
Unveiling Therapeutic Potential: Targeting Fusobacterium nucleatum’s Lipopolysaccharide Biosynthesis for Endodontic Infections—An In Silico Screening Study
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Identification of Inhibitors against Lipid A Biosynthetic Acyl Transferases
2.2. Toxicity and ADME Screening
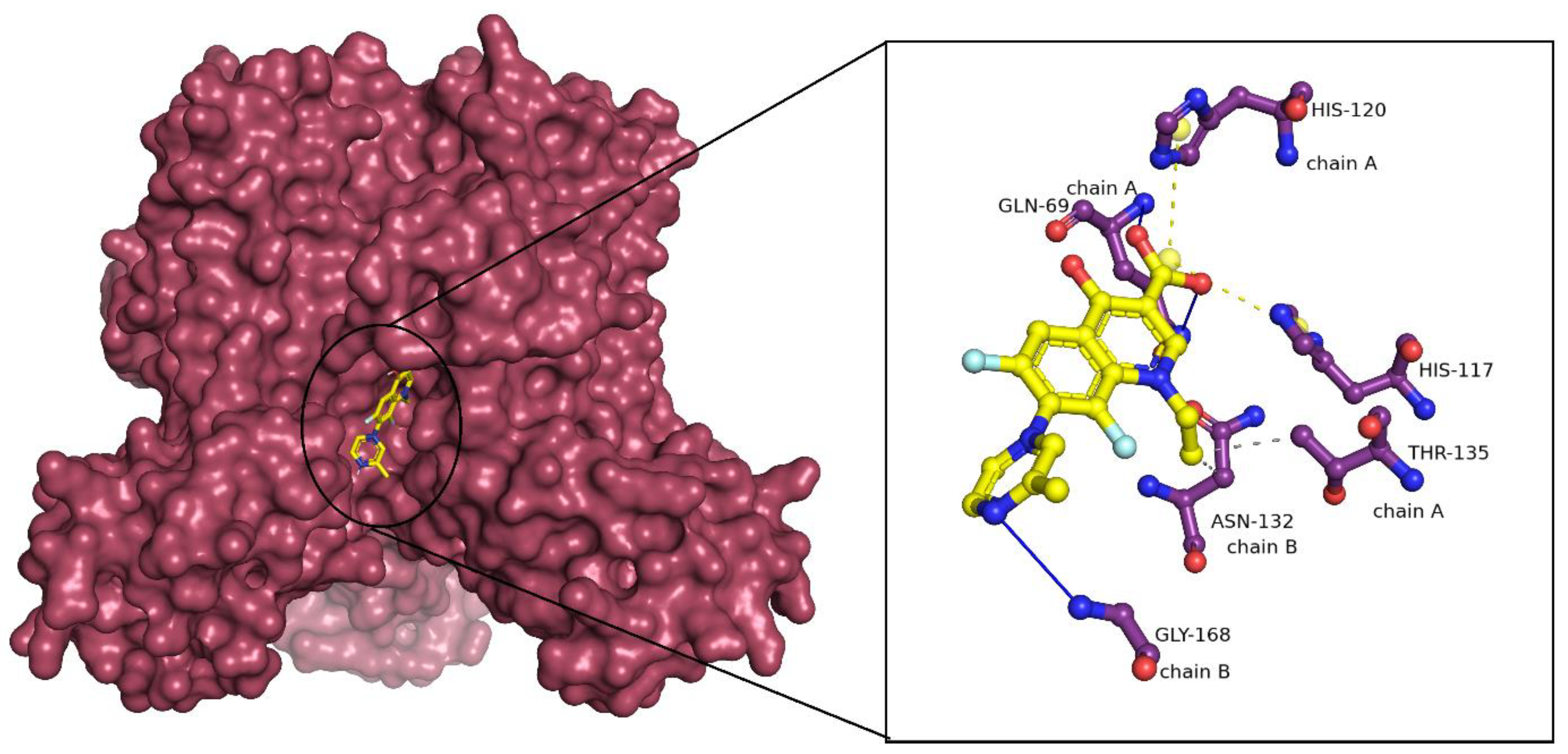
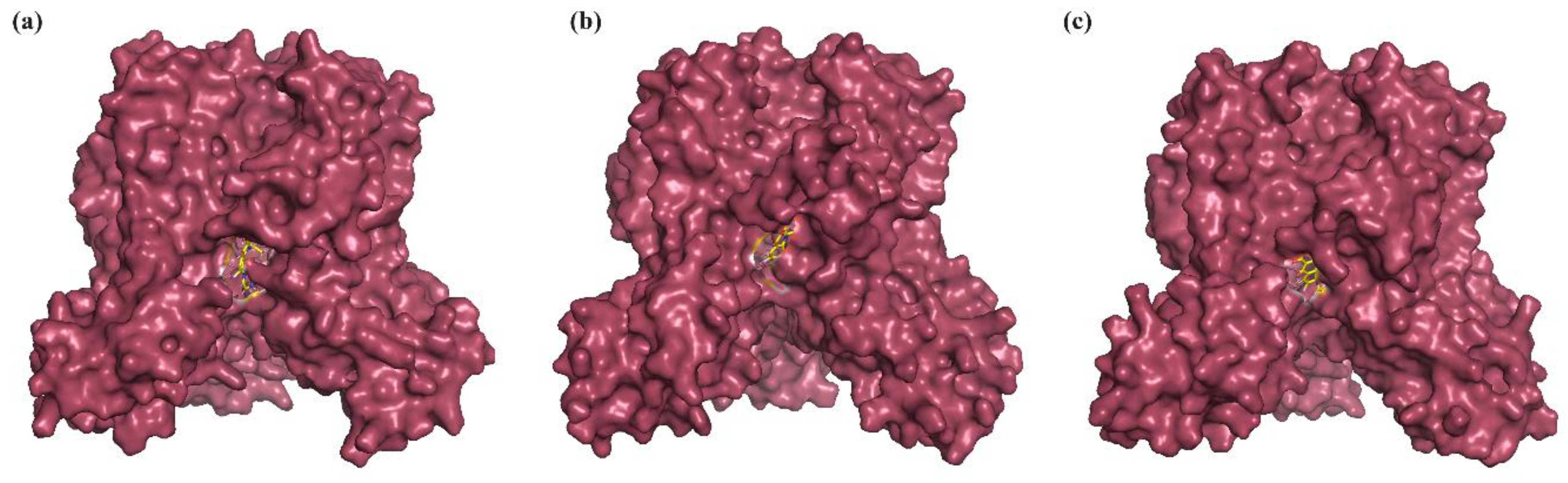
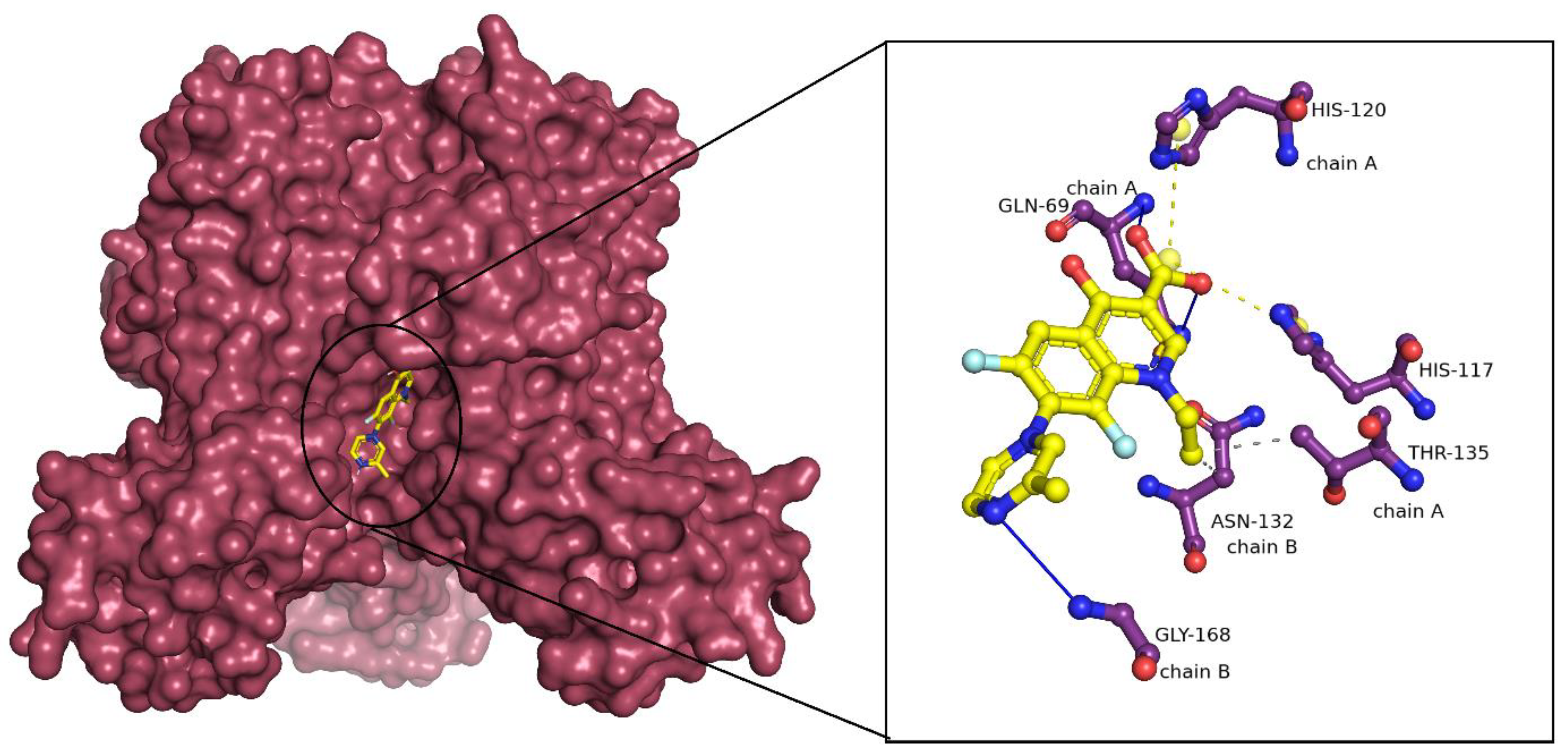
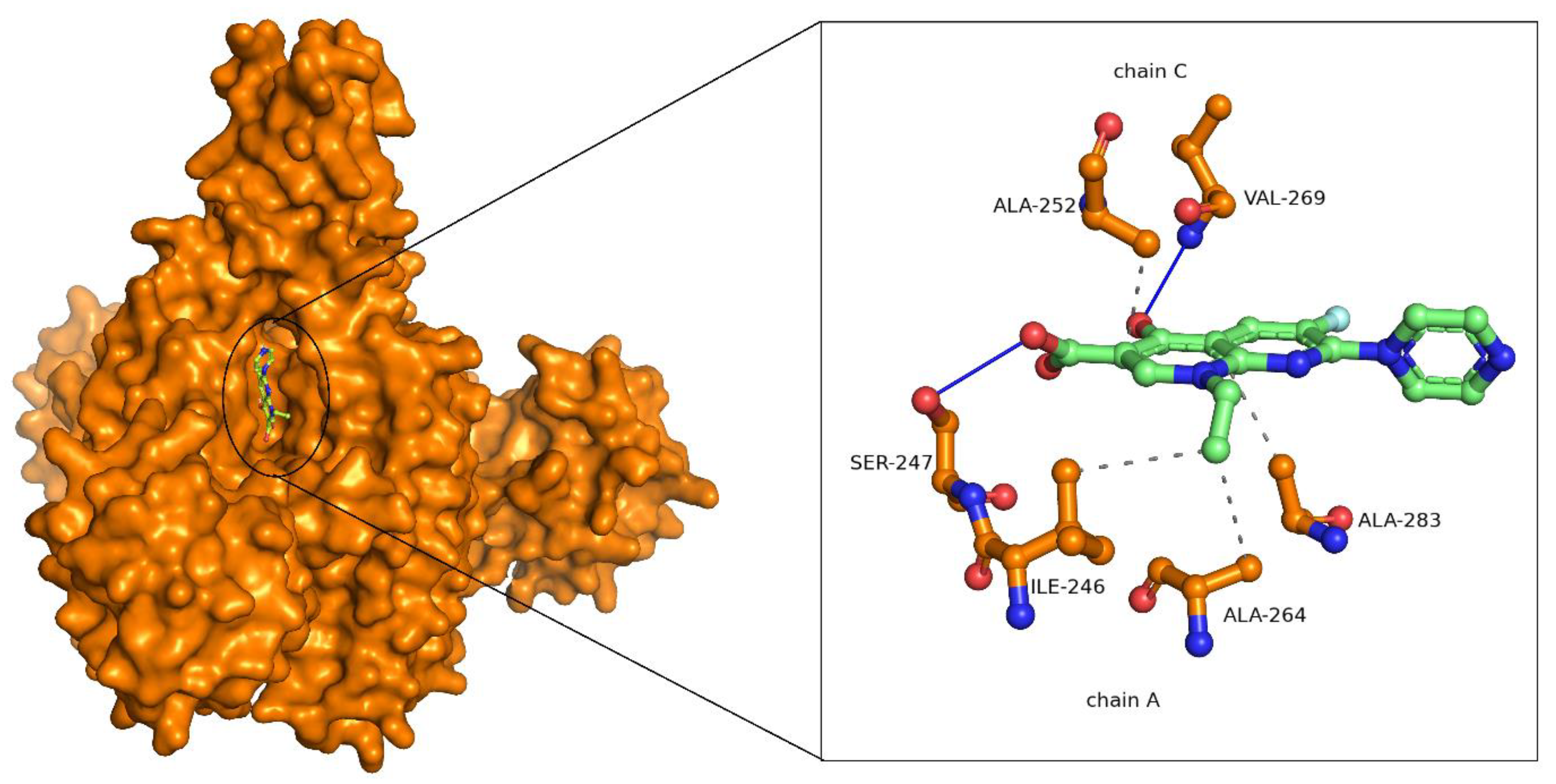
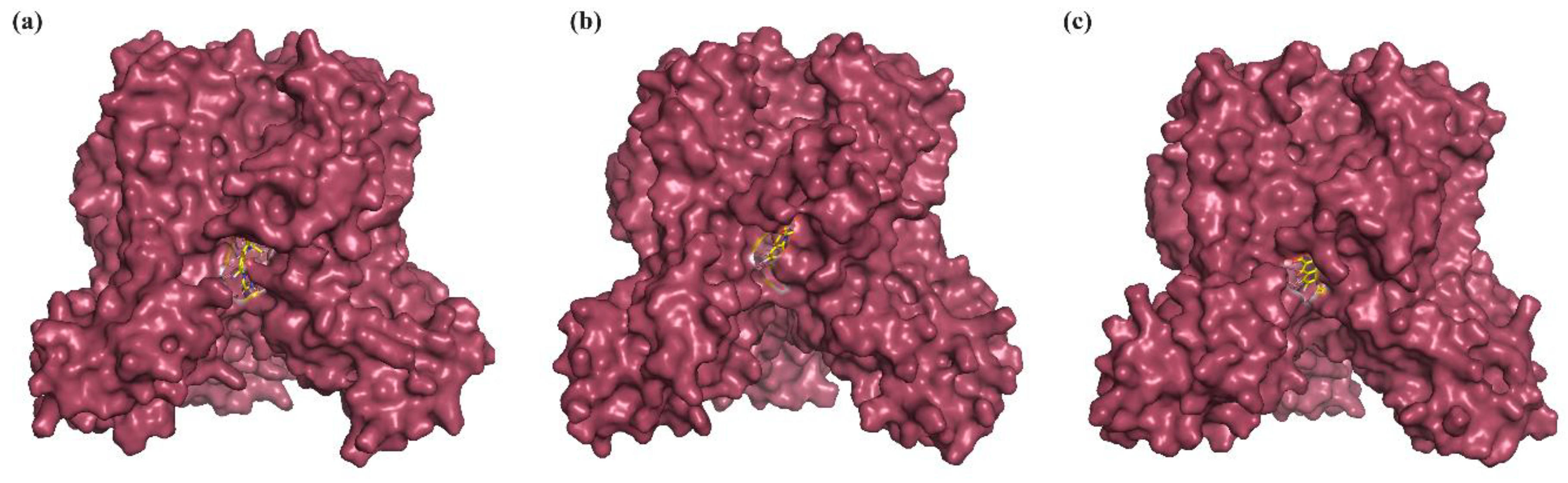
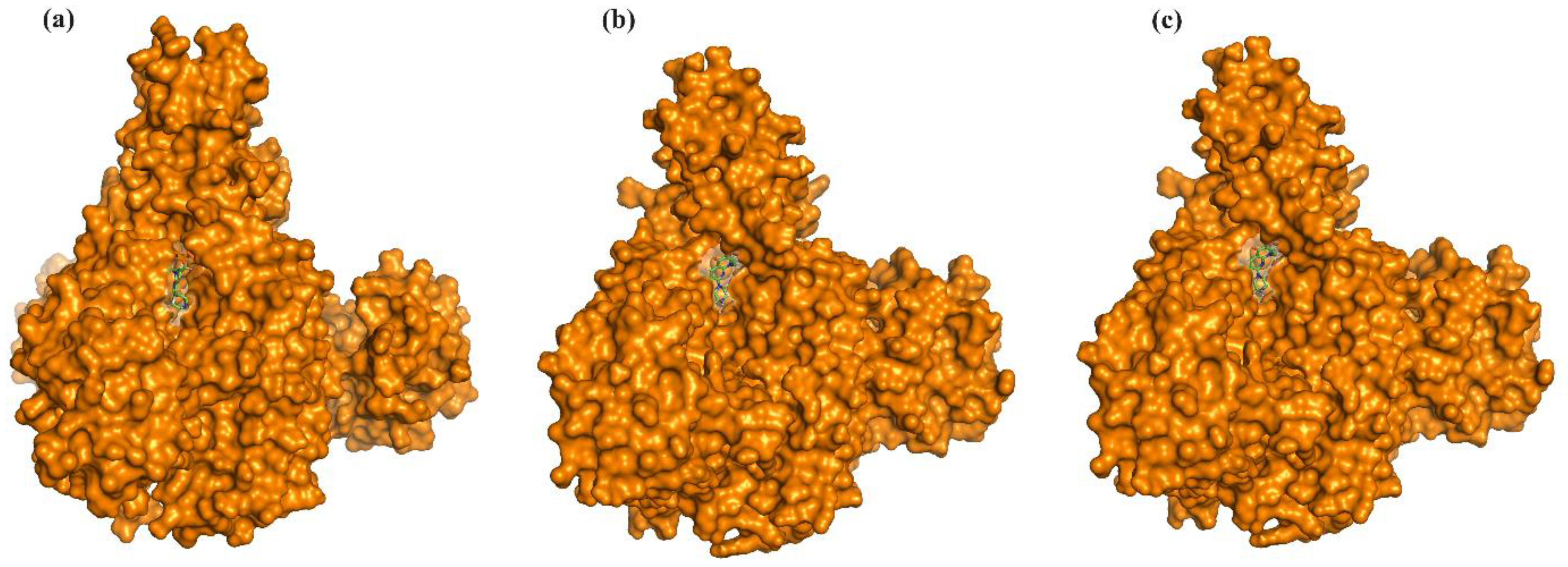
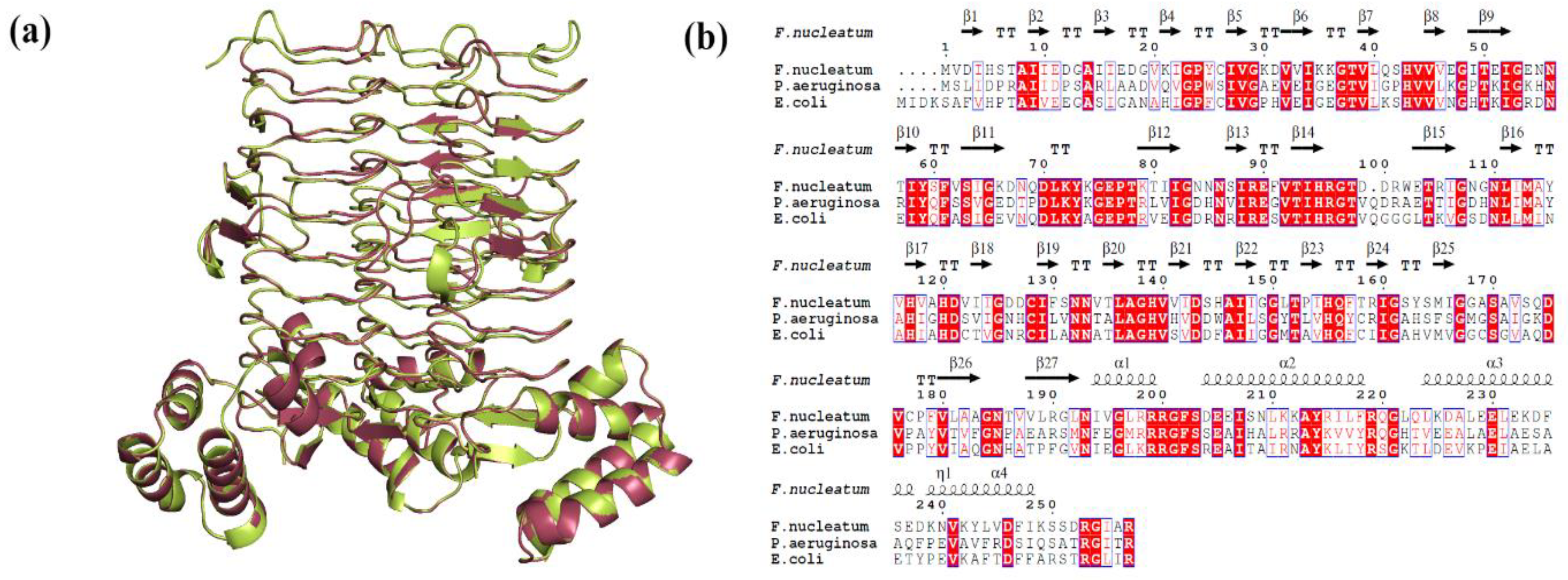
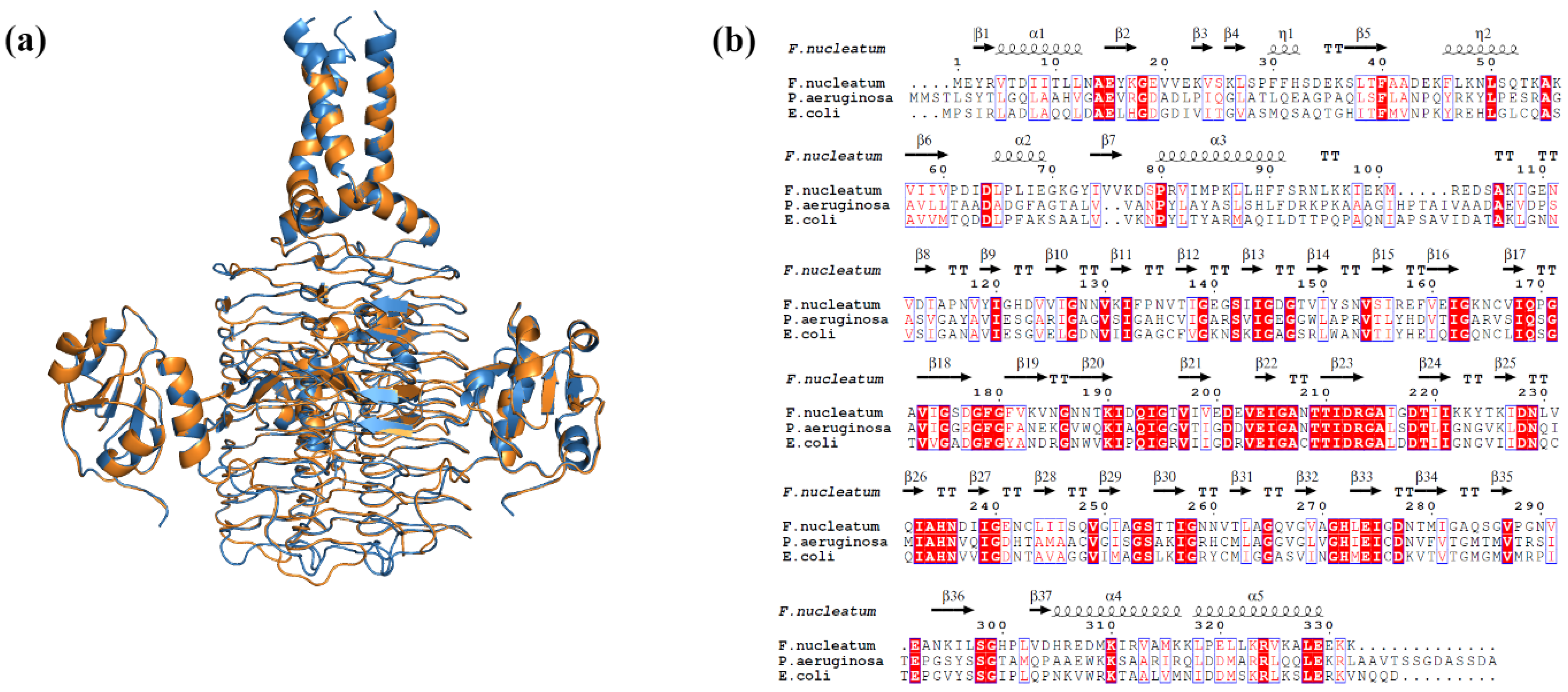
2.3. Binding Mode Analysis of Small Molecules with Acyl Transferases
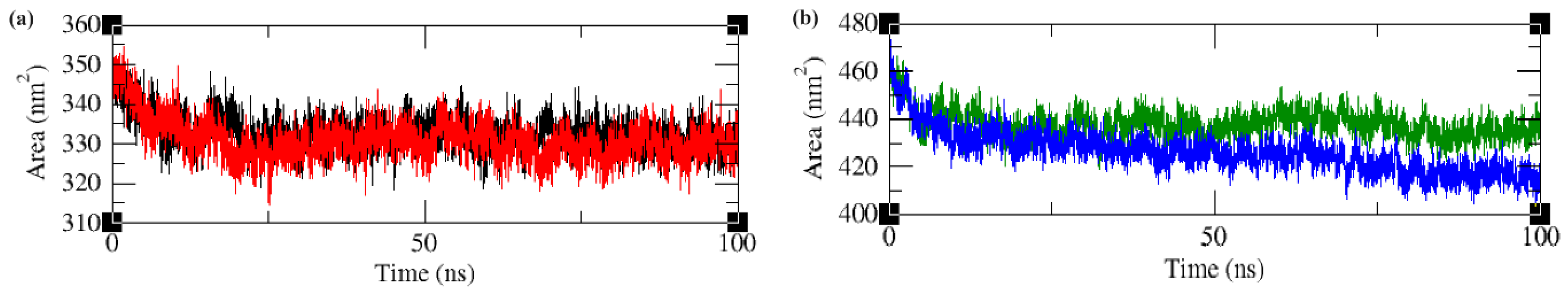
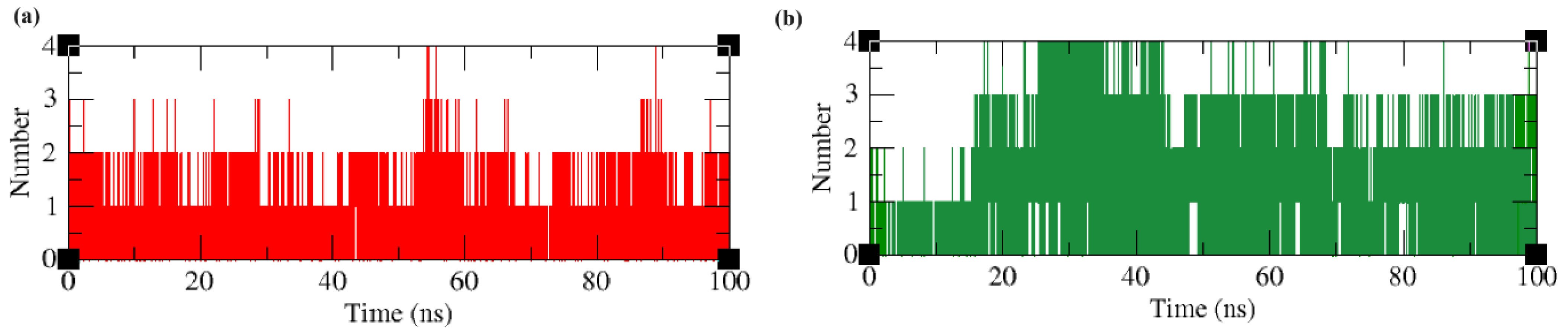
2.4. Molecular Dynamics and Simulation Analysis
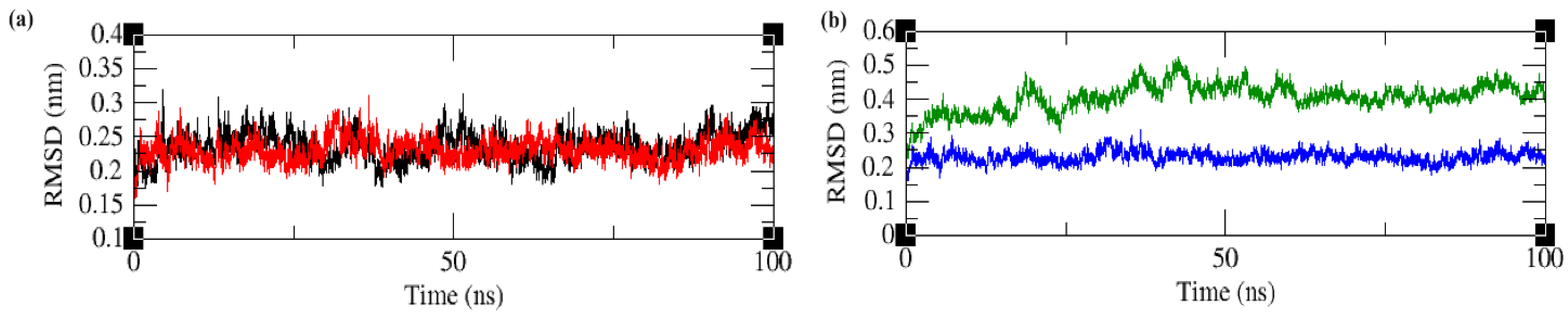
2.4.1. Root Mean Square Deviation (RMSD)
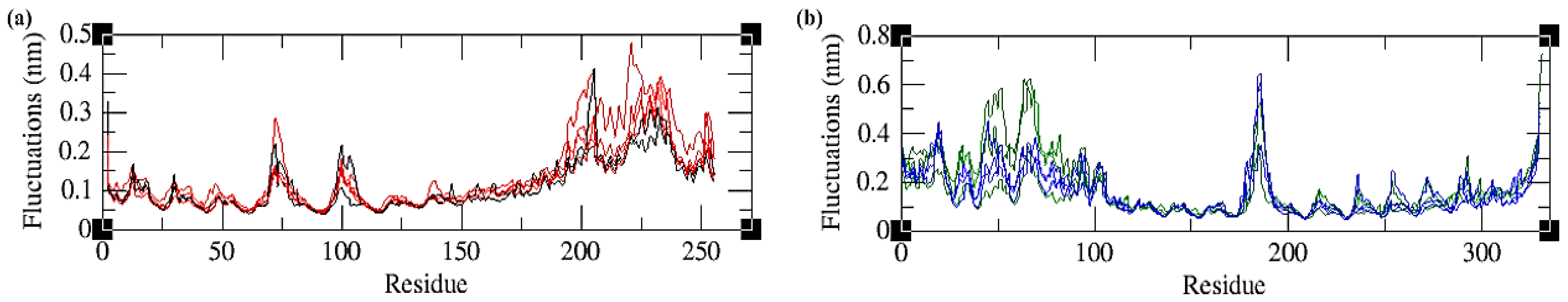
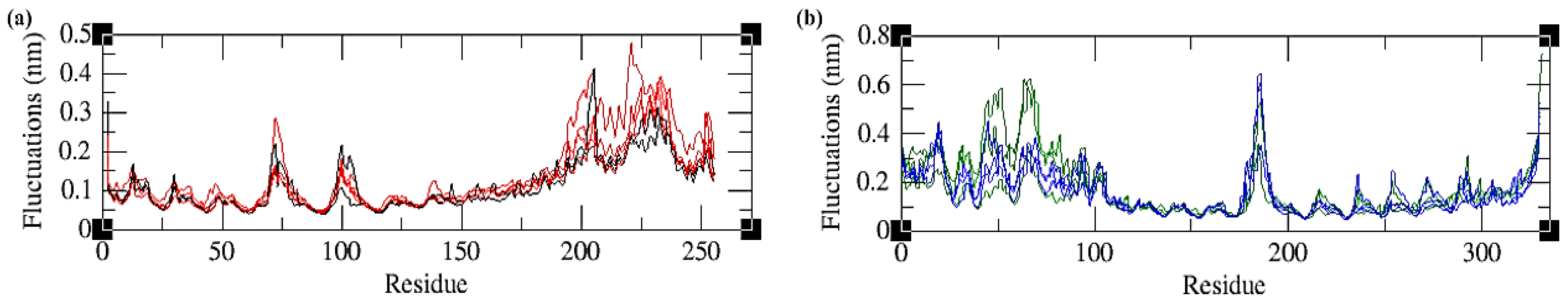
2.4.2. Root Mean Square Fluctuations (RMSFs)
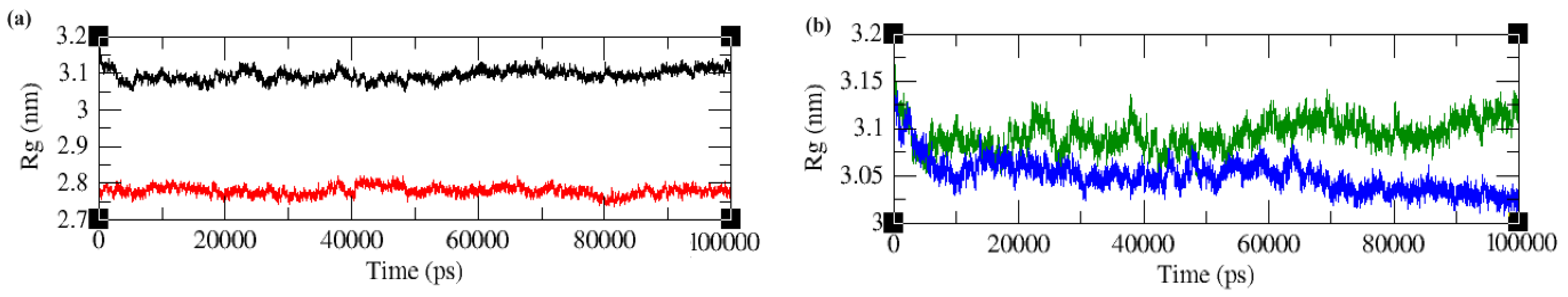
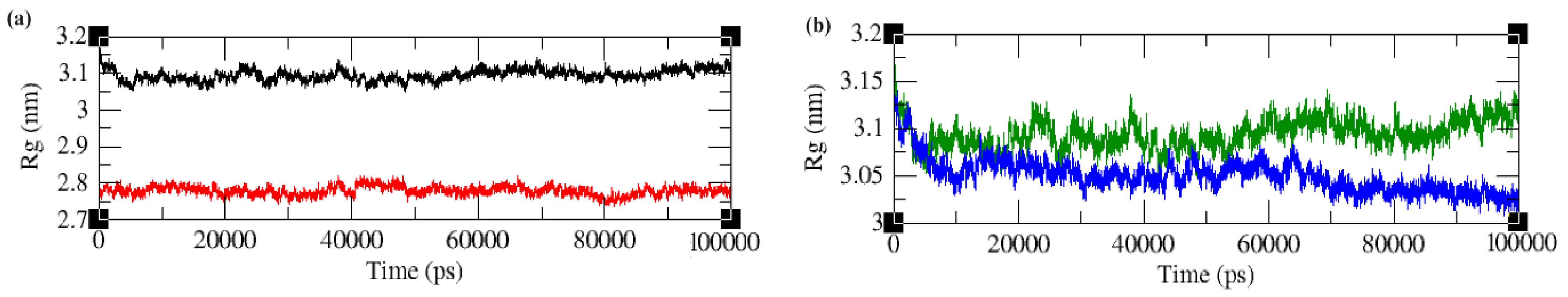
2.4.3. Radius of Gyration (Rg)
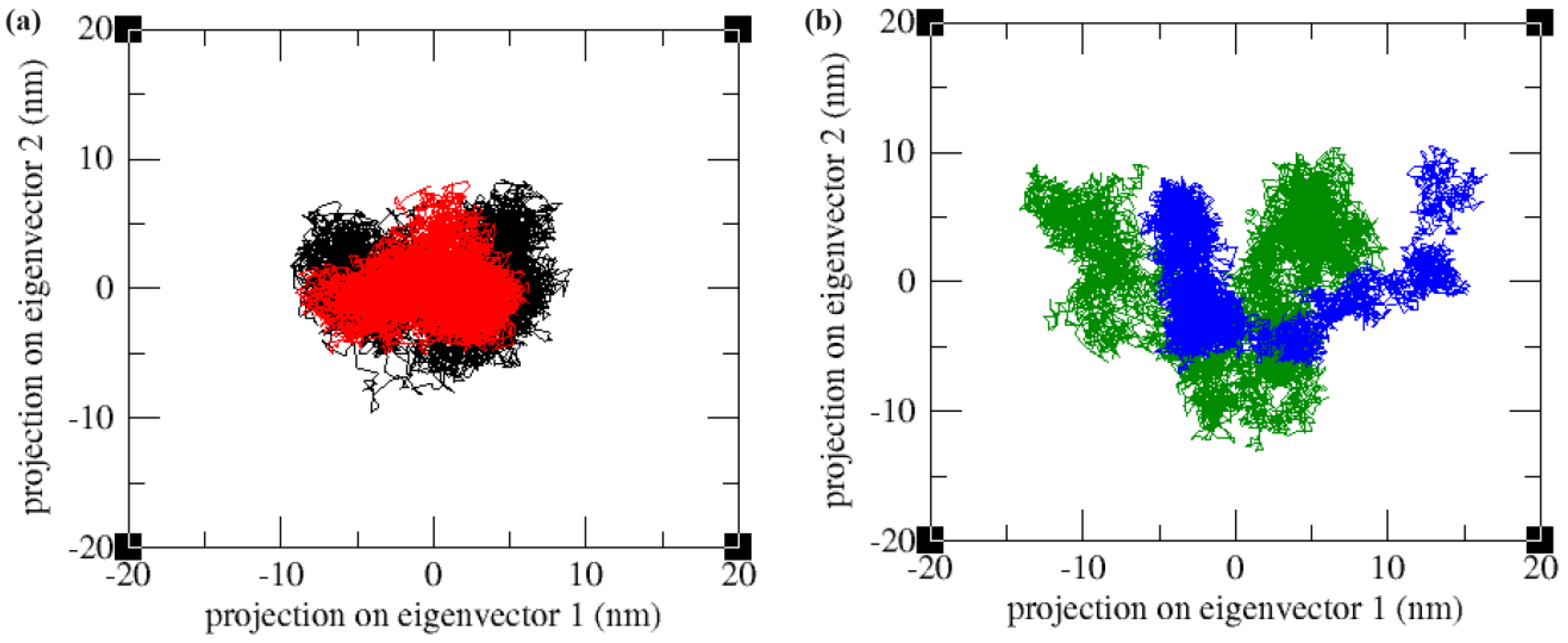
2.4.4. Essential Dynamics
2.4.5. Solvent Accessible Surface Area (SASA)
2.4.6. Intermolecular Hydrogen Bonds and Time Scale Analysis
2.4.7. Binding-Free Energy Calculations
3. Discussion
4. Materials and Methods
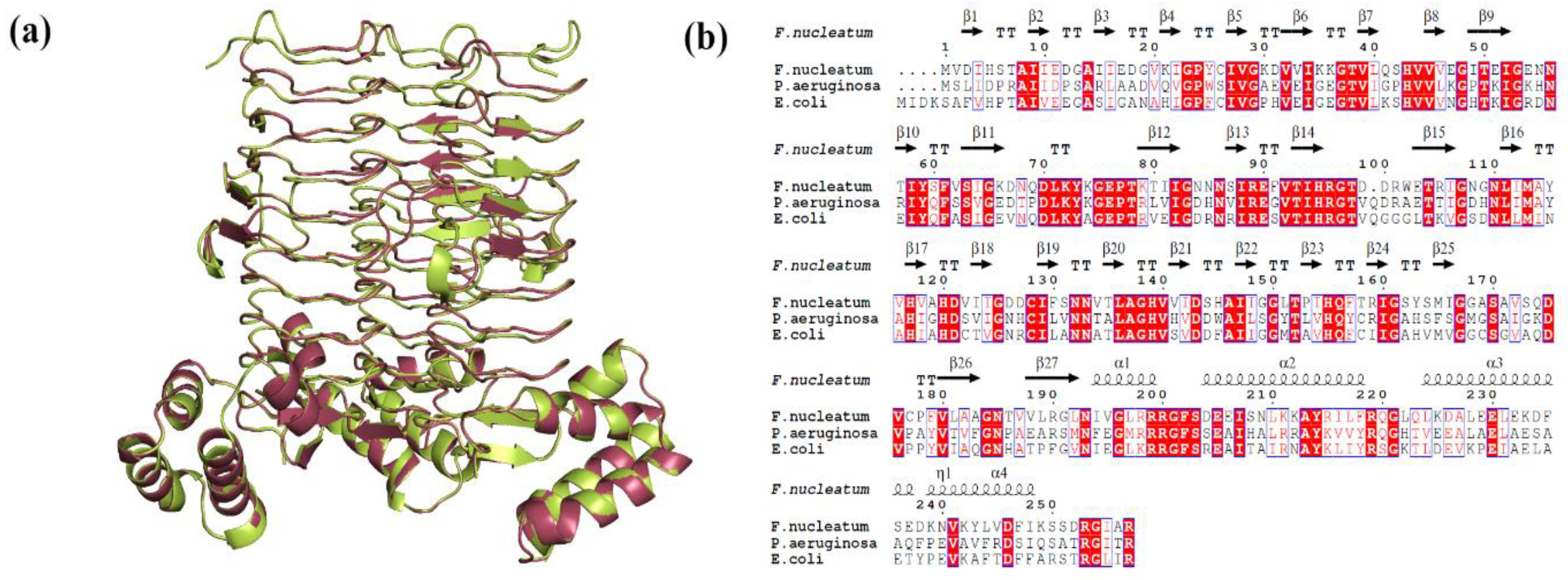
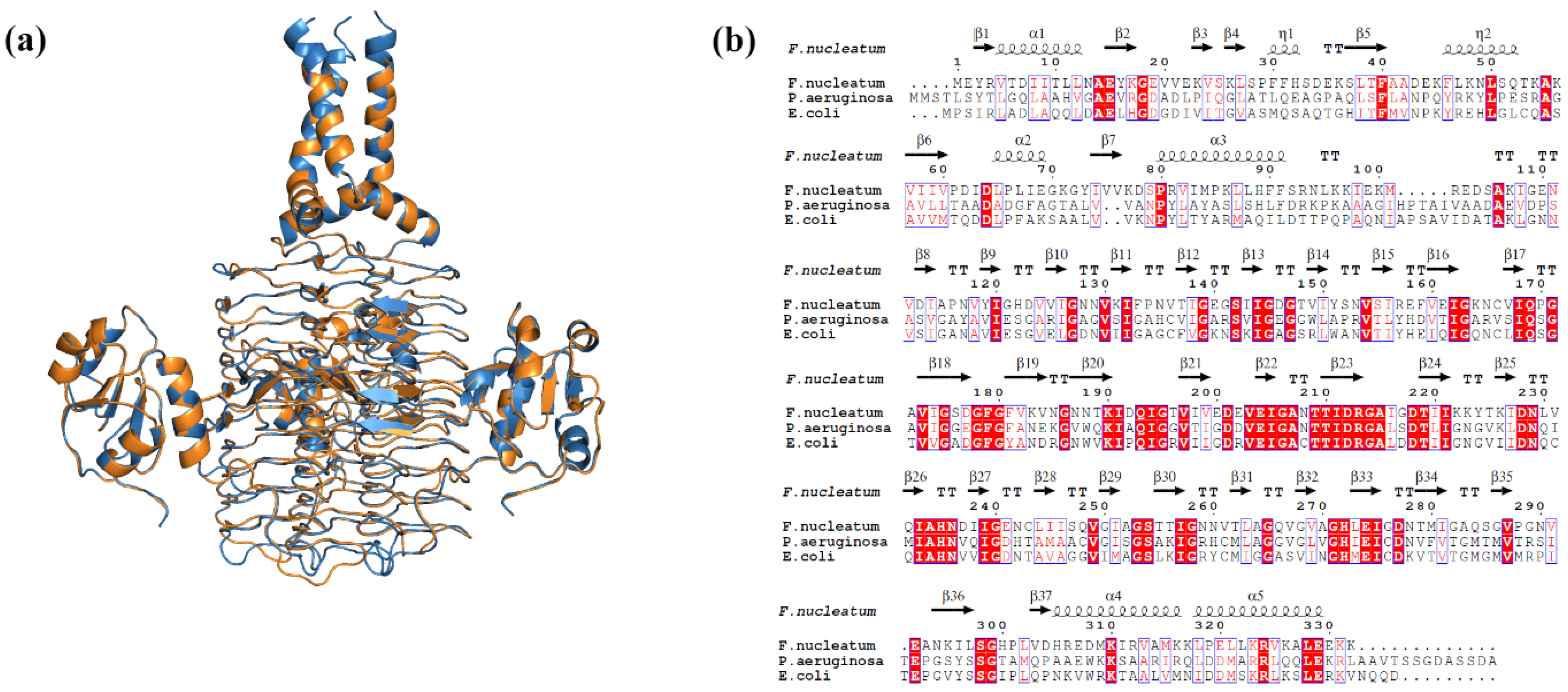
4.1. Proteins Structure Preparation
4.2. Identification of Inhibitors for Library Creation
4.3. Virtual Screening of LpxA/LpxD against In-House Compound Library
4.4. ADMET Screening and Activity Spectra
4.5. Molecular Dynamics and Simulation
4.6. MM/PBSA Based Binding Free Energy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Griffen, A.L.; Beall, C.J.; Campbell, J.H.; Firestone, N.D.; Kumar, P.S.; Yang, Z.K.; Podar, M.; Leys, E.J. Distinct and Complex Bacterial Profiles in Human Periodontitis and Health Revealed by 16S Pyrosequencing. ISME J. 2012, 6, 1176–1185. [Google Scholar] [CrossRef]
- Saygun, I.; Nizam, N.; Keskiner, I.; Bal, V.; Kubar, A.; Açıkel, C.; Serdar, M.; Slots, J. Salivary Infectious Agents and Periodontal Disease Status. J. Periodontal Res. 2011, 46, 235–239. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, X.; Li, J.; Aprecio, R.M.; Zhang, W.; Li, Y. Real-Time PCR Quantification of Six Periodontal Pathogens in Saliva Samples from Healthy Young Adults. Clin. Oral Investig. 2015, 19, 937–946. [Google Scholar] [CrossRef]
- Gomes, B.P.F.A.; Louzada, L.M.; Almeida-Gomes, R.F.; Pinheiro, E.T.; Sousa, E.L.R.; Jacinto, R.C.; Arruda-Vasconcelos, R. Investigation of Filifactor Alocis in Primary and in Secondary Endodontic Infections: A Molecular Study. Arch. Oral Biol. 2020, 118, 104826. [Google Scholar] [CrossRef]
- de Almeida Gomes, B.P.F.; Herrera, D.R. Etiologic Role of Root Canal Infection in Apical Periodontitis and Its Relationship with Clinical Symptomatology. Braz. Oral Res. 2018, 32, e69. [Google Scholar] [CrossRef]
- Ardila, C.M.; Granada, M.I.; Guzmán, I.C. Antibiotic Resistance of Subgingival Species in Chronic Periodontitis Patients. J. Periodontal Res. 2010, 45, 557–563. [Google Scholar] [CrossRef]
- Ansiliero, R.; Gelinski, J.M.L.N.; Samistraro, Q.L.; Baratto, C.M.; Almeida, C.A.; Locatelli, C. Pathogenic Microbial Profile and Antibiotic Resistance Associated with Periodontitis. Indian J. Microbiol. 2021, 61, 55–65. [Google Scholar] [CrossRef]
- Han, Y.W. Fusobacterium Nucleatum: A Commensal-Turned Pathogen. Curr. Opin. Microbiol. 2015, 23, 141–147. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Guan, Z.; Ingram, B.O.; Six, D.A.; Song, F.; Wang, X.; Zhao, J. Discovery of New Biosynthetic Pathways: The Lipid A Story. J. Lipid Res. 2009, 50, S103–S108. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid a modification systems in gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.F.; Rôças, I.N. Present Status and Future Directions: Microbiology of Endodontic Infections. Int. Endod. J. 2022, 55 (Suppl. S3), 512–530. [Google Scholar] [CrossRef]
- Yang, N.-Y.; Zhang, Q.; Li, J.-L.; Yang, S.-H.; Shi, Q. Progression of Periodontal Inflammation in Adolescents Is Associated with Increased Number of Porphyromonas Gingivalis, Prevotella Intermedia, Tannerella Forsythensis, and Fusobacterium Nucleatum. Int. J. Paediatr. Dent. 2014, 24, 226–233. [Google Scholar] [CrossRef]
- Kroeck, K.G.; Sacco, M.D.; Smith, E.W.; Zhang, X.; Shoun, D.; Akhtar, A.; Darch, S.E.; Cohen, F.; Andrews, L.D.; Knox, J.E.; et al. Discovery of Dual-Activity Small-Molecule Ligands of Pseudomonas Aeruginosa LpxA and LpxD Using SPR and X-ray Crystallography. Sci. Rep. 2019, 9, 15450. [Google Scholar] [CrossRef]
- King, D.E.; Malone, R.; Lilley, S.H. New Classification and Update on the Quinolone Antibiotics. Am. Fam. Physician 2000, 61, 2741–2748. [Google Scholar] [PubMed]
- Hoban, D.; Grabowski, M.; Koss, J.; Weselowski, V. Lomefloxacin, a New Difluoroquinolone: In Vitro Activity against Gram-Positive and Gram-Negative Bacteria. Diagn. Microbiol. Infect. Dis. 1989, 12, 77–82. [Google Scholar] [CrossRef]
- Leigh, D.A.; Tait, S.; Walsh, B. Antibacterial Activity of Lomefloxacin. J. Antimicrob. Chemother. 1991, 27, 589–598. [Google Scholar] [CrossRef]
- Henwood, J.M.; Monk, J.P. Enoxacin. A Review of Its Antibacterial Activity, Pharmacokinetic Properties and Therapeutic Use. Drugs 1988, 36, 32–66. [Google Scholar] [CrossRef] [PubMed]
- Jaber, L.A.; Bailey, E.M.; Rybak, M.J. Enoxacin: A New Fluoroquinolone. Clin. Pharm. 1989, 8, 97–107. [Google Scholar] [PubMed]
- Abe, F.C.; Kodaira, K.; Motta, C.d.C.B.; Barberato-Filho, S.; Silva, M.T.; Guimarães, C.C.; Martins, C.C.; Lopes, L.C. Antimicrobial Resistance of Microorganisms Present in Periodontal Diseases: A Systematic Review and Meta-Analysis. Front. Microbiol. 2022, 13, 961986. [Google Scholar] [CrossRef] [PubMed]
- Romano, K.P.; Hung, D.T. Targeting LPS Biosynthesis and Transport in Gram-Negative Bacteria in the Era of Multi-Drug Resistance. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119407. [Google Scholar] [CrossRef]
- Kralj, S.; Jukič, M.; Bren, U. Molecular Filters in Medicinal Chemistry. Encyclopedia 2023, 3, 501–511. [Google Scholar] [CrossRef]
- Wu, F.; Zhou, Y.; Li, L.; Shen, X.; Chen, G.; Wang, X.; Liang, X.; Tan, M.; Huang, Z. Computational Approaches in Preclinical Studies on Drug Discovery and Development. Front. Chem. 2020, 8, 726. [Google Scholar] [CrossRef]
- Shah, P.M.; Müller, S.; Kipp, J.; Stille, W. In Vitro Activity of Lomefloxacin (NY-198 or SC 47111). Diagn. Microbiol. Infect. Dis. 1989, 12, 97S–101S. [Google Scholar] [CrossRef] [PubMed]
- Muranaka, K.; Greenwood, D. The Response of Streptococcus Faecalis to Ciprofloxacin, Norfloxacin and Enoxacin. J. Antimicrob. Chemother. 1988, 21, 545–554. [Google Scholar] [CrossRef]
- Poveda Roda, R.; Bagan, J.V.; Sanchis Bielsa, J.M.; Carbonell Pastor, E. Antibiotic Use in Dental Practice. A Review. Med. Oral Patol. Oral Cir. Bucal 2007, 12, E186–E192. [Google Scholar]
- Pundir, S.; Martin, M.J.; O’Donovan, C. UniProt Protein Knowledgebase. Methods Mol. Biol. 2017, 1558, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 Update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Bursal, E.; Abdullah Yılmaz, M.; Aras, A.; Türkan, F.; Yildiko, Ü.; Kılıç, Ö.; Dey, A. Determination of Phenolic Content, Biological Activity, and Enzyme Inhibitory Properties with Molecular Docking Studies of Rumex nepalensis, an Endemic Medicinal Plant. J. Food Nutr. Res. 2021, 9, 114–123. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein–Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of Activity Spectra for Biologically Active Substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | MW | HBA | HBD | cLogP | DL |
|---|---|---|---|---|---|
| Lomefloxacin | 353.36 | 6 | 3 | 0.52 | 5.967 |
| Enoxacin | 322.33 | 6 | 3 | −0.16 | 36.456 |
| Ligand | GI Absorption | BBB Permeability | TPSA | CYP Inhibitor | PAINS Alert |
|---|---|---|---|---|---|
| Lomefloxacin | High | No | 76.04 | No | 0 |
| Enoxacin | High | No | 88.40 | No | 0 |
| Ligand | Activity | Pa | Pi |
|---|---|---|---|
| Lomefloxacin | Anti-infective | 0.889 | 0.004 |
| Antibacterial | 0.624 | 0.0008 | |
| Enoxacin | Antibiotic Naphthyridine-like | 0.606 | 0 |
| Antibacterial | 0.5 | 0.016 |
| Complex | ΔGbinding * | ΔEele * | ΔEvdw * | ΔEps * | ΔESASA * |
|---|---|---|---|---|---|
| LpxA–Lomefloxacin | −95.66 ± 13.20 | −13.23 ± 11.06 | −159.59 ± 9.65 | 94.85 ± 9.85 | −17.69 ± 1.40 |
| LpxD–Enoxacin | −101.13 ± 11.20 | −105.39 ± 16.96 | −166.62 ± 7.62 | 188.01 ± 11.21 | −17.13 ± 0.74 |
| Protein | Template PDB ID | Query Coverage (%) | Identity (%) | Procheck (%) | ERRAT Score 5 (%) | ProSA Z Score 6 | RMSD 7 (Å) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| A 1 | B 2 | C 3 | D 4 | |||||||
| LpxA | 7OKC | 98 | 51.37 | 88.7 | 10.0 | 1.3 | 0.0 | 94.33 | −7.28 | 0.110 |
| LpxD | 3EH0 | 98 | 33.53 | 83.9 | 14.8 | 0.7 | 0.6 | 82.32 | −6.6 | 0.129 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boreak, N.; Alrajab, E.A.; Nahari, R.A.; Najmi, L.E.; Masmali, M.A.; Ghawi, A.A.; Al Moaleem, M.M.; Alhazmi, M.Y.; Maqbul, A.A. Unveiling Therapeutic Potential: Targeting Fusobacterium nucleatum’s Lipopolysaccharide Biosynthesis for Endodontic Infections—An In Silico Screening Study. Int. J. Mol. Sci. 2024, 25, 4239. https://doi.org/10.3390/ijms25084239
Boreak N, Alrajab EA, Nahari RA, Najmi LE, Masmali MA, Ghawi AA, Al Moaleem MM, Alhazmi MY, Maqbul AA. Unveiling Therapeutic Potential: Targeting Fusobacterium nucleatum’s Lipopolysaccharide Biosynthesis for Endodontic Infections—An In Silico Screening Study. International Journal of Molecular Sciences. 2024; 25(8):4239. https://doi.org/10.3390/ijms25084239
Chicago/Turabian StyleBoreak, Nezar, Ethar Awad Alrajab, Rayan Ali Nahari, Loay Ebrahim Najmi, Muhannad Ali Masmali, Atiah Abdulrahman Ghawi, Mohammed M. Al Moaleem, Majed Yahya Alhazmi, and Abdulrahman Abdullah Maqbul. 2024. "Unveiling Therapeutic Potential: Targeting Fusobacterium nucleatum’s Lipopolysaccharide Biosynthesis for Endodontic Infections—An In Silico Screening Study" International Journal of Molecular Sciences 25, no. 8: 4239. https://doi.org/10.3390/ijms25084239