
Genetic Linkage between CAPN5 and TYR Variants in the Context of Albinism and Autosomal Dominant Neovascular Inflammatory Vitreoretinopathy Absence: A Case Report

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
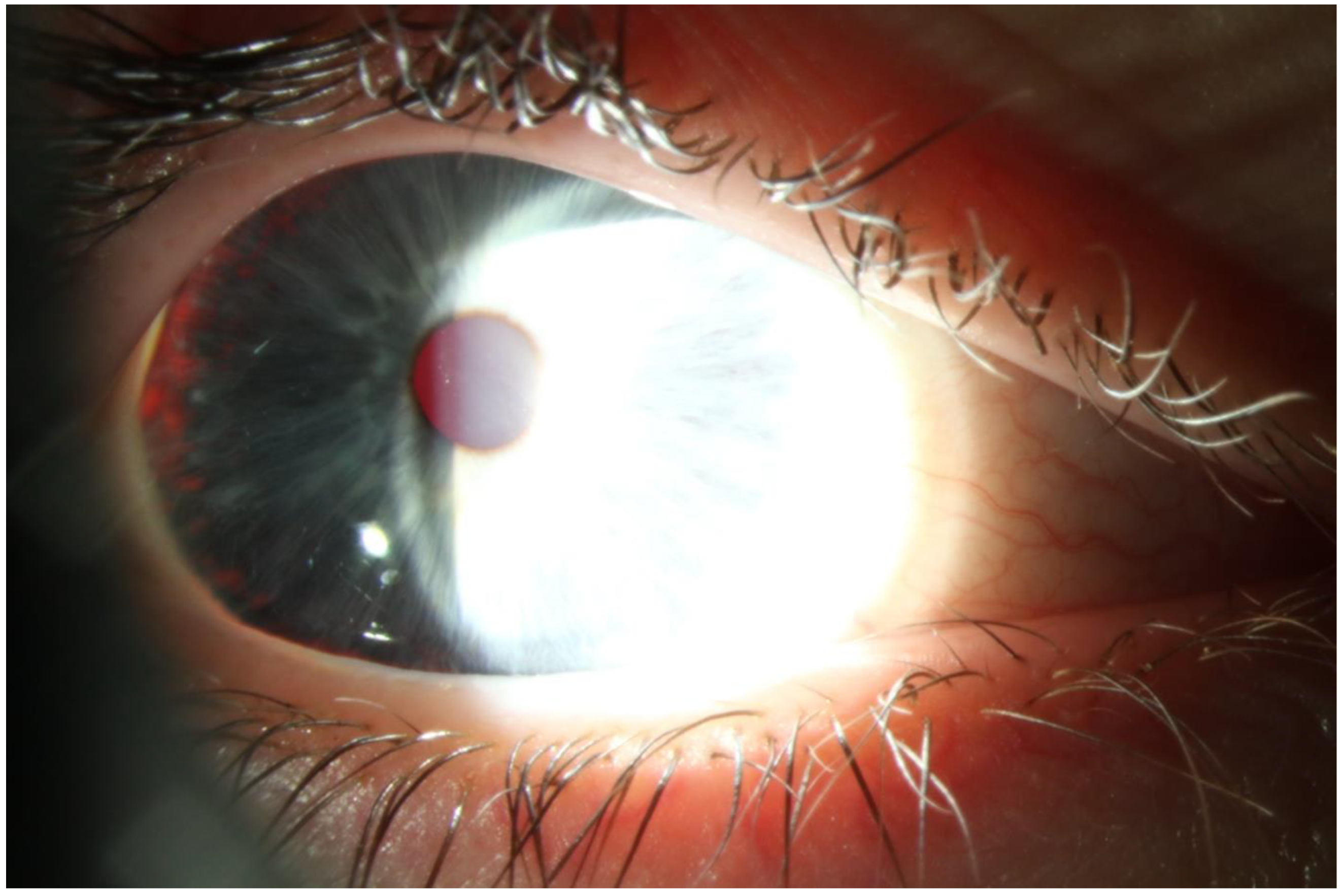
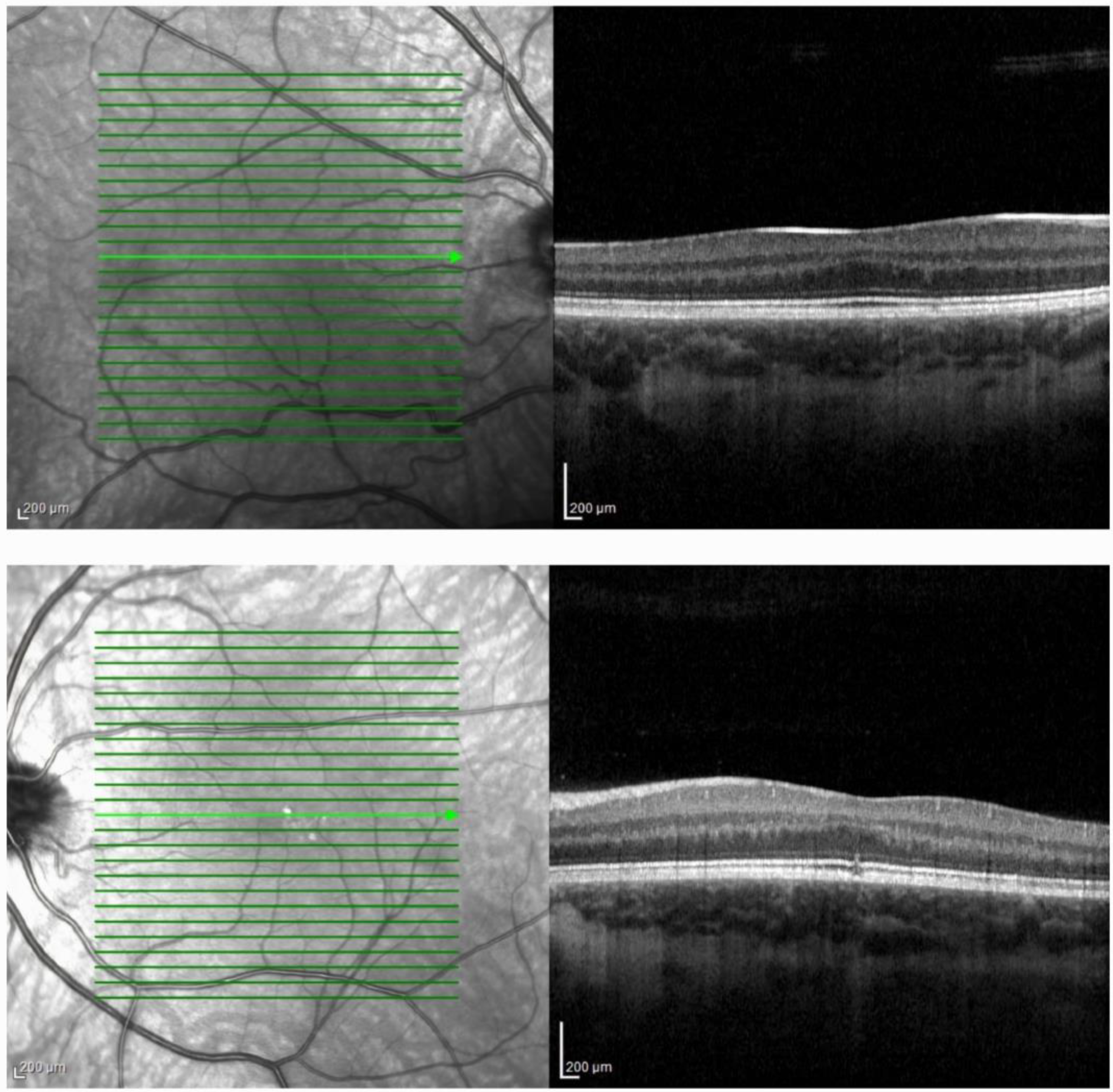
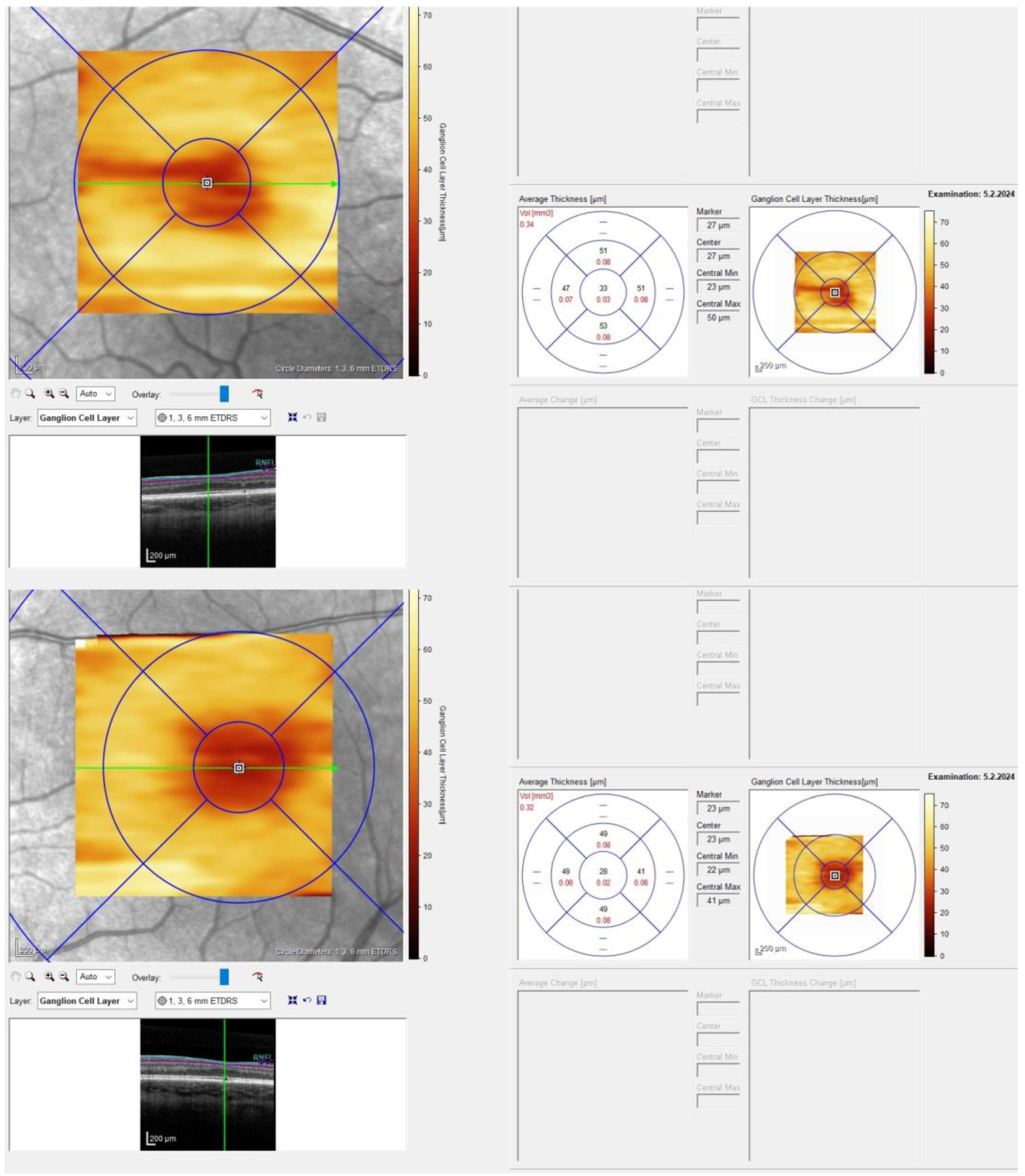
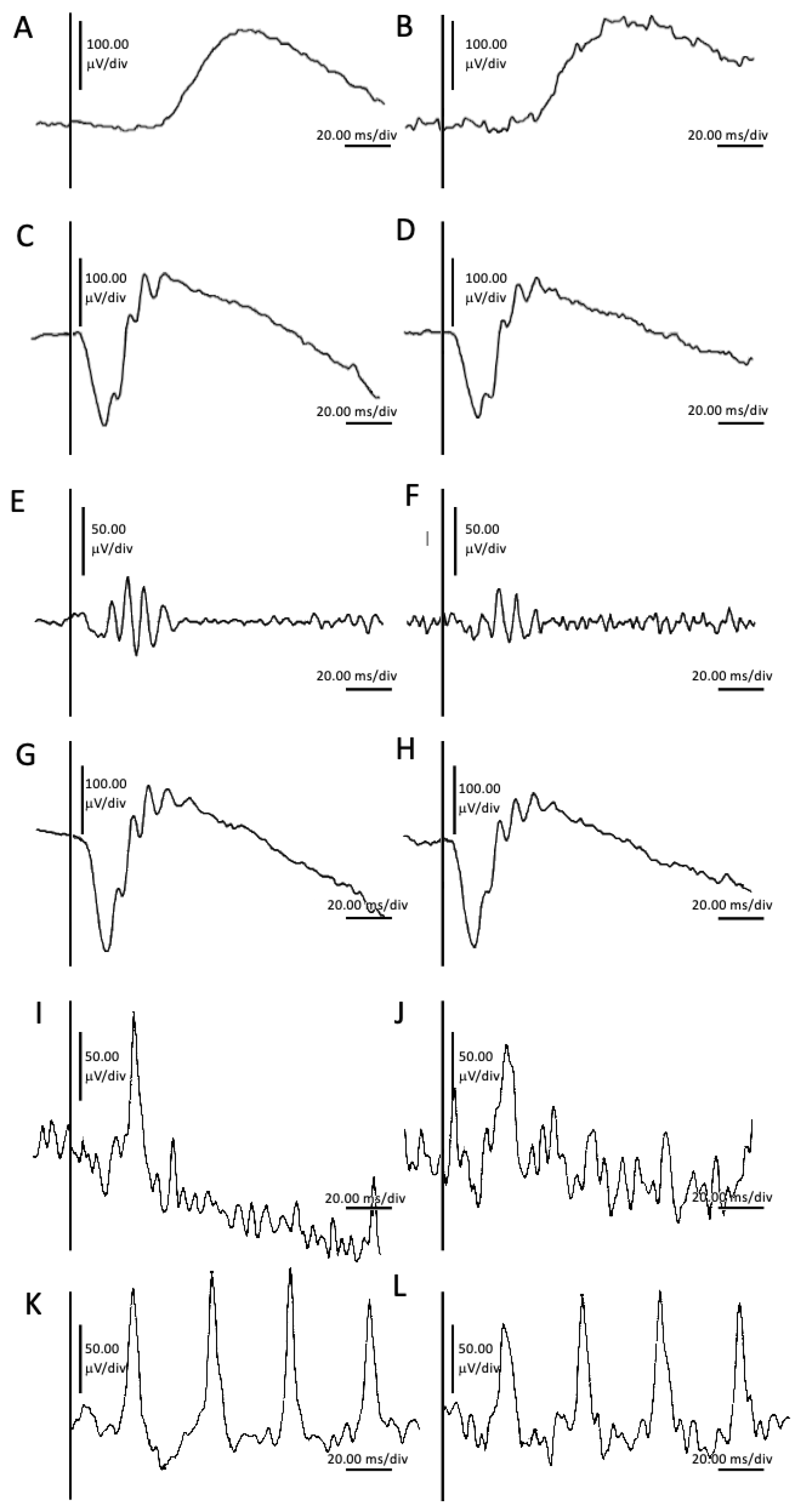
2. Case Presentation
Materials and Methods
3. Discussion
3.1. TYR
3.1.1. NM_000372.5(TYR):c.1205G>A (p.Arg402Gln)
3.1.2. NM_000372.5(TYR):c.1307G>C (p.Gly436Ala)
3.2. CAPN5
NM_004055.5(CAPN5):c.230A>G (p.Gln77Arg)
3.3. Albinism
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Yamaguchi, Y.; Brenner, M.; Hearing, V.J. The Regulation of Skin Pigmentation. J. Biol. Chem. 2007, 282, 27557–27561. [Google Scholar] [CrossRef]
- Grønskov, K.; Ek, J.; Brondum-Nielsen, K. Oculocutaneous albinism. Orphanet J. Rare Dis. 2007, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Suzuki, T. Oculocutaneous Albinism Type 4. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1510/ (accessed on 25 March 2024).
- Wang, N.; Hebert, D.N. Tyrosinase maturation through the mammalian secretory pathway: Bringing color to life. Pigment. Cell Res. 2006, 19, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Bijelic, A.; Pretzler, M.; Molitor, C.; Zekiri, F.; Rompel, A. The Structure of a Plant Tyrosinase from Walnut Leaves Reveals the Importance of “Substrate-Guiding Residues” for Enzymatic Specificity. Angew. Chem. Int. Ed. 2015, 54, 14677–14680. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.A.; Uwaydat, S.; Yunis, M.H.; Mansour, A.M. Foveal avascular zone in oculocutaneous albinism. BMJ Case Rep. 2021, 14, e240208. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.-W.; Spector, E.; Tsai, A.C.-H. Oculocutaneous albinism spectrum. Am. J. Med. Genet. Part A 2009, 149, 1590–1591. [Google Scholar] [CrossRef] [PubMed]
- Kruijt, C.C.; de Wit, G.C.; Bergen, A.A.; Florijn, R.J.; Schalij-Delfos, N.E.; van Genderen, M.M. The Phenotypic Spectrum of Albinism. Ophthalmology 2018, 125, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, V.C.; Kimura, A.E.; Folk, J.C.; Bennett, S.R.; Streb, L.M.; Nichols, B.E.; Stone, E.M. The gene for autosomal dominant neovascular inflammatory vitreoretinopathy maps to 11q13. Am. J. Hum. Genet. 1992, 51, A35. [Google Scholar]
- Stone, E.M.; Kimura, A.E.; Folk, J.C.; Bennett, S.R.; Nichols, B.E.; Streb, L.M.; Sheffield, V.C. Genetic linkage of autosomal dominant neovascular inflammatory vitreoretinopathy to chromosome 11q13. Hum. Mol. Genet. 1992, 1, 685–689. [Google Scholar] [CrossRef]
- Morell, R.; Spritz, R.A.; Ho, L.; Pierpont, J.; Guo, W.; Friedman, T.B.; Asher, J.H., Jr. Apparent digenic inheritance of Waar-denburg syndrome type 2 (WS2) and autosomal recessive ocular albinism (AROA). Hum. Mol. Genet. 1997, 5, 659–664. [Google Scholar] [CrossRef]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, V.B.; Skeie, J.M.; Bassuk, A.G.; Fingert, J.H.; Braun, T.A.; Daggett, H.T.; Folk, J.C.; Sheffield, V.C.; Stone, E.M. Cal-pain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet. 2012, 8, e1003001. [Google Scholar] [CrossRef] [PubMed]
- Roland Consult Electrophysiology and Imaging. Short Manual. Available online: https://www.roland-consult.de/downloads/#86-short-manual (accessed on 28 May 2024).
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Blueprint Genetics. Variant Classification. Available online: https://blueprintgenetics.com/variant-classification/ (accessed on 12 May 2024).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Oetting, W.S. The Tyrosinase Gene and Oculocutaneous Albinism Type 1 (OCA1): A Model for Understanding the Molecular Biology of Melanin Formation. Pigment. Cell Res. 2000, 13, 320–325. [Google Scholar] [CrossRef] [PubMed]
- King, R.A.; Pietsch, J.; Fryer, J.P.; Savage, S.; Brott, M.J.; Russell-Eggitt, I.; Summers, C.G.; Oetting, W.S. Tyrosinase gene mu-tations in oculocutaneous albinism 1 (OCA1): Definition of the phenotype. Hum. Genet. 2003, 13, 502–513. [Google Scholar] [CrossRef] [PubMed]
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/3779/ (accessed on 25 March 2024).
- Monfermé, S.; Lasseaux, E.; Duncombe-Poulet, C.; Hamel, C.; Defoort-Dhellemmes, S.; Drumare, I.; Zanlonghi, X.; Dollfus, H.; Perdomo, Y.; Bonneau, D.; et al. Mild form of oculocutaneous albinism type 1: Phenotypic analysis of compound heterozygous patients with the R402Q variant of the TYR gene. Br. J. Ophthalmol. 2018, 103, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.K.; Giebel, L.B.; Strunk, K.M.; Spritz, R.A. A polymorphism of the human tyrosinase gene is associated with tem-perature-sensitive enzymatic activity. Gene Expr. 1991, 1, 103–110. [Google Scholar] [PubMed]
- Giebel, L.B.; Tripathi, R.K.; King, R.A.; Spritz, R.A. A tyrosinase gene missense mutation in temperature-sensitive type I ocu-locutaneous albinism. A human homologue to the Siamese cat and the Himalayan mouse. J. Clin. Investig. 1991, 87, 1119–1122. [Google Scholar] [CrossRef]
- Oetting, W.S.; King, R.A. Analysis of Mutations in the Copper B Binding Region Associated With Type I (Tyrosinase-Related) Oculocutaneous Albinism. Pigment. Cell Res. 1992, 5, 274–278. [Google Scholar] [CrossRef]
- Dolinska, M.B.; Kus, N.J.; Farney, S.K.; Wingfield, P.T.; Brooks, B.P.; Sergeev, Y.V. Oculocutaneous albinism type 1: Link between mutations, tyrosinase conformational stability, and enzymatic activity. Pigment. Cell Melanoma Res. 2016, 30, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Hutton, S.M.; Spritz, R.A. A Comprehensive Genetic Study of Autosomal Recessive Ocular Albinism in Caucasian Patients. Investig. Ophthalmol. Vis. Sci. 2008, 49, 868–872. [Google Scholar] [CrossRef] [PubMed]
- ClinVar. Available online: https://preview.ncbi.nlm.nih.gov/clinvar/variation/882341/ (accessed on 26 March 2024).
- Lopez, V.M.; Decatur, C.L.; Stamer, W.D.; Lynch, R.M.; McKay, B.S. L-DOPA Is an Endogenous Ligand for OA1. PLoS Biol. 2008, 6, e236. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.R.; Folk, J.C.; Kimura, A.E.; Russell, S.R.; Stone, E.M.; Raphtis, E.M. Autosomal Dominant Neovascular Inflammatory Vitreoretinopathy. Ophthalmology 1990, 97, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Varsome. chr11-77093746-A-G (CAPN5:p.Q77R). Available online: https://varsome.com/variant/hg38/capn5%20c.230A%3EG?annotation-mode=germline (accessed on 21 May 2024).
- Li, A.S.; Velez, G.; Darbro, B.; Toral, M.A.; Yang, J.; Tsang, S.H.; Ferguson, P.J.; Folk, J.C.; Bassuk, A.G.; Mahajan, V.B. Whole-Exome Sequencing of Patients with Posterior Segment Uveitis. Arch. Ophthalmol. 2020, 221, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Bakker, R.; Wagstaff, E.L.; Kruijt, C.C.; Emri, E.; van Karnebeek, C.D.; Hoffmann, M.B.; Brooks, B.P.; Boon, C.J.; Montoliu, L.; van Genderen, M.M.; et al. The retinal pigmentation pathway in human albinism: Not so black and white. Prog. Retin. Eye Res. 2022, 91, 101091. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Nagy, J.A.; Pal, S.; Vasile, E.; Eckelhoefer, I.A.; Bliss, V.S.; Manseau, E.J.; Dasgupta, P.S.; Dvorak, H.F.; Mukhopadhyay, D. The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat. Med. 2001, 7, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Velez, G.; Bassuk, A.G.; Colgan, D.; Tsang, S.H.; Mahajan, V.B. Therapeutic drug repositioning using personalized proteomics of liquid biopsies. J. Clin. Investig. 2017, 2, e97818. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H. Foveal hypoplasia and optical coherence tomographic imaging. Taiwan J. Ophthalmol. 2018, 8, 181–188. [Google Scholar] [CrossRef]
- Lewis, R.A. Oculocutaneous Albinism Type 1—Retired Chapter, for Historical Reference Only. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1166/ (accessed on 27 March 2024).
- Brücher, V.C.; Heiduschka, P.; Grenzebach, U.; Eter, N.; Biermann, J. Distribution of macular ganglion cell layer thickness in foveal hypoplasia: A new diagnostic criterion for ocular albinism. PLoS ONE 2019, 14, e0224410. [Google Scholar] [CrossRef]
- Thomas, M.G.; Kumar, A.; Mohammad, S.; Proudlock, F.A.; Engle, E.C.; Andrews, C.; Chan, W.M.; Thomas, S.; Gottlob, I. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology 2011, 118, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Kuht, H.J.; Maconachie, G.D.E.; Han, J.; Kessel, L.; van Genderen, M.M.; McLean, R.J.; Hisaund, M.; Tu, Z.; Hertle, R.W.; Gronskov, K.; et al. Genotypic and Phenotypic Spectrum of Foveal Hypoplasia: A Multicenter Study. Ophthalmology 2022, 129, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Oetting, W.S.; Pietsch, J.; Brott, M.J.; Savage, S.; Fryer, J.P.; Summers, C.G.; King, R.A. The R402Q tyrosinase variant does not cause autosomal recessive ocular albinism. Am. J. Med. Genet. Part A 2009, 149, 466–469. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. dbSNP Short Genetic Variations. Available online: https://www.ncbi.nlm.nih.gov/snp/rs1591124579#hgvs_tab (accessed on 5 June 2024).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bjeloš, M.; Ćurić, A.; Bušić, M.; Rak, B.; Kuzmanović Elabjer, B. Genetic Linkage between CAPN5 and TYR Variants in the Context of Albinism and Autosomal Dominant Neovascular Inflammatory Vitreoretinopathy Absence: A Case Report. Int. J. Mol. Sci. 2024, 25, 6442. https://doi.org/10.3390/ijms25126442
Bjeloš M, Ćurić A, Bušić M, Rak B, Kuzmanović Elabjer B. Genetic Linkage between CAPN5 and TYR Variants in the Context of Albinism and Autosomal Dominant Neovascular Inflammatory Vitreoretinopathy Absence: A Case Report. International Journal of Molecular Sciences. 2024; 25(12):6442. https://doi.org/10.3390/ijms25126442
Chicago/Turabian StyleBjeloš, Mirjana, Ana Ćurić, Mladen Bušić, Benedict Rak, and Biljana Kuzmanović Elabjer. 2024. "Genetic Linkage between CAPN5 and TYR Variants in the Context of Albinism and Autosomal Dominant Neovascular Inflammatory Vitreoretinopathy Absence: A Case Report" International Journal of Molecular Sciences 25, no. 12: 6442. https://doi.org/10.3390/ijms25126442