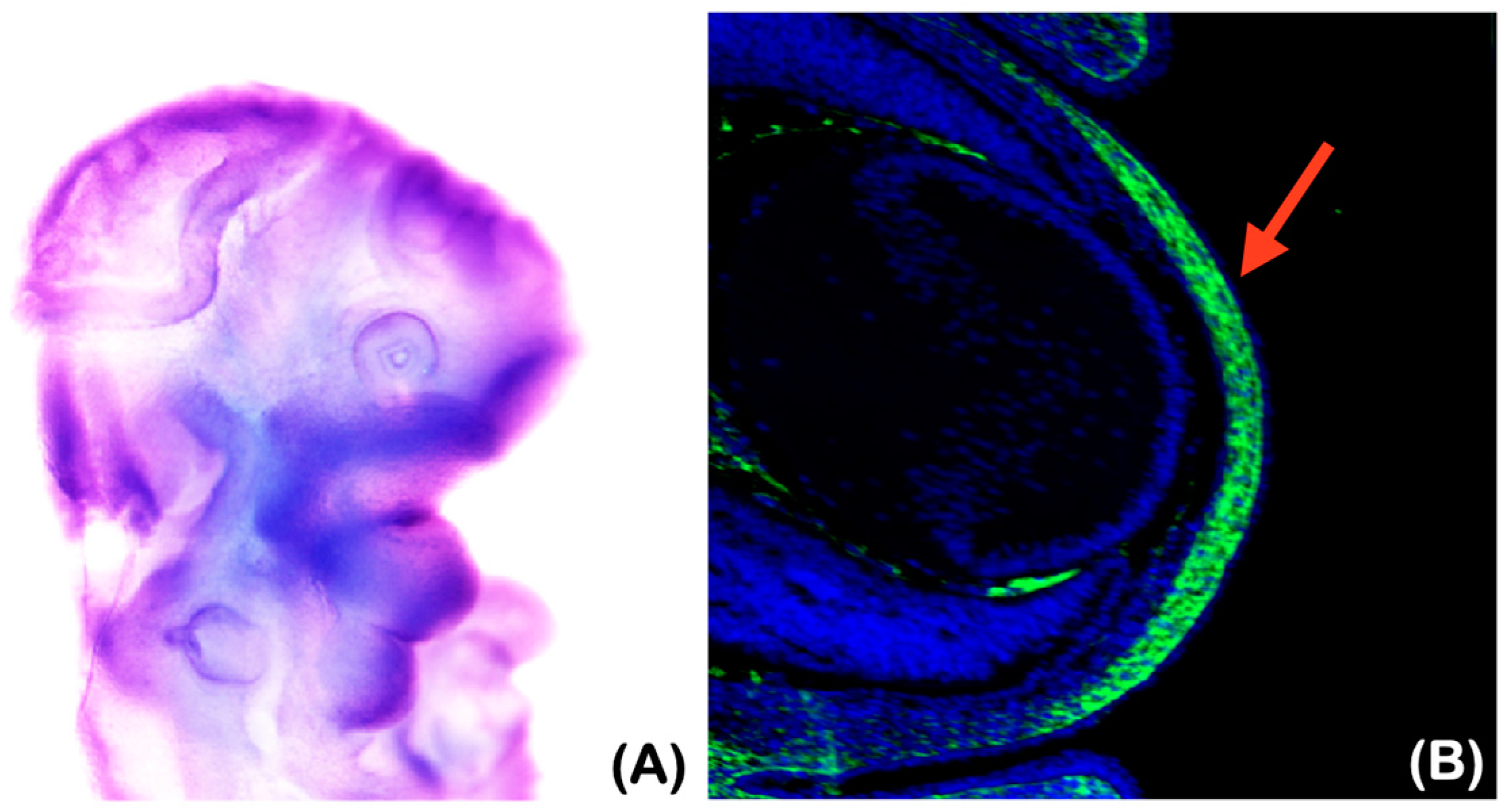
2.6.2. Underdeveloped Jaws and Corneal Defects
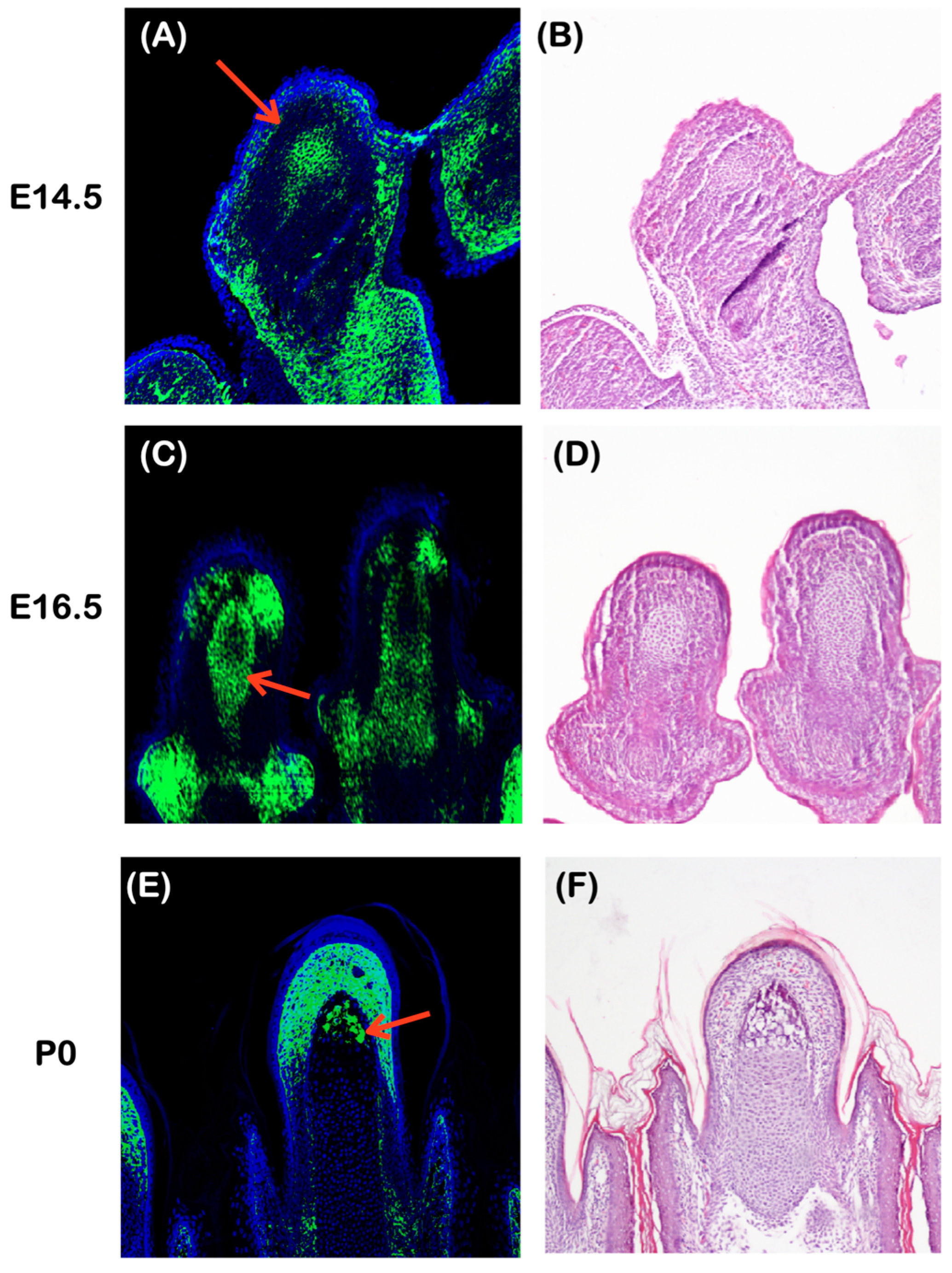
2.6.3. Plec Expression in the Developing Distal Phalanges
2.7. Mutant Protein Model
3. Materials and Methods
3.1. Whole Exome Sequencing and Bioinformatic Analysis
3.2. Histological Study
3.3. Immunohistochemistry of the Skin
3.4. Transmission Electron Microscopy of Skin Biopsy
3.5. Immunohistochemistry of Plec Expression in Mouse Embryo
3.6. Mutant Protein Modeling
4. Discussion
4.1. Genetic Variant in Plec and Congenital Insensitivity to Pain
4.2. Structure, Complexity, and Expression of Plectin
4.3. Functions of Plectin
4.4. Plectin in Central and Peripheral Nervous Systems
4.5. Genetic Variant in Plec and Corneal Ulceration and Scarring
4.6. Genetic Variant in Plec, Underdeveloped Jaw Bones, and Autoextraction of Teeth
4.7. Genetic Variant in Plec and Acro-Osteolysis (Amputation of Digits)
4.8. Genetic Variant in Plec, Staphylococcus aureus and Pastuerella multocida Infections, and Delayed Wound Healing
4.9. Plec Variant and Its Clinical Phenotypes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, L.; Chiu, I.M. Microbes and pain. PLoS Pathog. 2021, 17, e1009398. [Google Scholar] [CrossRef]
- Aguggia, M. Neurophysiology of pain. Neurol. Sci. 2003, 24, s57–s60. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Lischka, A.; Lassuthova, P.; Çakar, A.; Record, C.J.; Van Lent, J.; Baets, J.; Dohrn, M.F.; Senderek, J.; Lampert, A.; Bennett, D.L.; et al. Genetic pain loss disorders. Nat. Rev. Dis. Primers 2022, 8, 41. [Google Scholar] [CrossRef]
- Drissi, I.; Woods, W.A.; Woods, C.G. Understanding the genetic basis of congenital insensitivity to pain. Br. Med. Bull. 2020, 133, 65–78. [Google Scholar] [CrossRef]
- Lischka, A.; Eggermann, K.; Record, C.J.; Dohrn, M.F.; Laššuthová, P.; Kraft, F.; Begemann, M.; Dey, D.; Eggermann, T.; Beijer, D.; et al. Genetic landscape of congenital insensitivity to pain and hereditary sensory and autonomic neuropathies. Brain 2023, 146, 4880–4890. [Google Scholar] [CrossRef]
- Habib, A.M.; Matsuyama, A.; Okorokov, A.L.; Santana-Varela, S.; Bras, J.T.; Aloisi, A.M.; Emery, E.C.; Bogdanov, Y.D.; Follenfant, M.; Gossage, S.J.; et al. A novel human pain insensitivity disorder caused by a point mutation in ZFHX2. Brain 2018, 141, 365–376. [Google Scholar] [CrossRef]
- Winter, L.; Türk, M.; Harter, P.N.; Mittelbronn, M.; Kornblum, C.; Norwood, F.; Jungbluth, H.; Thiel, C.T.; Schlötzer-Schrehardt, U.; Schröder, R. Downstream effects of Plectin mutations in epidermolysis bullosa simplex with muscular dystrophy. Acta Neuropathol. Commun. 2016, 4, 44. [Google Scholar] [CrossRef]
- Potokar, M.; Jorgačevski, J. Plectin in the Central Nervous System and a Putative Role in Brain Astrocytes. Cells 2021, 10, 2353. [Google Scholar] [CrossRef]
- Wiche, G. Plectin in Health and Disease. Cells 2022, 11, 1412. [Google Scholar] [CrossRef]
- Vahidnezhad, H.; Youssefian, L.; Harvey, N.; Tavasoli, A.R.; Saeidian, A.H.; Sotoudeh, S.; Varghaei, A.; Mahmoudi, H.; Mansouri, P.; Mozafari, N.; et al. Mutation update: The spectra of Plec sequence variants and related Plectinopathies. Hum. Mutat. 2022, 43, 1706–1731. [Google Scholar] [CrossRef]
- Martin, P. Tissue patterning in the developing mouse limb. Int. J. Dev. Biol. 1990, 34, 323–336. [Google Scholar]
- Hu, J.; He, L. Patterning mechanisms controlling digit development. J. Genet. Genom. 2008, 35, 517–524. [Google Scholar] [CrossRef]
- Ortega, E.; Manso, J.A.; Buey, R.M.; Carballido, A.M.; Carabias, A.; Sonnenberg, A.; de Pereda, J.M. The Structure of the Plakin Domain of Plectin Reveals an Extended Rod-like Shape. J. Biol. Chem. 2016, 291, 18643–18662. [Google Scholar] [CrossRef]
- Ketema, M.; Secades, P.; Kreft, M.; Nahidiazar, L.; Janssen, H.; Jalink, K.; de Pereda, J.M.; Sonnenberg, A. The rod domain is not essential for the function of Plectin in maintaining tissue integrity. Mol. Biol. Cell 2015, 26, 2402–2417. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Auwera, G.A.V.d.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018, 201178. [Google Scholar] [CrossRef]
- Måns, M. Genmod. 2018. Available online: https://github.com/moonso/genmod (accessed on 8 January 2024).
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Guzmán-Vega, F.J.; González-Álvarez, A.C.; Peña-Guerra, K.A.; Cardona-Londoño, K.J.; Arold, S.T. Leveraging AI Advances and Online Tools for Structure-Based Variant Analysis. Curr. Protoc. 2023, 3, e857. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Valencia, R.G.; Mihailovska, E.; Winter, L.; Bauer, K.; Fischer, I.; Walko, G.; Jorgacevski, J.; Potokar, M.; Zorec, R.; Wiche, G. Plectin dysfunction in neurons leads to tau accumulation on microtubules affecting neuritogenesis, organelle trafficking, pain sensitivity and memory. Neuropathol. Appl. Neurobiol. 2021, 47, 73–95. [Google Scholar] [CrossRef]
- Sonnenberg, A.; Liem, R.K. Plakins in development and disease. Exp. Cell Res. 2007, 313, 2189–2203. [Google Scholar] [CrossRef]
- Rief, M.; Pascual, J.; Saraste, M.; Gaub, H.E. Single molecule force spectroscopy of spectrin repeats: Low unfolding forces in helix bundles. J. Mol. Biol. 1999, 286, 553–561. [Google Scholar] [CrossRef]
- Castañón, M.J.; Walko, G.; Winter, L.; Wiche, G. Plectin-intermediate filament partnership in skin, skeletal muscle, and peripheral nerve. Histochem. Cell. Biol. 2013, 140, 33–53. [Google Scholar] [CrossRef]
- Winter, L.; Wiche, G. The many faces of Plectin and Plectinopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 77–93. [Google Scholar] [CrossRef]
- Fuchs, P.; Zörer, M.; Reipert, S.; Rezniczek, G.A.; Propst, F.; Walko, G.; Fischer, I.; Bauer, J.; Leschnik, M.W.; Lüscher, B.; et al. Targeted inactivation of a developmentally regulated neural Plectin isoform (Plectin 1c) in mice leads to reduced motor nerve conduction velocity. J. Biol. Chem. 2009, 284, 26502–26509. [Google Scholar] [CrossRef]
- Perrot, R.; Berges, R.; Bocquet, A.; Eyer, J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol. Neurobiol. 2008, 38, 27–65. [Google Scholar] [CrossRef]
- Dubey, M.; Hoda, S.; Chan, W.K.; Pimenta, A.; Ortiz, D.D.; Shea, T.B. Reexpression of vimentin in differentiated neuroblastoma cells enhances elongation of axonal neurites. J. Neurosci. Res. 2004, 78, 245–249. [Google Scholar] [CrossRef]
- Helfand, B.T.; Chou, Y.H.; Shumaker, D.K.; Goldman, R.D. Intermediate filament proteins participate in signal transduction. Trends Cell Biol. 2005, 15, 568–570. [Google Scholar] [CrossRef]
- Mimura, T.; Amano, S.; Fukuoka, S.; Honda, N.; Arita, R.; Ochiai, M.; Yanagisawa, M.; Usui, T.; Ono, K.; Araki, F.; et al. In vivo confocal microscopy of hereditary sensory and autonomic neuropathy. Curr. Eye Res. 2008, 33, 940–945. [Google Scholar] [CrossRef]
- Elsana, B.; Gradstein, L.; Imtirat, A.; Yagev, R.; Barrett, C.; Ling, G.; Abu Tailakh, M.; Baidousi, A.; Tsumi, E. Ocular manifestations of congenital insensitivity to pain: A long-term follow-up. Br. J. Ophthalmol. 2022, 106, 1217–1221. [Google Scholar] [CrossRef]
- Elsana, B.; Imtirat, A.; Yagev, R.; Gradstein, L.; Majdalani, P.; Iny, O.; Parvari, R.; Tsumi, E. Ocular manifestations among patients with congenital insensitivity to pain due to variants in PRDM12 and SCN9A genes. Am. J. Med. Genet. A 2022, 188, 3463–3468. [Google Scholar] [CrossRef]
- Amano, S.; Fukuoka, S.; Usui, T.; Honda, N.; Ideta, R.; Ochiai, M.; Yamagami, S.; Araie, M.; Awaya, Y. Ocular manifestations of congenital insensitivity to pain with anhidrosis. Am. J. Ophthalmol. 2006, 141, 472–477. [Google Scholar] [CrossRef]
- Zhang, S.; Malik Sharif, S.; Chen, Y.C.; Valente, E.M.; Ahmed, M.; Sheridan, E.; Bennett, C.; Woods, G. Clinical features for diagnosis and management of patients with PRDM12 congenital insensitivity to pain. J. Med. Genet. 2016, 53, 533–535. [Google Scholar] [CrossRef]
- Jarade, E.F.; El-Sheikh, H.F.; Tabbara, K.F. Indolent corneal ulcers in a patient with congenital insensitivity to pain with anhidrosis: A case report and literature review. Eur. J. Ophthalmol. 2002, 12, 60–65. [Google Scholar] [CrossRef]
- Nilsen, K.B.; Nicholas, A.K.; Woods, C.G.; Mellgren, S.I.; Nebuchennykh, M.; Aasly, J. Two novel SCN9A mutations causing insensitivity to pain. Pain 2009, 143, 155–158. [Google Scholar] [CrossRef]
- Cox, J.J.; Sheynin, J.; Shorer, Z.; Reimann, F.; Nicholas, A.K.; Zubovic, L.; Baralle, M.; Wraige, E.; Manor, E.; Levy, J.; et al. Congenital insensitivity to pain: Novel SCN9A missense and in-frame deletion mutations. Hum. Mutat. 2010, 31, E1670–E1686. [Google Scholar] [CrossRef]
- Guven, Y.; Altunoglu, U.; Aktoren, O.; Uyguner, Z.O.; Kayserili, H.; Kaewkahya, M.; Kantaputra, P.N. Twins with hereditary sensory and autonomic neuropathy type IV with preserved periodontal sensation. Eur. J. Med. Genet. 2014, 57, 240–246. [Google Scholar] [CrossRef]
- Yiş, U.; Mademan, I.; Kavukçu, S.; Baets, J. A novel NTRK1 mutation in a patient with congenital insensitivity to pain with anhidrosis. Acta Neurol. Belg. 2015, 115, 509–511. [Google Scholar] [CrossRef]
- López-Cortés, A.; Zambrano, A.K.; Guevara-Ramírez, P.; Echeverría, B.A.; Guerrero, S.; Cabascango, E.; Pérez-Villa, A.; Armendáriz-Castillo, I.; García-Cárdenas, J.M.; Yumiceba, V.; et al. Clinical, genomics and networking analyses of a high-altitude native American Ecuadorian patient with congenital insensitivity to pain with anhidrosis: A case report. BMC Med. Genom. 2020, 13, 113. [Google Scholar] [CrossRef]
- Zhu, R.; Zhu, Y.; Xu, M.; Gu, Z. Ophthalmic findings of congenital insensitivity to pain with anhidrosis with a novel neurotrophic tyrosine kinase receptor type 1 gene mutation: A case report. Front. Med. 2022, 9, 955929. [Google Scholar] [CrossRef]
- Thrush, D.C. Congenital insensitivity to pain. A clinical, genetic and neurophysiological study of four children from the same family. Brain 1973, 96, 369–386. [Google Scholar] [CrossRef]
- Brahim, J.S.; Roberts, M.W.; McDonald, H.D. Oral and maxillofacial complications associated with congenital sensory neuropathy with anhydrosis: Report of two cases. J. Oral. Maxillofac. Surg. 1987, 45, 331–334. [Google Scholar] [CrossRef]
- Bodzioch, M.; Lapicka, K.; Aslanidis, C.; Kacinski, M.; Schmitz, G. Two novel mutant alleles of the gene encoding neurotrophic tyrosine kinase receptor type 1 (NTRK1) in a patient with congenital insensitivity to pain with anhidrosis: A splice junction mutation in intron 5 and cluster of four mutations in exon 15. Hum. Mutat. 2001, 17, 72. [Google Scholar] [CrossRef]
- Schalka, M.M.; Corrêa, M.S.; Ciamponi, A.L. Congenital insensitivity-to-pain with anhidrosis (CIPA): A case report with 4-year follow-up. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2006, 101, 769–773. [Google Scholar] [CrossRef]
- Safari, A.; Khaledi, A.A.; Vojdani, M. Congenital Insensitivity to Pain with Anhidrosis (CIPA): A Case Report. Iran Red. Crescent. Med. J. 2011, 13, 134–138. [Google Scholar]
- Gao, L.; Guo, H.; Ye, N.; Bai, Y.; Liu, X.; Yu, P.; Xue, Y.; Ma, S.; Wei, K.; Jin, Y.; et al. Oral and craniofacial manifestations and two novel missense mutations of the NTRK1 gene identified in the patient with congenital insensitivity to pain with anhidrosis. PLoS ONE 2013, 8, e66863. [Google Scholar] [CrossRef]
- Iftikhar, S.; Javed, M.A. Seeing is not always believing: Congenital insensitivity to pain with anhidrosis mimicking leprosy. Mayo Clin. Proc. 2013, 88, e153–e154. [Google Scholar] [CrossRef]
- Xue, X.M.; Liu, Y.Q.; Pang, P.; Sun, C.F. Congenital Loss of Permanent Teeth in a Patient with Congenital Insensitivity to Pain with Anhidrosis due to 2 Novel Mutations in the NTRK1 Gene. J. Oral Maxillofac. Surg. 2018, 76, e2581–e2582. [Google Scholar] [CrossRef]
- Kim, J.S.; Woo, Y.J.; Kim, G.M.; Kim, C.J.; Ma, J.S.; Hwang, T.J.; Lee, M.C. Congenital insensitivity to pain with anhidrosis: A case report. J. Korean Med. Sci. 1999, 14, 460–464. [Google Scholar] [CrossRef]
- Ma, A.; Turner, A. A life without pain: Congenital insensitivity to pain due to compound heterozygous SCN9A mutation. J. Paediatr. Child Health 2012, 48, 285–286. [Google Scholar] [CrossRef]
- Kalaskar, R.; Kalaskar, A. Hereditary sensory and autonomic neuropathy type V: Report of a rare case. Contemp. Clin. Dent. 2015, 6, 103–106. [Google Scholar] [CrossRef]
- Prabhu, S.; Fortier, K.; Newsome, L.; Reebye, U.N. Office-Based Anesthetic and Oral Surgical Management of a Child with Hereditary Sensory Autonomic Neuropathy Type IV: A Case Report. Anesth. Prog. 2018, 65, 181–186. [Google Scholar] [CrossRef]
- Pinsky, L.; DiGeorge, A.M. Congenital familial sensory neuropathy with anhidrosis. J. Pediatr. 1966, 68, 1–13. [Google Scholar] [CrossRef]
- Person, J.R.; Rogers, R.S.; Rhodes, K.H. Congenital sensory neuropathy: Report of an atypical case. Arch. Dermatol. 1977, 113, 954–957. [Google Scholar] [CrossRef]
- Pavone, L.; Huttenlocher, P.; Siciliano, L.; Micali, G.; Rizzo, R.; Anastasi, M.; Maimone, D.; Woolmann, R. Two brothers with a variant of hereditary sensory neuropathy. Neuropediatrics 1992, 23, 92–95. [Google Scholar] [CrossRef]
- Rosemberg, S.; Marie, S.K.; Kliemann, S. Congenital insensitivity to pain with anhidrosis (hereditary sensory and autonomic neuropathy type IV). Pediatr. Neurol. 1994, 11, 50–56. [Google Scholar] [CrossRef]
- Ismail, E.A.; Al-Shammari, N.; Anim, J.T.; Moosa, A. Congenital insensitivity to pain with anhidrosis: Lack of eccrine sweat gland innervation confirmed. J. Child. Neurol. 1998, 13, 243–246. [Google Scholar] [CrossRef]
- Erdem, T.L.; Ozcan, I.; Ilgüy, D.; Sirin, S. Hereditary sensory and autonomic neuropathy: Review and a case report with dental implications. J. Oral. Rehabil. 2000, 27, 180–183. [Google Scholar] [CrossRef]
- Nolano, M.; Crisci, C.; Santoro, L.; Barbieri, F.; Casale, R.; Kennedy, W.R.; Wendelschafer-Crabb, G.; Provitera, V.; Di Lorenzo, N.; Caruso, G. Absent innervation of skin and sweat glands in congenital insensitivity to pain with anhidrosis. Clin. Neurophysiol. 2000, 111, 1596–1601. [Google Scholar] [CrossRef]
- Toscano, E.; della Casa, R.; Mardy, S.; Gaetaniello, L.; Sadile, F.; Indo, Y.; Pignata, C.; Andria, G. Multisystem involvement in congenital insensitivity to pain with anhidrosis (CIPA), a nerve growth factor receptor(Trk A)-related disorder. Neuropediatrics 2000, 31, 39–41. [Google Scholar] [CrossRef]
- Indo, Y.; Mardy, S.; Miura, Y.; Moosa, A.; Ismail, E.A.; Toscano, E.; Andria, G.; Pavone, V.; Brown, D.L.; Brooks, A.; et al. Congenital insensitivity to pain with anhidrosis (CIPA): Novel mutations of the TRKA (NTRK1) gene, a putative uniparental disomy, and a linkage of the mutant TRKA and PKLR genes in a family with CIPA and pyruvate kinase deficiency. Hum. Mutat. 2001, 18, 308–318. [Google Scholar] [CrossRef]
- Sztriha, L.; Lestringant, G.G.; Hertecant, J.; Frossard, P.M.; Masouyé, I. Congenital insensitivity to pain with anhidrosis. Pediatr. Neurol. 2001, 25, 63–66. [Google Scholar] [CrossRef]
- Gupta, B. Congenital insensitivity of pain with anhidrosis. Indian J. Pediatr. 2003, 70, 109–111. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Vargiami, E.; Economou, M.; Gombakis, N. Self-mutilation and mental retardation: Clues to congenital insensitivity to pain with anhidrosis. J. Pediatr. 2004, 144, 284. [Google Scholar] [CrossRef]
- Karande, S.; Satam, N. Hereditary sensory and autonomic neuropathy type IV. Indian Pediatr. 2005, 42, 608–609. [Google Scholar]
- Tunçbilek, G.; Oztekin, C.; Kayikçioğlu, A. Calcaneal ulcer in a child with congenital insensitivity to pain syndrome. Scand. J. Plast. Reconstr. Surg. Hand Surg. 2005, 39, 180–183. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, A.B.; Lin, Z.; Zhou, J.N. Congenital insensitivity to pain with anhidrosis and progressing acro-osteolysis: A case report with 7-year follow-up. Chin. Med. J. 2006, 119, 2134–2137. [Google Scholar] [CrossRef]
- Canbay, O.; Kose, E.A.; Celebi, N.; Karagoz, A.H.; Ozgen, S. Anesthesia for congenital insensitivity to pain with anhidrosis. Paediatr. Anaesth. 2007, 17, 190–192. [Google Scholar] [CrossRef]
- Tüysüz, B.; Bayrakli, F.; DiLuna, M.L.; Bilguvar, K.; Bayri, Y.; Yalcinkaya, C.; Bursali, A.; Ozdamar, E.; Korkmaz, B.; Mason, C.E.; et al. Novel NTRK1 mutations cause hereditary sensory and autonomic neuropathy type IV: Demonstration of a founder mutation in the Turkish population. Neurogenetics 2008, 9, 119–125. [Google Scholar] [CrossRef]
- John, D.; Thomas, M.; Jacob, P. Neurotrophic keratitis and congenital insensitivity to pain with anhidrosis—A case report with 10-year follow-up. Cornea 2011, 30, 100–102. [Google Scholar] [CrossRef]
- Labib, S.; Adnane Berdai, M.; Abourazzak, S.; Hida, M.; Harandou, M. Congenital insensitivity to pain with anhydrosis: Report of a family case. Pan. Afr. Med. J. 2011, 9, 33. [Google Scholar] [CrossRef]
- Ali, N.; Sharma, S.; Sharma, S.; Kamal, Y.; Sharma, S. Congenital Insensitivity to Pain with Anhidrosis (HSAN Type IV), Extremely Rare Syndrome that Can Be Easily Missed by Bone and Joint Surgeons: A Case Report. Iran. J. Pediatr. 2012, 22, 559–563. [Google Scholar]
- Prashanth, G.P.; Kamate, M. A case of hereditary sensory autonomic neuropathy type IV. Ann. Indian Acad. Neurol. 2012, 15, 134–136. [Google Scholar] [CrossRef]
- Ramaraj, S.M.; Durga, D. Congenital Insensitivity to Pain with Anhidrosis in Twin Sisters with Sensorineural Deafness. Indian J. Pediatr. 2015, 82, 755–756. [Google Scholar] [CrossRef]
- Azadvari, M.; Emami Razavi, S.Z.; Kazemi, S. Hereditary Sensory and Autonomic Neuropathy Type IV in 9 Year Old Boy: A Case Report. Iran J. Child Neurol. 2016, 10, 83–85. [Google Scholar]
- Eregowda, N.I.; Yadav, S.; Parameshwarappa, P.; Basavraj, R.K. A Girl with No Pain: Congenital Insensitivity To Pain and Anhidrosis (HSAN) Type IV—A Case Report. J. Clin. Diagn. Res. 2016, 10, Zl01–Zl02. [Google Scholar] [CrossRef]
- Nabiyev, V.; Kara, A.; Aksoy, M.C. Multidisciplinary assessment of congenital insensitivity to pain syndrome. Childs Nerv. Syst. 2016, 32, 1741–1744. [Google Scholar] [CrossRef]
- Ofluoglu, D.; Altin, N.; Yaman, E.; Tuna İnce, E.B.; Aytepe, Z.; Tanyeri, H. Oral manifestations and prosthetic rehabilitation in hereditary sensory and autonomic neuropathy (HSAN) type IV: A case report. J. Istanb. Univ. Fac. Dent. 2016, 50, 49–53. [Google Scholar] [CrossRef]
- Othman, S.A.; Malik, A.A. Congenital insensitivity to pain with anhidrosis in Sudanese children. Sudan. J. Paediatr. 2016, 16, 80–85. [Google Scholar]
- Wang, Q.L.; Guo, S.; Duan, G.; Ying, Y.; Huang, P.; Liu, J.Y.; Zhang, X. Phenotypes and Genotypes in Five Children with Congenital Insensitivity to Pain with Anhidrosis. Pediatr. Neurol. 2016, 61, 63–69. [Google Scholar] [CrossRef]
- Wang, T.; Li, H.; Xiang, J.; Wei, B.; Zhang, Q.; Zhu, Q.; Liu, M.; Sun, M.; Li, H. Identification of a novel nonsense mutation of the neurotrophic tyrosine kinase receptor type 1 gene in two siblings with congenital insensitivity to pain with anhidrosis. J. Int. Med. Res. 2017, 45, 549–555. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J.; Liu, G.; Cao, W.; Liu, S.; Chen, Y.; Zuo, Y.; Chen, W.; Chen, J.; Zhang, Y.; et al. Phenotypic heterogeneity of intellectual disability in patients with congenital insensitivity to pain with anhidrosis: A case report and literature review. J. Int. Med. Res. 2018, 46, 2445–2457. [Google Scholar] [CrossRef]
- Hermans, F.; Hemeryck, L.; Lambrichts, I.; Bronckaers, A.; Vankelecom, H. Intertwined Signaling Pathways Governing Tooth Development: A Give-and-Take Between Canonical Wnt and Shh. Front. Cell Dev. Biol. 2021, 9, 758203. [Google Scholar] [CrossRef]
- Hayakawa, I.; Kubota, M. Digital Amputation by Congenital Insensitivity to Pain with Anhidrosis. J. Pediatr. 2019, 208, 290. [Google Scholar] [CrossRef]
- Liu, S.Z.; Zhou, X.; Song, A.; Wang, Y.P.; Liu, Y. Onychomadesis and phalanx osteolysis in congenital insensitivity to pain with anhidrosis. Rheumatology 2020, 59, 2648–2649. [Google Scholar] [CrossRef]
- Kim, W.; Guinot, A.; Marleix, S.; Chapuis, M.; Fraisse, B.; Violas, P. Hereditary sensory and autonomic neuropathy type IV and orthopaedic complications. Orthop. Traumatol. Surg. Res. 2013, 99, 881–885. [Google Scholar] [CrossRef]
- Gucev, Z.; Tasic, V.; Bogevska, I.; Laban, N.; Saveski, A.; Polenakovic, M.; Plaseska-Karanfilska, D.; Komlosi, K.; Winter, J.; Schweiger, S.; et al. Heterotopic ossifications and Charcot joints: Congenital insensitivity to pain with anhidrosis (CIPA) and a novel NTRK1 gene mutation. Eur. J. Med. Genet. 2020, 63, 103613. [Google Scholar] [CrossRef]
- Schwartzlow, C.; Kazamel, M. Hereditary Sensory and Autonomic Neuropathies: Adding More to the Classification. Curr. Neurol. Neurosci. Rep. 2019, 19, 52. [Google Scholar] [CrossRef]
- Boldt, K.; van Reeuwijk, J.; Lu, Q.; Koutroumpas, K.; Nguyen, T.M.; Texier, Y.; van Beersum, S.E.; Horn, N.; Willer, J.R.; Mans, D.A.; et al. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun. 2016, 7, 11491. [Google Scholar] [CrossRef]
- Rezniczek, G.A.; Winter, L.; Walko, G.; Wiche, G. Functional and Genetic Analysis of Plectin in Skin and Muscle. Methods Enzym. 2016, 569, 235–259. [Google Scholar] [CrossRef]
- Žugec, M.; Furlani, B.; Castañon, M.J.; Rituper, B.; Fischer, I.; Broggi, G.; Caltabiano, R.; Barbagallo, G.M.V.; Di Rosa, M.; Tibullo, D.; et al. Plectin plays a role in the migration and volume regulation of astrocytes: A potential biomarker of glioblastoma. J. Biomed. Sci. 2024, 31, 14. [Google Scholar] [CrossRef]
- Lafreniere, R.G.; MacDonald, M.L.; Dube, M.P.; MacFarlane, J.; O’Driscoll, M.; Brais, B.; Meilleur, S.; Brinkman, R.R.; Dadivas, O.; Pape, T.; et al. Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the Study of Canadian Genetic Isolates. Am. J. Hum. Genet. 2004, 74, 1064–1073. [Google Scholar] [CrossRef]
- Yuan, J.H.; Hashiguchi, A.; Yoshimura, A.; Sakai, N.; Takahashi, M.P.; Ueda, T.; Taniguchi, A.; Okamoto, S.; Kanazawa, N.; Yamamoto, Y.; et al. WNK1/HSN2 founder mutation in patients with hereditary sensory and autonomic neuropathy: A Japanese cohort study. Clin. Genet. 2017, 92, 659–663. [Google Scholar] [CrossRef]
- Edvardson, S.; Cinnamon, Y.; Jalas, C.; Shaag, A.; Maayan, C.; Axelrod, F.B.; Elpeleg, O. Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann. Neurol. 2012, 71, 569–572. [Google Scholar] [CrossRef]
- Manganelli, F.; Parisi, S.; Nolano, M.; Tao, F.; Paladino, S.; Pisciotta, C.; Tozza, S.; Nesti, C.; Rebelo, A.P.; Provitera, V.; et al. Novel mutations in dystonin provide clues to the pathomechanisms of HSAN-VI. Neurology 2017, 88, 2132–2140. [Google Scholar] [CrossRef]
- Fortugno, P.; Angelucci, F.; Cestra, G.; Camerota, L.; Ferraro, A.S.; Cordisco, S.; Uccioli, L.; Castiglia, D.; De Angelis, B.; Kurth, I.; et al. Recessive mutations in the neuronal isoforms of DST, encoding dystonin, lead to abnormal actin cytoskeleton organization and HSAN type VI. Hum. Mutat. 2019, 40, 106–114. [Google Scholar] [CrossRef]
- Pérez-Gómez, R.; Haro, E.; Fernández-Guerrero, M.; Bastida, M.F.; Ros, M.A. Role of Hox genes in regulating digit patterning. Int. J. Dev. Biol. 2018, 62, 797–805. [Google Scholar] [CrossRef]
- Bastida, M.F.; Pérez-Gómez, R.; Trofka, A.; Zhu, J.; Rada-Iglesias, A.; Sheth, R.; Stadler, H.S.; Mackem, S.; Ros, M.A. The formation of the thumb requires direct modulation of Gli3 transcription by Hoxa13. Proc. Natl. Acad. Sci. USA 2020, 117, 1090–1096. [Google Scholar] [CrossRef]
- Kin, N.W.; Sanders, V.M. It takes nerve to tell T and B cells what to do. J. Leukoc. Biol. 2006, 79, 1093–1104. [Google Scholar] [CrossRef]
- Hepburn, L.; Prajsnar, T.K.; Klapholz, C.; Moreno, P.; Loynes, C.A.; Ogryzko, N.V.; Brown, K.; Schiebler, M.; Hegyi, K.; Antrobus, R.; et al. Innate immunity. A Spaetzle-like role for nerve growth factor β in vertebrate immunity to Staphylococcus aureus. Science 2014, 346, 641–646. [Google Scholar] [CrossRef]
- Piorunek, M.; Brajer-Luftmann, B.; Walkowiak, J. Pasteurella Multocida Infection in Humans. Pathogens 2023, 12, 1210. [Google Scholar] [CrossRef]
- Ridge, K.M.; Eriksson, J.E.; Pekny, M.; Goldman, R.D. Roles of vimentin in health and disease. Genes Dev. 2022, 36, 391–407. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kantaputra, P.; Daroontum, T.; Kitiyamas, K.; Piyakhunakorn, P.; Kawasaki, K.; Sathienkijkanchai, A.; Wasant, P.; Vatanavicharn, N.; Yasanga, T.; Kaewgahya, M.; et al. Homozygosity for a Rare Plec Variant Suggests a Contributory Role in Congenital Insensitivity to Pain. Int. J. Mol. Sci. 2024, 25, 6358. https://doi.org/10.3390/ijms25126358
Kantaputra P, Daroontum T, Kitiyamas K, Piyakhunakorn P, Kawasaki K, Sathienkijkanchai A, Wasant P, Vatanavicharn N, Yasanga T, Kaewgahya M, et al. Homozygosity for a Rare Plec Variant Suggests a Contributory Role in Congenital Insensitivity to Pain. International Journal of Molecular Sciences. 2024; 25(12):6358. https://doi.org/10.3390/ijms25126358
Chicago/Turabian StyleKantaputra, Piranit, Teerada Daroontum, Kantapong Kitiyamas, Panat Piyakhunakorn, Katsushige Kawasaki, Achara Sathienkijkanchai, Pornswan Wasant, Nithiwat Vatanavicharn, Thippawan Yasanga, Massupa Kaewgahya, and et al. 2024. "Homozygosity for a Rare Plec Variant Suggests a Contributory Role in Congenital Insensitivity to Pain" International Journal of Molecular Sciences 25, no. 12: 6358. https://doi.org/10.3390/ijms25126358