
Impact of Phosphorylation at Various Sites on the Active Pocket of Human Ferrochelatase: Insights from Molecular Dynamics Simulations
 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
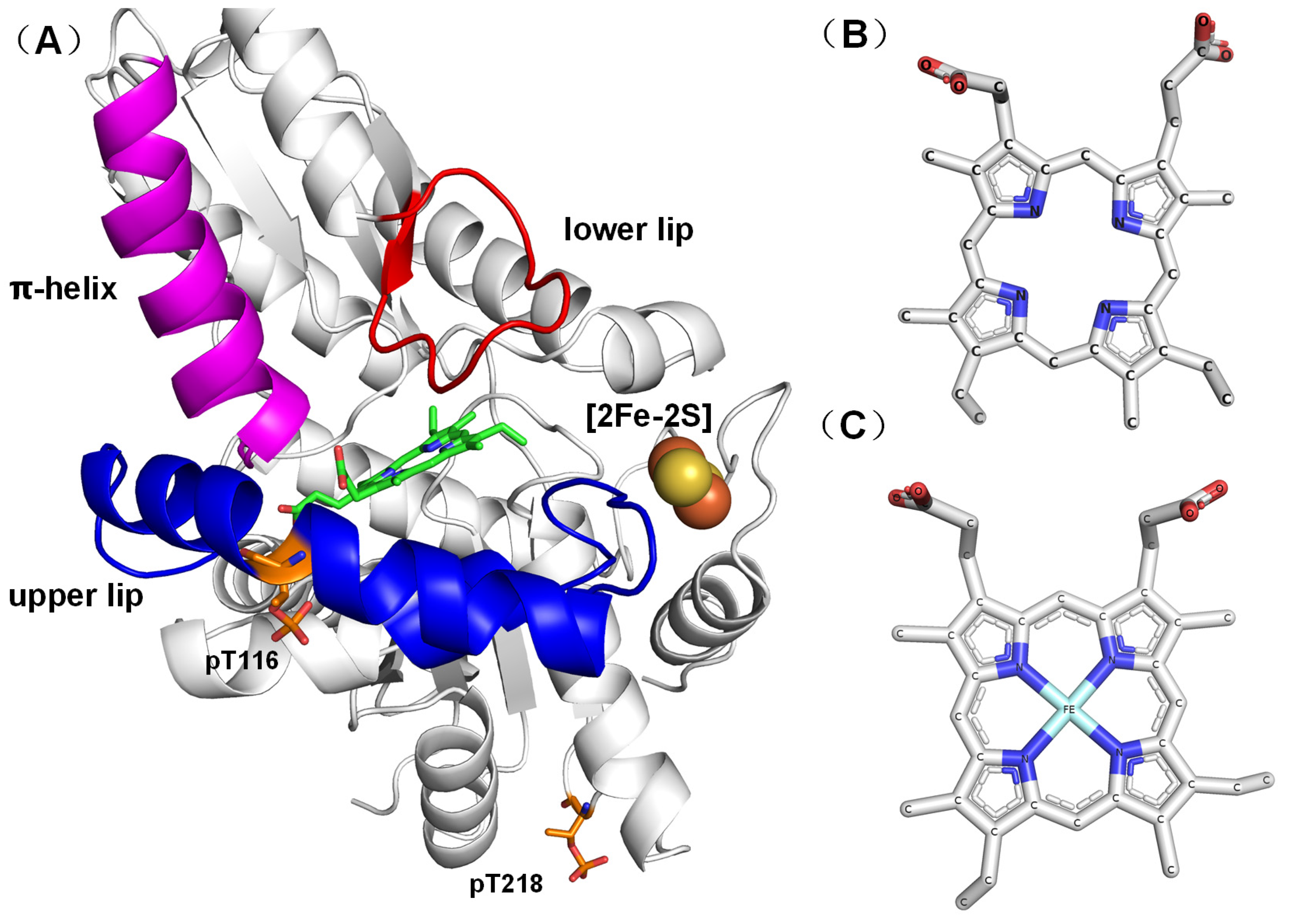
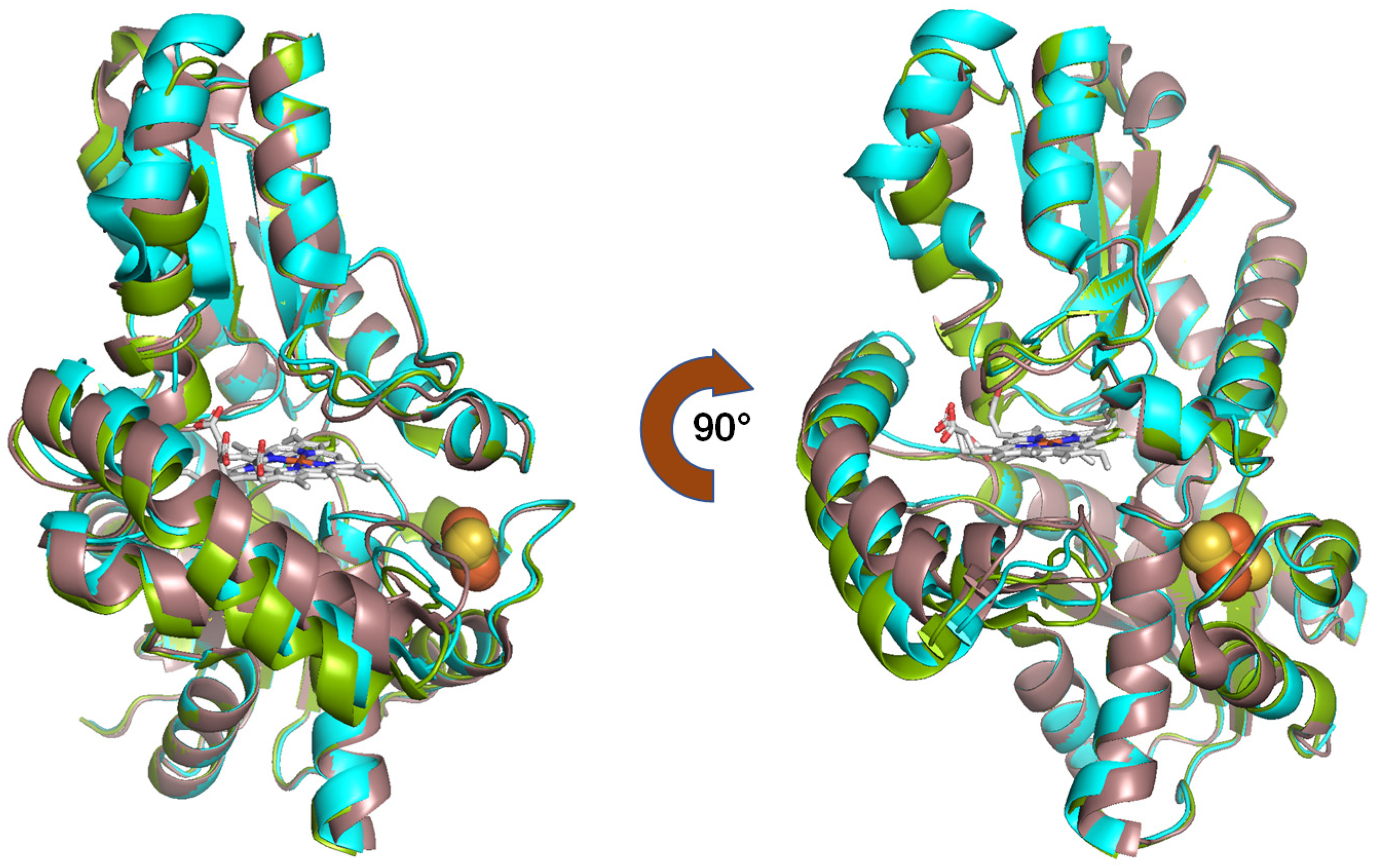
2.1. Crystal Structure
2.2. Identification of FECH Phosphorylation Sites
2.3. Structural Analysis of Phosphorylated FECH
2.4. Residue Interactions of the Active Pocket
2.5. Binding Free Energy Analysis
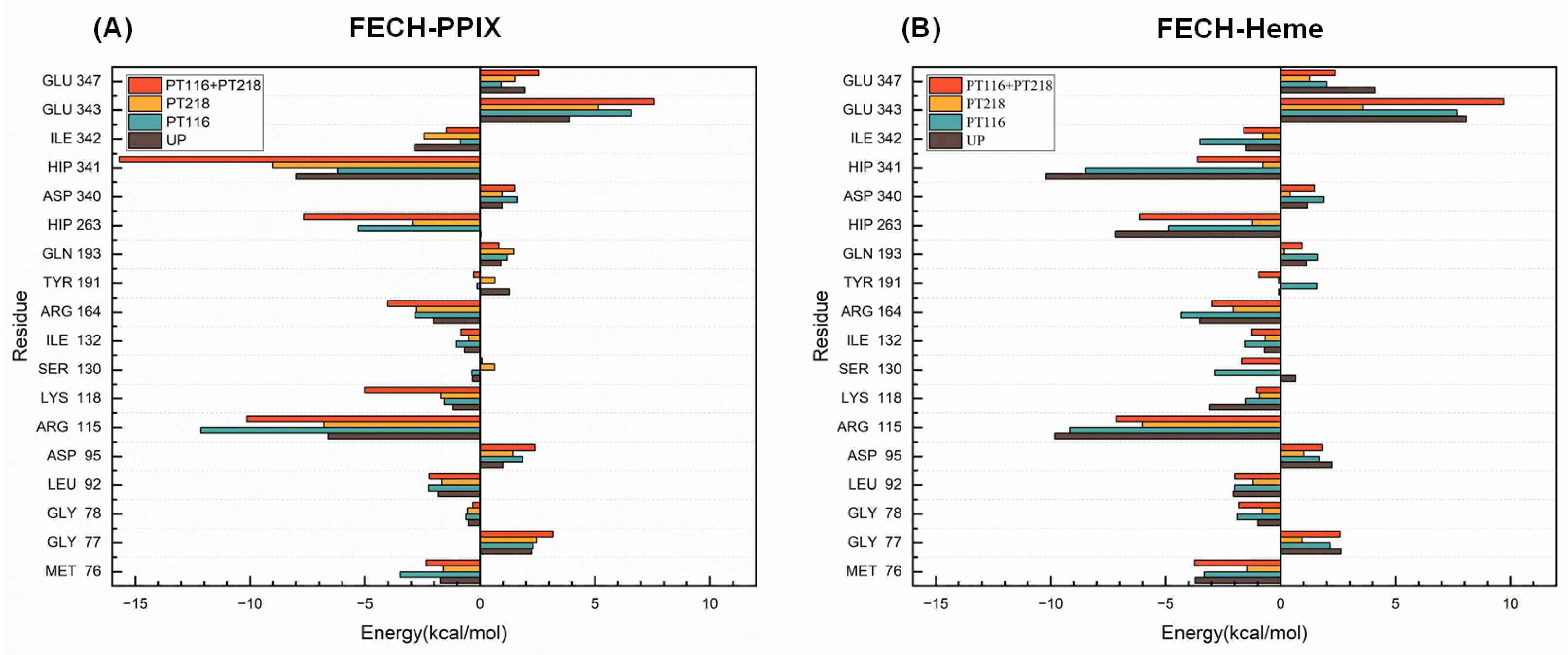
2.6. Per-Residue Free Energy Decomposition Analysis
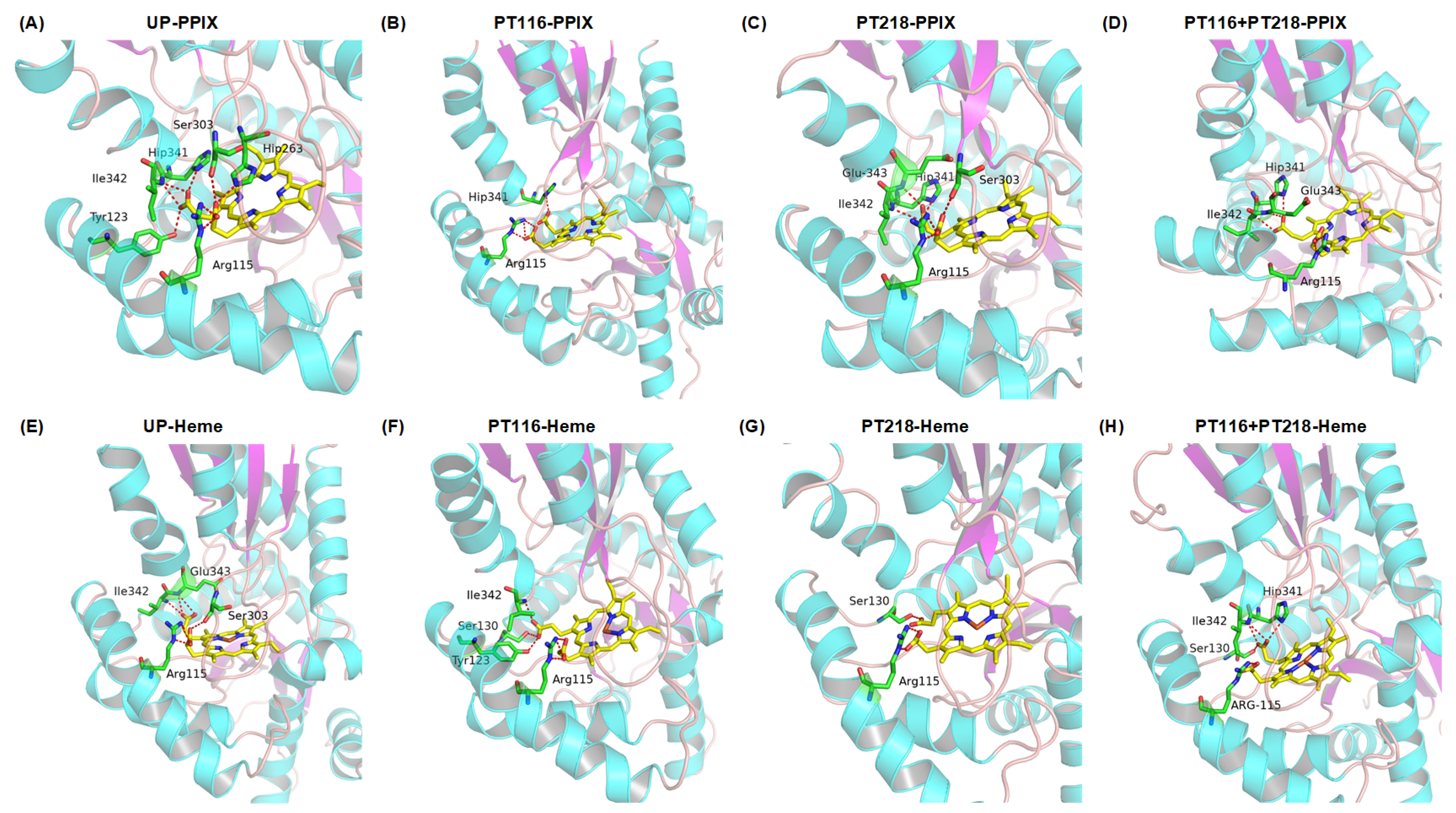
2.7. Protein–Substrate Interactions
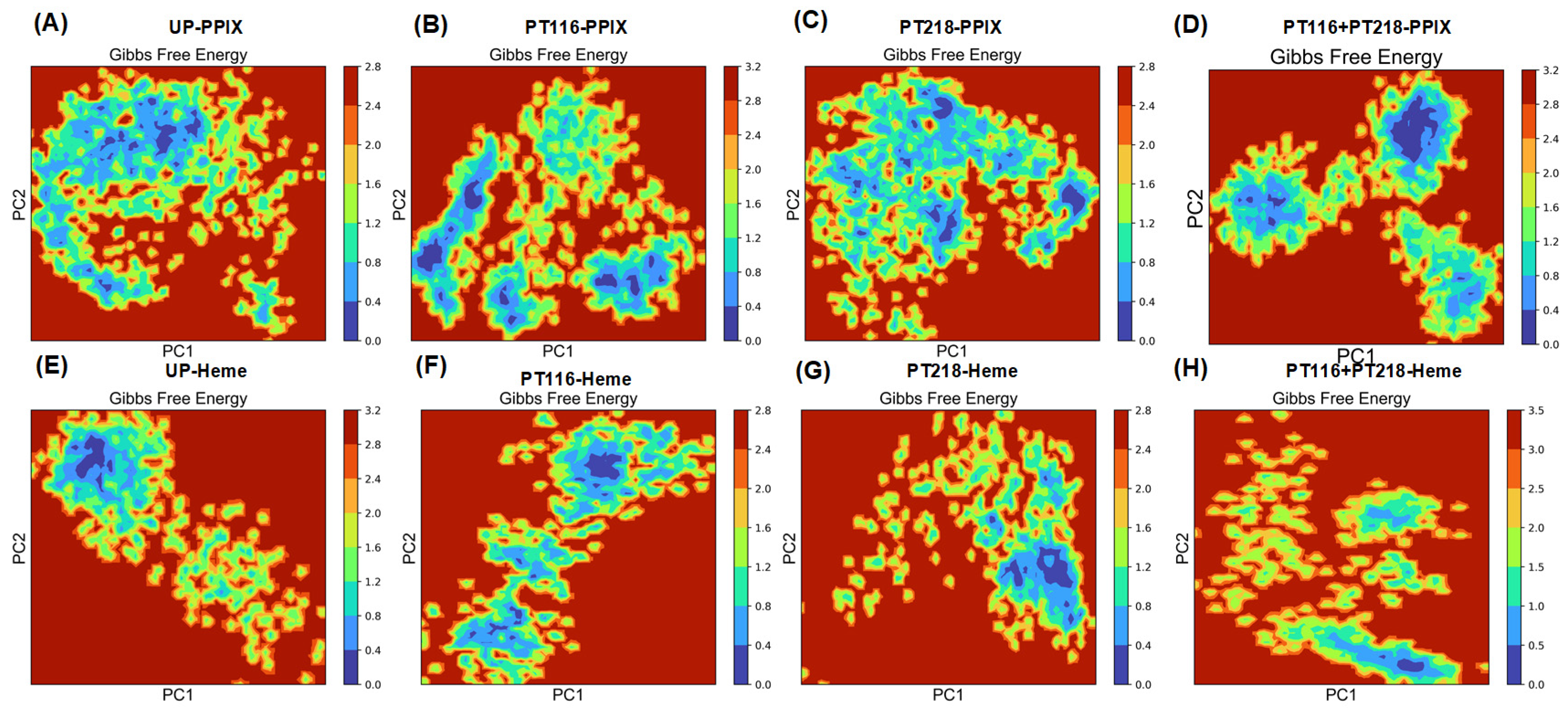
2.8. Principal Component Analysis
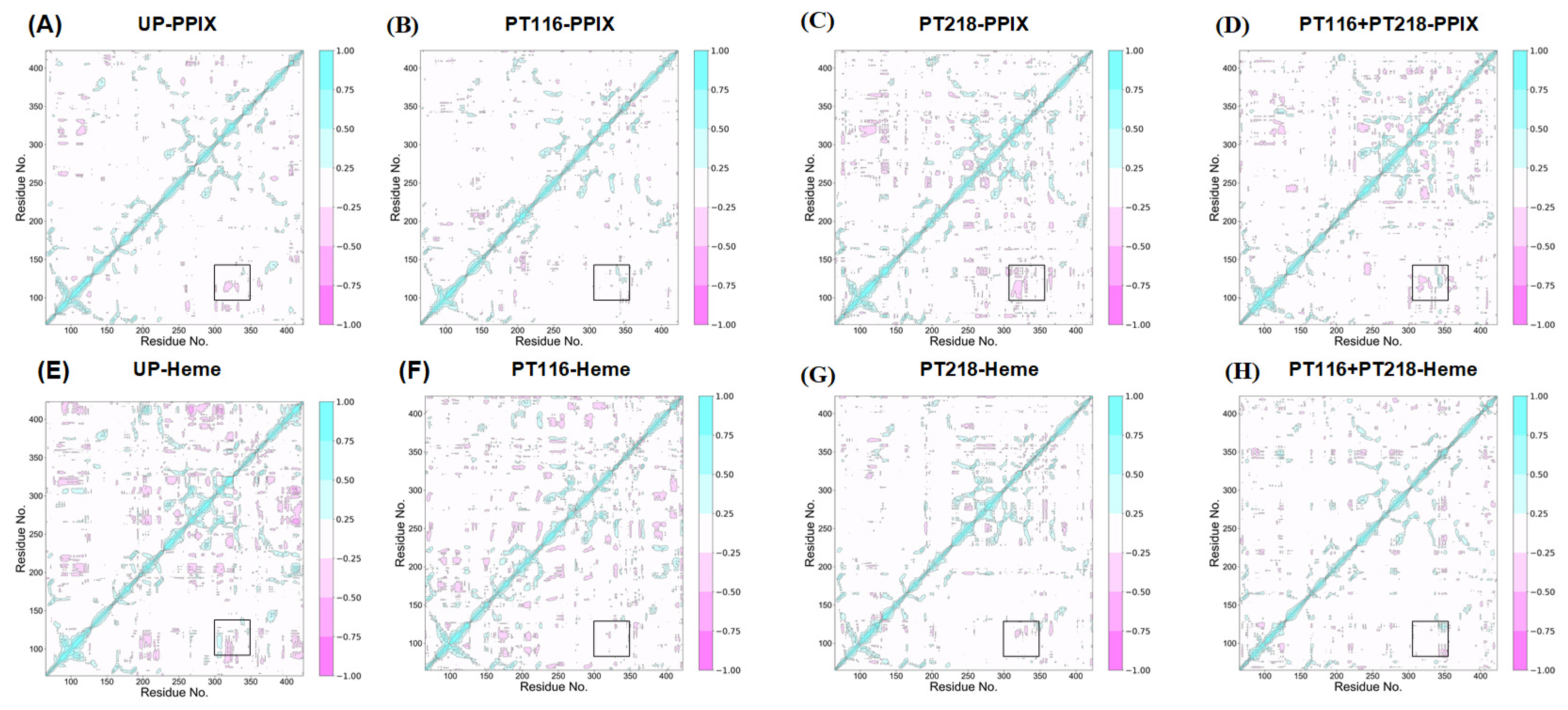
2.9. Dynamic Cross-Correlation Matrix
3. Materials and Methods
3.1. Cloning, Expression, and Purification of Phosphorylated Human FECH
3.2. Mass Spectrometry Analysis of Purified FECH Protein
3.3. System Modeling
3.4. Binding Free Energy Calculations
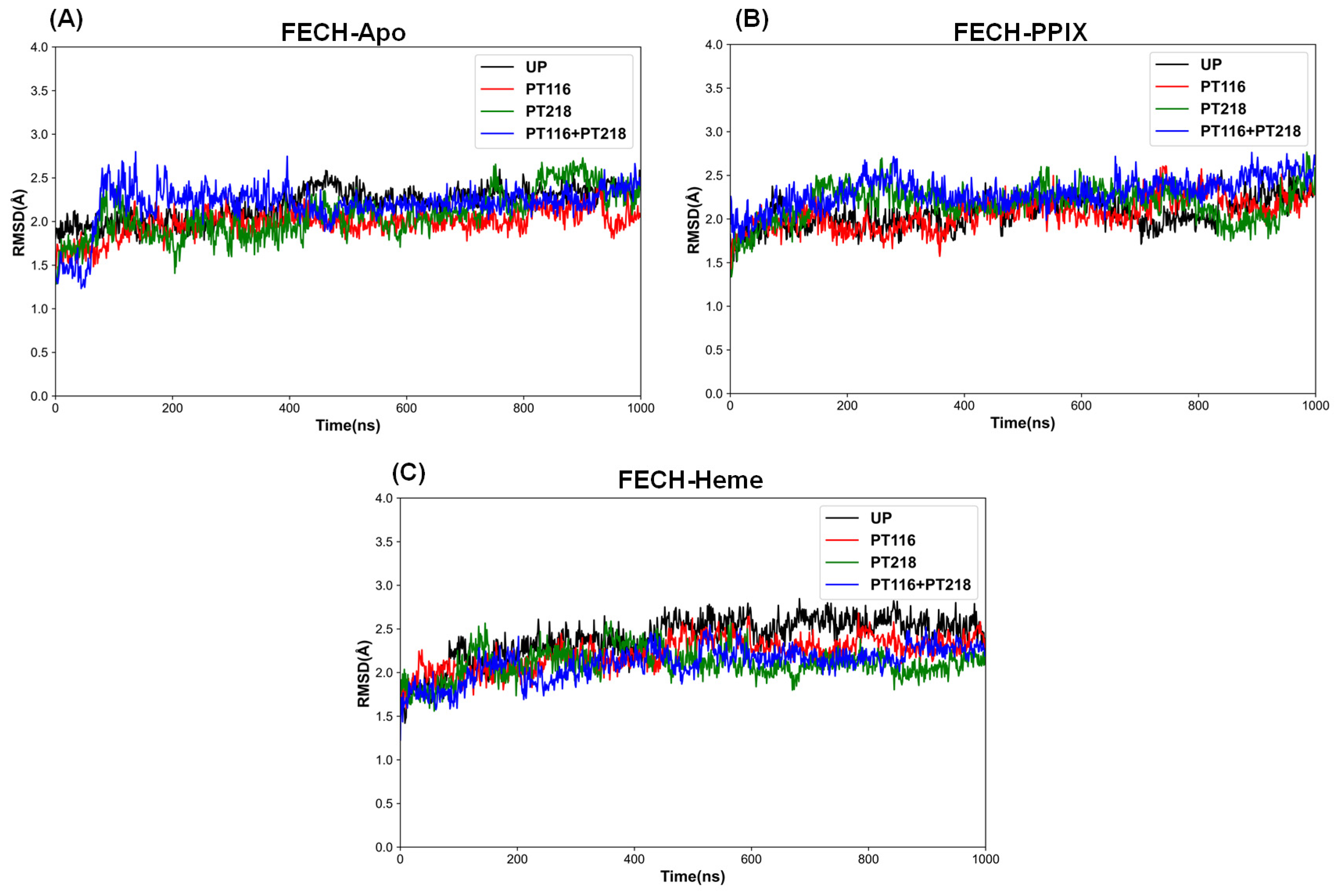
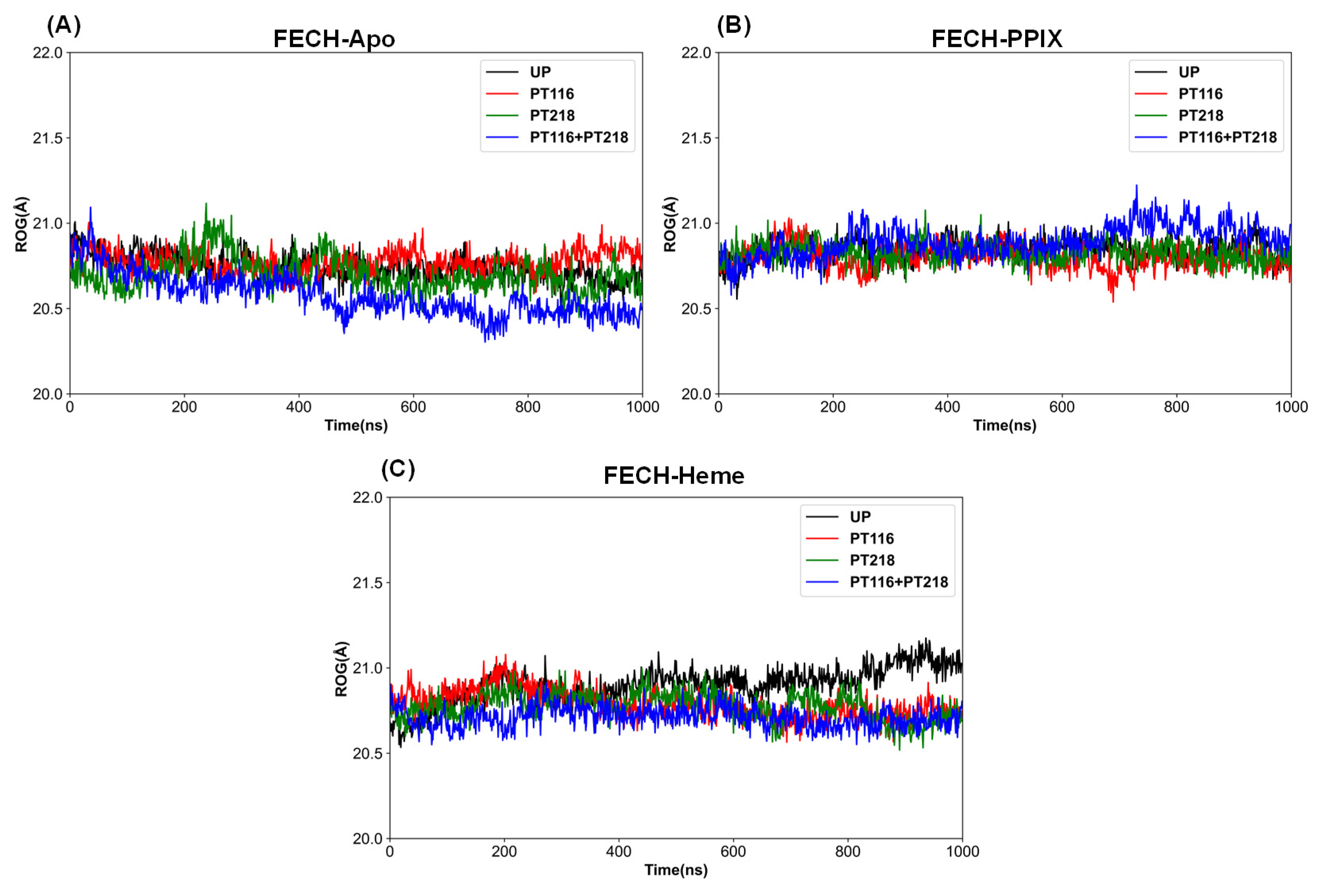
3.5. Molecular Dynamics Trajectory Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burris, T.P. Nuclear Hormone Receptors for Heme: REV-ERBalpha and REV-ERBbeta Are Ligand-Regulated Components of the Mammalian Clock. Mol. Endocrinol. 2008, 22, 1509–1520. [Google Scholar] [CrossRef]
- Faller, M.; Matsunaga, M.; Yin, S.; Loo, J.A.; Guo, F. Heme Is Involved in microRNA Processing. Nat. Struct. Mol. Biol. 2007, 14, 23–29. [Google Scholar] [CrossRef]
- Hu, R.-G.; Wang, H.; Xia, Z.; Varshavsky, A. The N-End Rule Pathway Is a Sensor of Heme. Proc. Natl. Acad. Sci. USA 2008, 105, 76–81. [Google Scholar] [CrossRef]
- Shen, J.; Sheng, X.; Chang, Z.; Wu, Q.; Wang, S.; Xuan, Z.; Li, D.; Wu, Y.; Shang, Y.; Kong, X.; et al. Iron Metabolism Regulates P53 Signaling through Direct Heme-P53 Interaction and Modulation of P53 Localization, Stability, and Function. Cell Rep. 2014, 7, 180–193. [Google Scholar] [CrossRef]
- Burton, M.J.; Kapetanaki, S.M.; Chernova, T.; Jamieson, A.G.; Dorlet, P.; Santolini, J.; Moody, P.C.E.; Mitcheson, J.S.; Davies, N.W.; Schmid, R.; et al. A Heme-Binding Domain Controls Regulation of ATP-Dependent Potassium Channels. Proc. Natl. Acad. Sci. USA 2016, 113, 3785–3790. [Google Scholar] [CrossRef]
- Sahoo, N.; Goradia, N.; Ohlenschläger, O.; Schönherr, R.; Friedrich, M.; Plass, W.; Kappl, R.; Hoshi, T.; Heinemann, S.H. Heme Impairs the Ball-and-Chain Inactivation of Potassium Channels. Proc. Natl. Acad. Sci. USA 2013, 110, E4036–E4044. [Google Scholar] [CrossRef]
- Al-Karadaghi, S.; Franco, R.; Hansson, M.; Shelnutt, J.A.; Isaya, G.; Ferreira, G.C. Chelatases: Distort to Select? Trends Biochem. Sci. 2006, 31, 135–142. [Google Scholar] [CrossRef]
- Medlock, A.E.; Dailey, H.A. New Avenues of Heme Synthesis Regulation. Int. J. Mol. Sci. 2022, 23, 7467. [Google Scholar] [CrossRef]
- Dailey, H.A.; Medlock, A.E. A Primer on Heme Biosynthesis. Biol. Chem. 2022, 403, 985–1003. [Google Scholar] [CrossRef]
- Obi, C.D.; Bhuiyan, T.; Dailey, H.A.; Medlock, A.E. Ferrochelatase: Mapping the Intersection of Iron and Porphyrin Metabolism in the Mitochondria. Front. Cell Dev. Biol. 2022, 10, 894591. [Google Scholar] [CrossRef]
- Medlock, A.E.; Dailey, T.A.; Ross, T.A.; Dailey, H.A.; Lanzilotta, W.N. A π-Helix Switch Selective for Porphyrin Deprotonation and Product Release in Human Ferrochelatase. J. Mol. Biol. 2007, 373, 1006–1016. [Google Scholar] [CrossRef]
- Balwani, M.; Doheny, D.; Bishop, D.F.; Nazarenko, I.; Yasuda, M.; Dailey, H.A.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.; Bonkovsky, H.L.; et al. Loss-of-Function Ferrochelatase and Gain-of-Function Erythroid-Specific 5-Aminolevulinate Synthase Mutations Causing Erythropoietic Protoporphyria and X-Linked Protoporphyria in North American Patients Reveal Novel Mutations and a High Prevalence of X-Linked Protoporphyria. Mol. Med. 2013, 19, 26–29. [Google Scholar] [CrossRef]
- Wensink, D.; Wagenmakers, M.A.E.M.; Qi, H.; Wilson, J.H.P.; Langendonk, J.G. Objective Light Exposure Measurements and Circadian Rhythm in Patients with Erythropoietic Protoporphyria: A Case-Control Study. Mol. Genet. Metab. 2022, 135, 215–220. [Google Scholar] [CrossRef]
- Dailey, H.A.; Meissner, P.N. Erythroid Heme Biosynthesis and Its Disorders. CSH. Perspect. Med. 2013, 3, a011676. [Google Scholar] [CrossRef]
- Chen, F.-P.; Risheg, H.; Liu, Y.; Bloomer, J. Ferrochelatase Gene Mutations in Erythropoietic Protoporphyria: Focus on Liver Disease. Cell. Mol. Biol. 2002, 48, 83–89. [Google Scholar]
- Medlock, A.; Swartz, L.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Substrate Interactions with Human Ferrochelatase. Proc. Natl. Acad. Sci. USA 2007, 104, 1789–1793. [Google Scholar] [CrossRef]
- Medlock, A.E.; Carter, M.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Product Release Rather than Chelation Determines Metal Specificity for Ferrochelatase. J. Mol. Biol. 2009, 393, 308–319. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-Translational Modifications in Signal Integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- Theillet, F.-X.; Smet-Nocca, C.; Liokatis, S.; Thongwichian, R.; Kosten, J.; Yoon, M.-K.; Kriwacki, R.W.; Landrieu, I.; Lippens, G.; Selenko, P. Cell Signaling, Post-Translational Protein Modifications and NMR Spectroscopy. J. Biomol. NMR 2012, 54, 217–236. [Google Scholar] [CrossRef]
- Chung, J.; Wittig, J.G.; Ghamari, A.; Maeda, M.; Dailey, T.A.; Bergonia, H.; Kafina, M.D.; Coughlin, E.E.; Minogue, C.E.; Hebert, A.S.; et al. Erythropoietin Signaling Regulates Heme Biosynthesis. eLife 2017, 6, e24767. [Google Scholar] [CrossRef]
- Chung, J.; Chen, C.; Paw, B.H. Heme Metabolism and Erythropoiesis. Curr. Opin. Hematol. 2012, 19, 156–162. [Google Scholar] [CrossRef]
- Tanaka, T.; Nangaku, M. Recent Advances and Clinical Application of Erythropoietin and Erythropoiesis-Stimulating Agents. Exp. Cell. Res. 2012, 318, 1068–1073. [Google Scholar] [CrossRef]
- Feng, G.; Zhang, X.; Li, Y.; Wang, R. Analysis of the Binding Sites on BAX and the Mechanism of BAX Activators through Extensive Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 5208–5222. [Google Scholar] [CrossRef]
- Maloney, R.C.; Zhang, M.; Liu, Y.; Jang, H.; Nussinov, R. The Mechanism of Activation of MEK1 by B-Raf and KSR1. Cell. Mol. Life Sci. 2022, 79, 281. [Google Scholar] [CrossRef]
- Duff, N.; Peters, B. Polymorph Specific RMSD Local Order Parameters for Molecular Crystals and Nuclei: α-, β-, and γ-Glycine. J. Chem. Phys. 2011, 135, 134101. [Google Scholar] [CrossRef]
- Kokkinidis, M.; Glykos, N.M.; Fadouloglou, V.E. Protein Flexibility and Enzymatic Catalysis. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 87, pp. 181–218. ISBN 978-0-12-398312-1. [Google Scholar]
- Kuzmanic, A.; Zagrovic, B. Determination of Ensemble-Average Pairwise Root Mean-Square Deviation from Experimental B-Factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef]
- Wang, S.; Peng, J.; Ma, J.; Xu, J. Protein Secondary Structure Prediction Using Deep Convolutional Neural Fields. Sci. Rep. 2016, 6, 18962. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Ju, J.; Shen, Y. Investigation by MD Simulation of the Key Residues Related to Substrate-Binding and Heme-Release in Human Ferrochelatase. J. Mol. Model. 2013, 19, 2509–2518. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, Y.; Chen, S. RNA –Ligand Molecular Docking: Advances and Challenges. WIREs Comput. Mol. Sci. 2022, 12, e1571. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. In eLS; Wiley: Hoboken, NJ, USA, 2010; ISBN 978-0-470-01617-6. [Google Scholar]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized Hydrophobic Interactions and Hydrogen Bonding at the Target-Ligand Interface Leads the Pathways of Drug-Designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Sakaino, M.; Ishigaki, M.; Ohgari, Y.; Kitajima, S.; Masaki, R.; Yamamoto, A.; Taketani, S. Dual Mitochondrial Localization and Different Roles of the Reversible Reaction of Mammalian Ferrochelatase. FEBS J. 2009, 276, 5559–5570. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. In Protein Dynamics; Livesay, D.R., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1084, pp. 193–226. ISBN 978-1-62703-657-3. [Google Scholar]
- Granato, D.; Santos, J.S.; Escher, G.B.; Ferreira, B.L.; Maggio, R.M. Use of Principal Component Analysis (PCA) and Hierarchical Cluster Analysis (HCA) for Multivariate Association between Bioactive Compounds and Functional Properties in Foods: A Critical Perspective. Trends Food Sci. Technol. 2018, 72, 83–90. [Google Scholar] [CrossRef]
- Yu, H.; Dalby, P.A. A. A Beginner’s Guide to Molecular Dynamics Simulations and the Identification of Cross-Correlation Networks for Enzyme Engineering. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 643, pp. 15–49. ISBN 978-0-12-821149-6. [Google Scholar]
- Dailey, H.A.; Sellers, V.M.; Dailey, T.A. Mammalian Ferrochelatase. Expression and Characterization of Normal and Two Human Protoporphyric Ferrochelatases. J. Biol. Chem. 1994, 269, 390–395. [Google Scholar] [CrossRef]
- Zhang, M.S.; Brunner, S.F.; Huguenin-Dezot, N.; Liang, A.D.; Schmied, W.H.; Rogerson, D.T.; Chin, J.W. Biosynthesis and Genetic Encoding of Phosphothreonine through Parallel Selection and Deep Sequencing. Nat. Methods 2017, 14, 729–736. [Google Scholar] [CrossRef]
- Burden, A.E.; Wu, C.; Dailey, T.A.; Busch, J.L.; Dhawan, I.K.; Rose, J.P.; Wang, B.; Dailey, H.A. Human Ferrochelatase: Crystallization, Characterization of the [2Fe-2S] Cluster and Determination That the Enzyme Is a Homodimer. Biochim. Biophys. Acta 1999, 1435, 191–197. [Google Scholar] [CrossRef]
- Bern, M.; Kil, Y.J.; Becker, C. Byonic: Advanced Peptide and Protein Identification Software. Curr. Protoc. Bioinform. 2012, 13, 13.20.1–13.20.14. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK Prediction and the Preparation of Biomolecular Structures for Atomistic Molecular Modeling and Simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Sigfridsson, E.; Ryde, U. The Importance of Porphyrin Distortions for the Ferrochelatase Reaction. J. Biol. Inorg. Chem. 2003, 8, 273–282. [Google Scholar] [CrossRef]
- Lee, T.-S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, T.D.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Xiong, Y.; Shabane, P.S.; Onufriev, A.V. Melting Points of OPC and OPC3 Water Models. ACS Omega 2020, 5, 25087–25094. [Google Scholar] [CrossRef]
- Homeyer, N.; Horn, A.H.C.; Lanig, H.; Sticht, H. AMBER Force-Field Parameters for Phosphorylated Amino Acids in Different Protonation States: Phosphoserine, Phosphothreonine, Phosphotyrosine, and Phosphohistidine. J. Mol. Model. 2006, 12, 281–289. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An Analysis of the Accuracy of Langevin and Molecular Dynamics Algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. S 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Srivastava, H.K.; Sastry, G.N. Molecular Dynamics Investigation on a Series of HIV Protease Inhibitors: Assessing the Performance of MM-PBSA and MM-GBSA Approaches. J. Chem. Inf. Model. 2012, 52, 3088–3098. [Google Scholar] [CrossRef]
- Kar, P.; Seel, M.; Hansmann, U.H.E.; Höfinger, S. Dispersion Terms and Analysis of Size- and Charge Dependence in an Enhanced Poisson−Boltzmann Approach. J. Phys. Chem. B 2007, 111, 8910–8918. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Ichiye, T.; Karplus, M. Collective Motions in Proteins: A Covariance Analysis of Atomic Fluctuations in Molecular Dynamics and Normal Mode Simulations. Proteins 1991, 11, 205–217. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; De Groot, B.L.; Van Aalten, D.M.F.; Berendsen, H.J.C. An Efficient Method for Sampling the Essential Subspace of Proteins. J. Biomol. Struct. Dyn. 1996, 13, 615–625. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R Package for the Comparative Analysis of Protein Structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a Platform for Computational Drug Design. WIREs Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Relation between Free Energy Landscapes of Proteins and Dynamics. J. Chem. Theory Comput. 2010, 6, 583–595. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | Phosphorylated Sites | Mean RMSD(SD) (Å) |
|---|---|---|
| FECH-Apo | UP | 2.19 (0.19) |
| PT116 | 1.98 (0.15) | |
| PT218 | 2.07 (0.26) | |
| PT116 + PT218 | 2.19 (0.24) | |
| FECH-PPIX | UP | 1.79 (0.17) |
| PT116 | 2.06 (0.18) | |
| PT218 | 2.17 (0.20) | |
| PT116 + PT218 | 2.30 (0.17) | |
| FECH-Heme | UP | 2.39 (0.25) |
| PT116 | 2.22 (0.18) | |
| PT218 | 2.10 (0.17) | |
| PT116 + PT218 | 2.11 (0.19) |
| System | Phosphorylation Site | Residue Pairs Mean Distance (SD) (Å) | |||
|---|---|---|---|---|---|
| P102–P107 | R115–E343 | R115–E347 | K118–E351 | ||
| FECH-Apo | UP | 6.56 (1.49) | 9.78 (2.28) | 8.10 (2.11) | 7.29 (1.88) |
| PT116 | 7.32 (1.61) | 10.17 (2.24) | 7.85 (2.28) | 6.93 (2.53) | |
| PT218 | 7.34 (3.90) | 11.55 (2.40) | 10.03 (2.51) | 7.10 (2.51) | |
| PT116 + PT218 | 5.00 (2.36) | 10.59 (1.89) | 9.10 (1.81) | 5.65 (2.16) | |
| FECH-PPIX | UP | 6.05 (1.38) | 12.7 (0.73) | 7.13 (0.83) | 5.56 (1.48) |
| PT116 | 6.70 (1.42) | 9.82 (3.39) | 8.16 (1.20) | 6.76 (1.69) | |
| PT218 | 5.64 (1.42) | 10.13 (2.27) | 7.82 (0.84) | 6.16 (1.47) | |
| PT116 + PT218 | 4.68 (1.21) | 7.23 (1.03) | 10.09 (1.12) | 7.98 (1.76) | |
| FECH-Heme | UP | 6.84 (1.45) | 11.60 (1.12) | 7.10 (1.02) | 5.91 (2.25) |
| PT116 | 6.43 (1.38) | 8.31 (0.58) | 8.41 (0.81) | 6.88 (2.16) | |
| PT218 | 6.08 (1.43) | 8.34 (2.50) | 7.68 (1.05) | 5.77 (1.48) | |
| PT116 + PT218 | 8.17 (1.53) | 9.82 (2.95) | 8.23 (0.90) | 7.59 (2.59) | |
| Component | |||||||
|---|---|---|---|---|---|---|---|
| UP-PPIX | −65.86 (1.98) | −396.00 (10.41) | 412.53 (10.88) | −6.07 (0.18) | −461.86 (12.34) | 406.47 (10.71) | −55.39 (1.76) |
| PT116-PPIX | −71.70 (0.29) | −317.58 (1.96) | 338.30 (1.69) | −6.51 (0.01) | −389.28 (1.97) | 331.79 (1.69) | −57.49 (0.71) |
| PT218-PPIX | −57.22 (3.06) | −306.88 (14.65) | 325.08 (15.62) | −5.12 (17.67) | −364.10 (15.35) | 319.96 (2.45) | −44.14 (1.45) |
| PT116 + PT218-PPIX | −72.58 (0.33) | −312.86 (1.74) | 319.78 (1.55) | −6.39 (0.01) | −385.44 (1.76) | 313.39 (1.55) | −72.05 (0.76) |
| UP-Heme | −64.22 (2.16) | −374.72 (10.80) | 378.33 (10.90) | −5.88 (0.20) | −438.94 (12.91) | 372.45 (10.71) | −66.49 (2.31) |
| PT116-Heme | −72.15 (1.51) | −292.46 (5.50) | 304.26 (5.74) | −6.25 (0.13) | −364.61 (6.94) | 298.01 (5.62) | −66.60 (1.52) |
| PT218-Heme | −46.09 (3.62) | −220.75 (14.66) | 241.00 (16.29) | −4.05 (0.32) | −266.84 (18.26) | 236.95 (15.97) | −29.89 (2.37) |
| PT116 + PT218-Heme | −69.21 (1.93) | −250.66 (6.54) | 275.07 (7.15) | −6.01 (0.16) | −319.87 (8.38) | 269.07 (6.99) | −50.81 (1.65) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.; Lin, Y.; Obi, C.D.; Zhao, P.; Dailey, H.A.; Medlock, A.E.; Shen, Y. Impact of Phosphorylation at Various Sites on the Active Pocket of Human Ferrochelatase: Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2024, 25, 6360. https://doi.org/10.3390/ijms25126360
Guo M, Lin Y, Obi CD, Zhao P, Dailey HA, Medlock AE, Shen Y. Impact of Phosphorylation at Various Sites on the Active Pocket of Human Ferrochelatase: Insights from Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2024; 25(12):6360. https://doi.org/10.3390/ijms25126360
Chicago/Turabian StyleGuo, Mingshan, Yuhong Lin, Chibuike David Obi, Peng Zhao, Harry A. Dailey, Amy E. Medlock, and Yong Shen. 2024. "Impact of Phosphorylation at Various Sites on the Active Pocket of Human Ferrochelatase: Insights from Molecular Dynamics Simulations" International Journal of Molecular Sciences 25, no. 12: 6360. https://doi.org/10.3390/ijms25126360