
Mitochondrial DNA Instability Supersedes Parkin Mutations in Driving Mitochondrial Proteomic Alterations and Functional Deficits in Polg Mutator Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
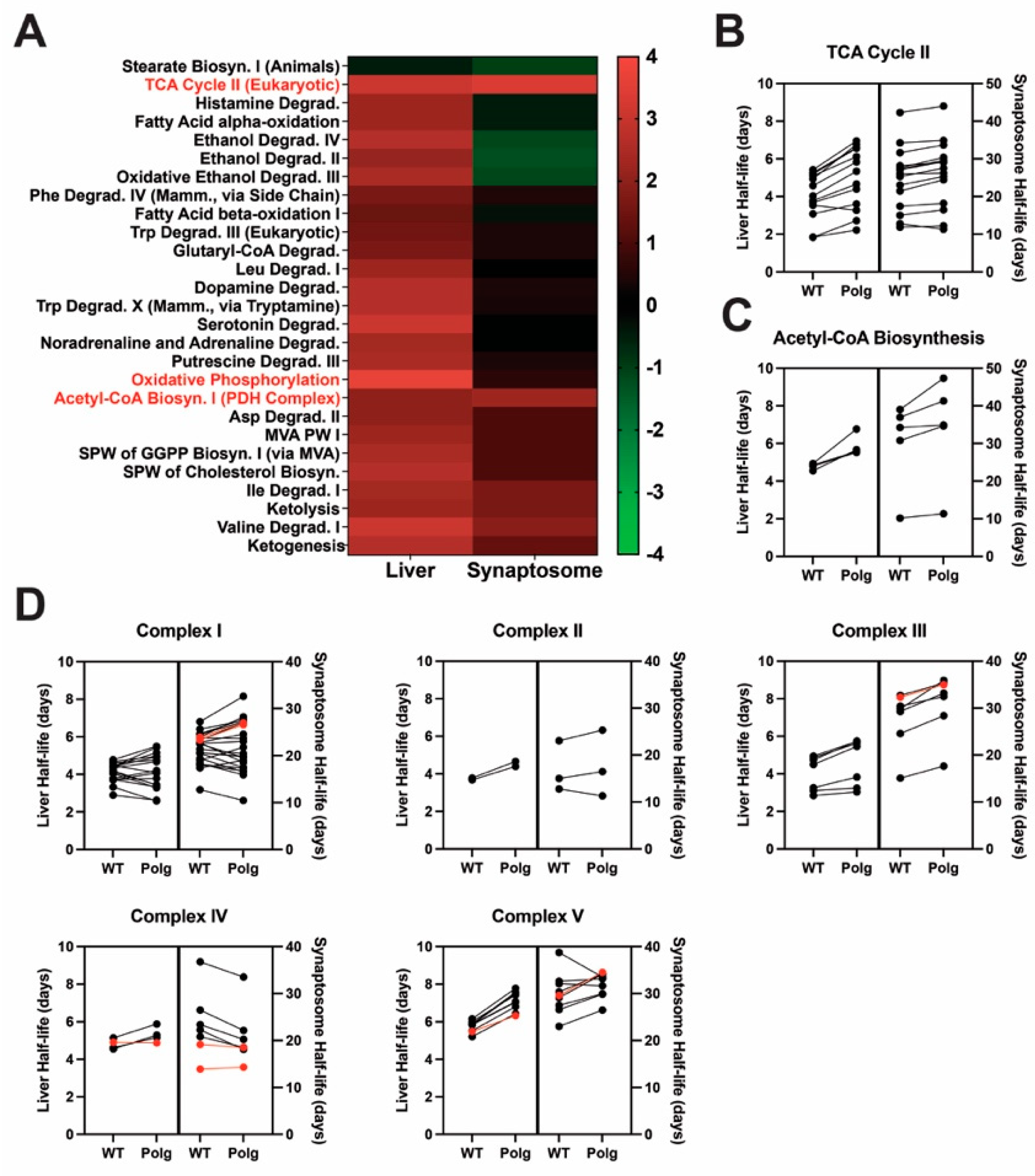
2.1. Liver Mitochondrial but Not Synaptosomal Protein Turnover Rates Are Affected by mtDNA Fidelity
2.2. Loss of mtDNA Fidelity Alters Dynamics of Proteins of Metabolism and Bioenergetics
2.3. Impaired mtDNA Fidelity Influences Oxidative Phosphorylation and Electron Transport Protein Expression
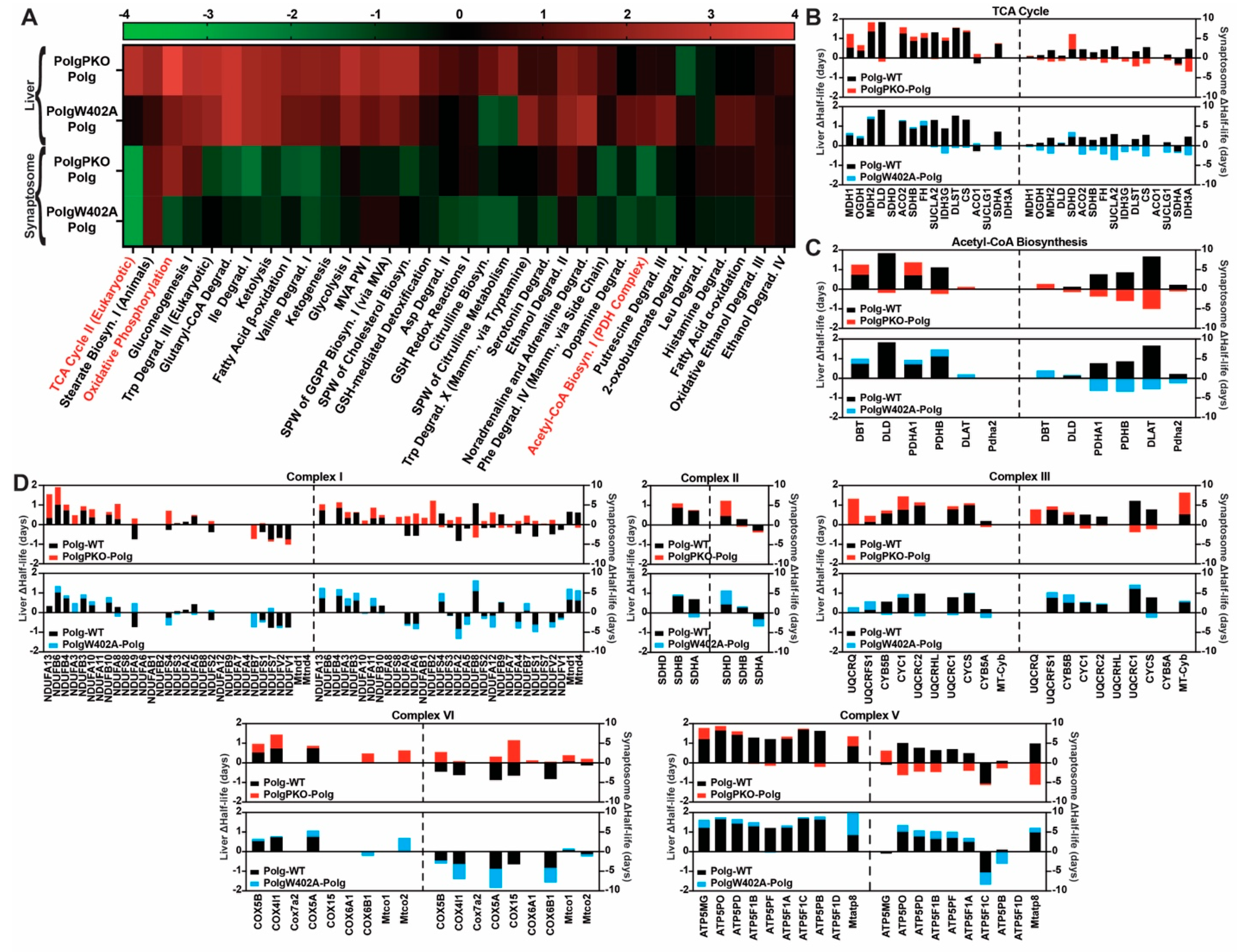
2.4. Influence of Parkin Expression or Activity on Proteome Dynamics in Polg Animals
2.4.1. Parkin Deficiency Reduces Turnover of Liver Mitochondrial Proteins
2.4.2. ParkinW402A Enhances Turnover of Synaptic Mitochondrial Proteins
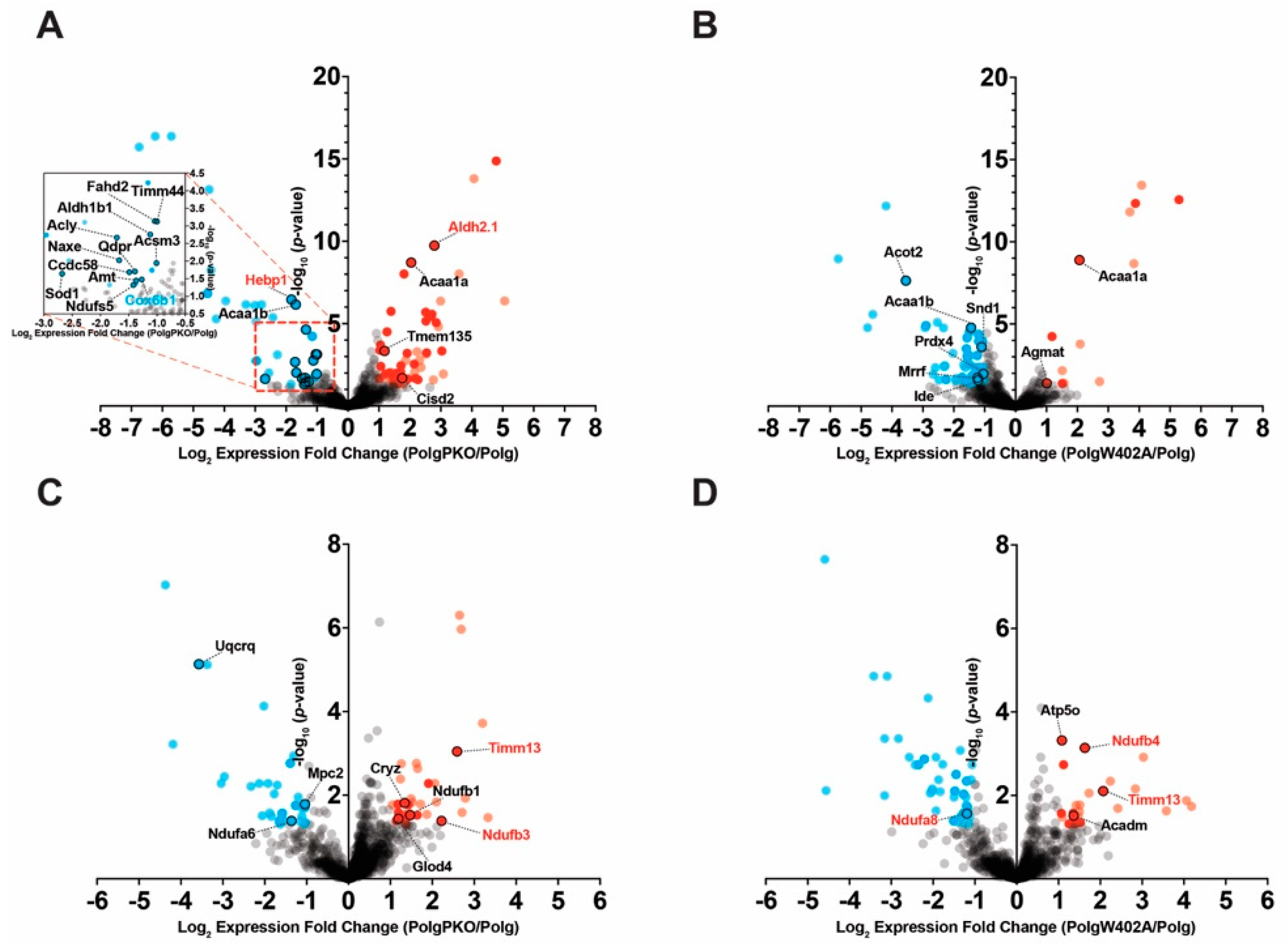
2.5. Polg Is the Primary Driver of Altered Proteostasis and Tissue, Specifically Modified by Parkin
2.6. Parkin’s Influence on Protein Expression Is Greater in Liver Mitochondria Than Synaptosomes
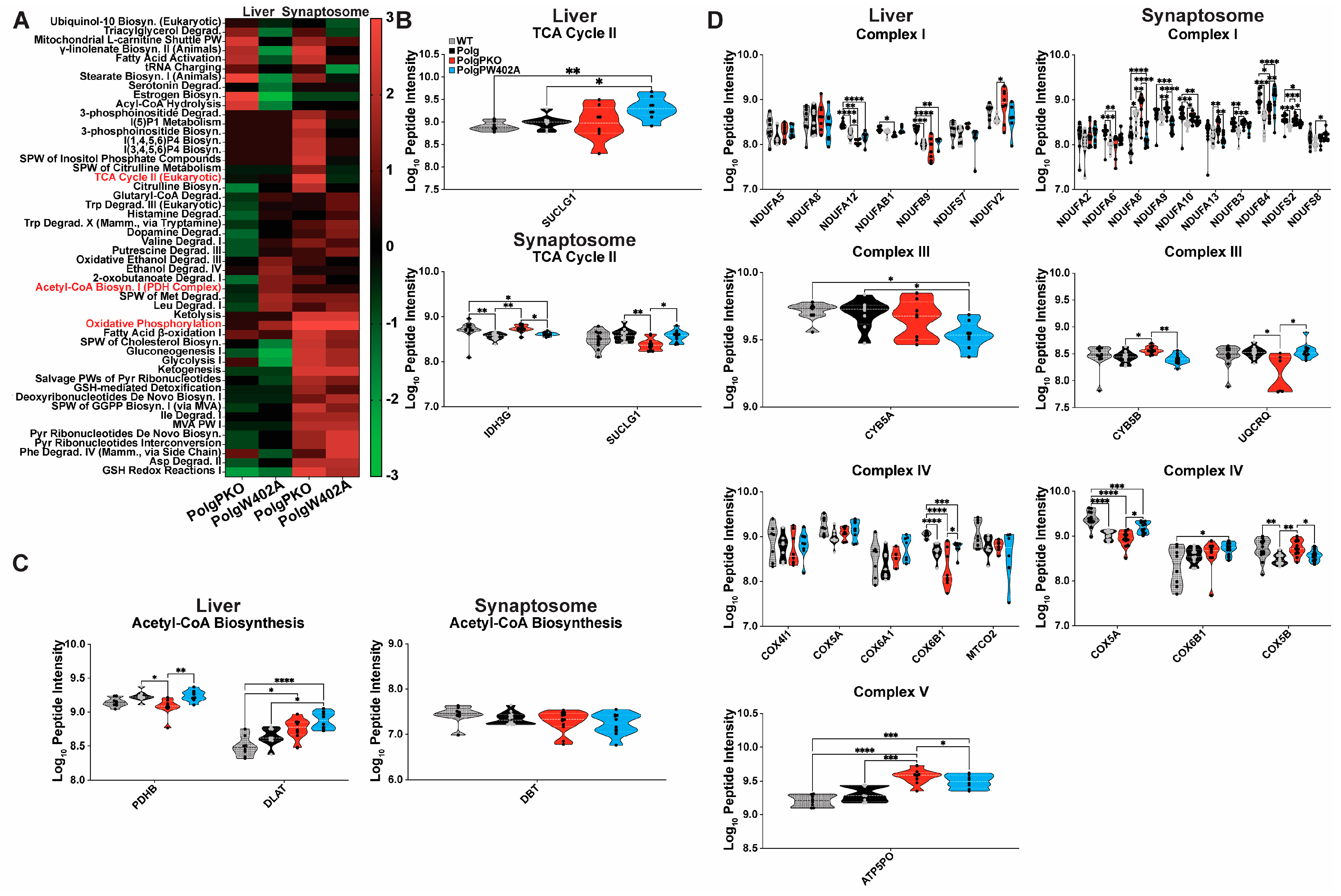
2.7. Pathway Analysis Reveals Parkin Fine Tunes Polg-Driven Protein Abundance Effects
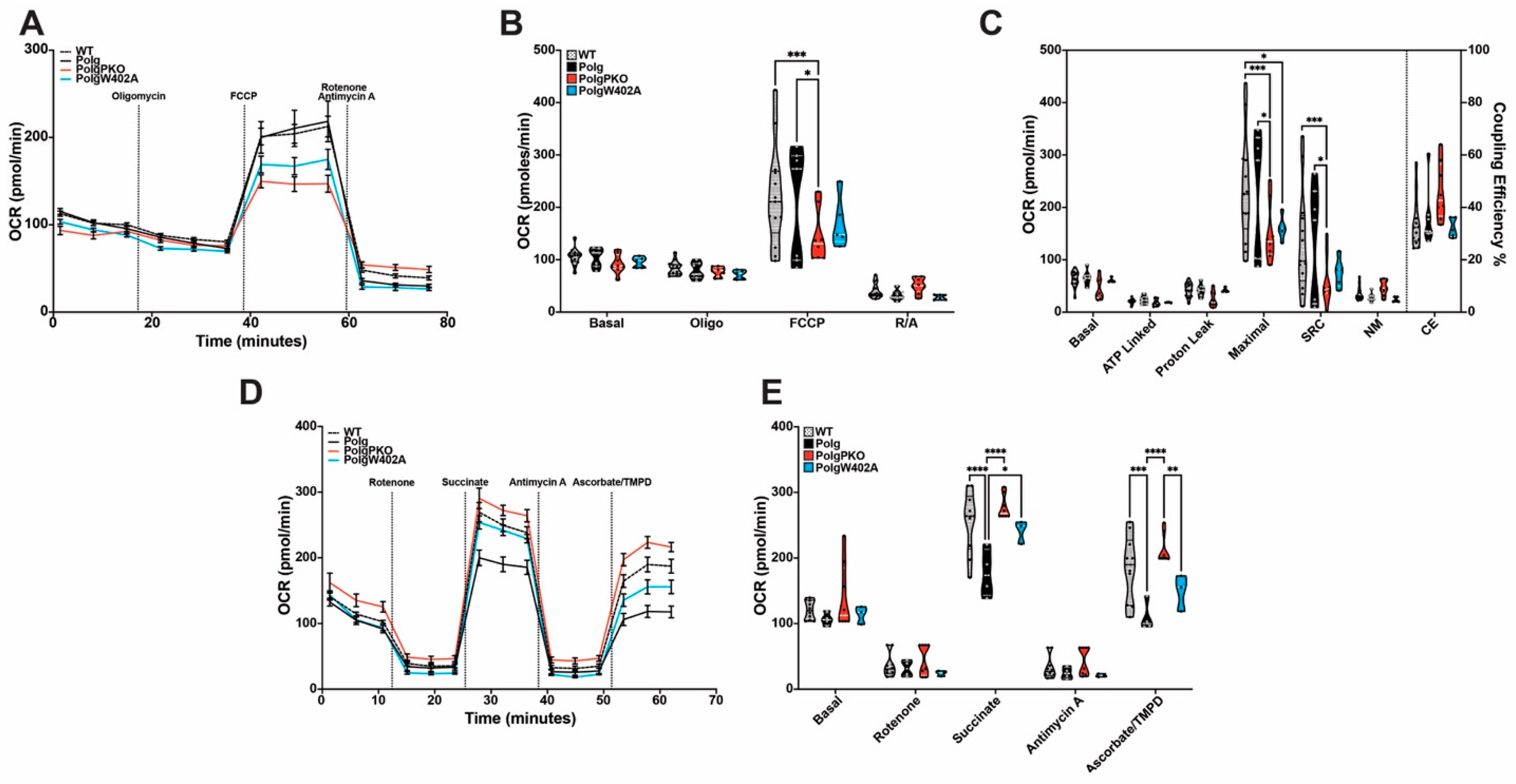
2.8. Polg Mutator Mice Exhibit Tissue-Specific Bioenergetic Capacity Impacted by Parkin Status
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Metabolic Labeling with Heavy Leucine
4.3. Subcellular Fractionation
4.3.1. Liver Mitochondria
4.3.2. Heart Ventricular Mitochondria
4.3.3. Synaptic Terminals (Synaptosomes)
4.4. Seahorse Metabolic Flux
4.4.1. Bioenergetic Analysis of Synaptosomes
4.4.2. Bioenergetic Analysis of Liver Mitochondria
4.5. Mass Spectrometry
4.5.1. Mass Spectrometry Sample Prep
4.5.2. Mass Spectrometry Acquisition
Liquid Chromatography
Data Acquisition
4.5.3. Mass Spectrometry Analysis
Abundance
Turnover
4.6. Pathway Analysis
4.7. Western Blot Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glancy, B.; Balaban, R.S. Role of Mitochondrial Ca2+ in the Regulation of Cellular Energetics. Biochemistry 2012, 51, 2959. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Jaiswal, M. Mitochondrial Calcium at the Synapse. Mitochondrion 2021, 59, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The Role of Mitochondrial Function and Cellular Bioenergetics in Ageing and Disease. Br. J. Dermatol. 2013, 169, 1. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Marmisolle, I.; Tarallo, D.; Quijano, C. Mitochondrial Bioenergetics and Dynamics in Secretion Processes. Front. Endocrinol. 2020, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.; Bowman, A.; Boulton, S.J.; Manning, P.; Birch-Machin, M.A. A Role for Human Mitochondrial Complex II in the Production of Reactive Oxygen Species in Human Skin. Redox Biol. 2014, 2, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Cartier, E.; Boyman, L.; Verhoeven, N.; Polster, B.M.; Huang, W.; Kane, M.; Jonathan Lederer, W.; Karbowski, M. Parkin-Independent Mitophagy via Drp1-Mediated Outer Membrane Severing and Inner Membrane Ubiquitination. J. Cell Biol. 2021, 220, e202006043. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Höke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-Independent Mitophagy Requires Drp1 and Maintains the Integrity of Mammalian Heart and Brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Yu, M.; Wang, C.; Xu, Z. Leucine-Rich Repeat Kinase 2 Disturbs Mitochondrial Dynamics via Dynamin-like Protein. J. Neurochem. 2012, 122, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of Mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9. [Google Scholar] [CrossRef]
- Youle, R.J.; Bliek, A.M. van der Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062. [Google Scholar] [CrossRef]
- King, L.; Plun-Favreau, H. Chapter 5—Mitophagy. In Parkinson’s Disease: Molecular Mechanisms Underlying Pathology; Academic Press: Cambridge, MA, USA, 2017; pp. 139–177. [Google Scholar] [CrossRef]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Zündorf, G.; Reiser, G. Calcium Dysregulation and Homeostasis of Neural Calcium in the Molecular Mechanisms of Neurodegenerative Diseases Provide Multiple Targets for Neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial Dysfunction in Neurological Disorders: Exploring Mitochondrial Transplantation. npj Regen. Med. 2020, 5, 22. [Google Scholar] [CrossRef]
- Vincow, E.S.; Merrihew, G.; Thomas, R.E.; Shulman, N.J.; Beyer, R.P.; MacCoss, M.J.; Pallanck, L.J. The PINK1-Parkin Pathway Promotes Both Mitophagy and Selective Respiratory Chain Turnover in Vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 6400–6405. [Google Scholar]
- Pathak, D.; Berthet, A.; Nakamura, K. Energy Failure: Does It Contribute to Neurodegeneration? Ann. Neurol. 2013, 74, 506–516. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, Pathophysiological Roles, and Analysis. Biol. Chem. 2012, 393, 547. [Google Scholar] [CrossRef]
- Koyano, F.; Matsuda, N. Molecular Mechanisms Underlying PINK1 and Parkin Catalyzed Ubiquitylation of Substrates on Damaged Mitochondria. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2791–2796. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin Is Phosphorylated by PINK1 to Activate Parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Koyano, F.; Yamano, K.; Kosako, H.; Tanaka, K.; Matsuda, N. Parkin Recruitment to Impaired Mitochondria for Nonselective Ubiquitylation Is Facilitated by MITOL. J. Biol. Chem. 2019, 294, 10300–10314. [Google Scholar] [CrossRef]
- Iguchi, M.; Kujuro, Y.; Okatsu, K.; Koyano, F.; Kosako, H.; Kimura, M.; Suzuki, N.; Uchiyama, S.; Tanaka, K.; Matsuda, N. Parkin-Catalyzed Ubiquitin-Ester Transfer Is Triggered by PINK1-Dependent Phosphorylation. J. Biol. Chem. 2013, 288, 22019–22032. [Google Scholar] [CrossRef] [PubMed]
- Eckl, E.-M.; Ziegemann, O.; Krumwiede, L.; Fessler, E.; Jae, L.T. Sensing, Signaling and Surviving Mitochondrial Stress. Cell. Mol. Life Sci. 2021, 78, 5925–5951. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN Signalling in Neurodegeneration and Neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, Y.; Hou, X.; Tao, Z.; Ren, H.; Wang, G. Dependence of PINK1 Accumulation on Mitochondrial Redox System. Aging Cell 2020, 19, e13211. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Martínez-Torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Gonzalez, A.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; et al. Binding to Serine 65-phosphorylated Ubiquitin Primes Parkin for Optimal PINK 1-dependent Phosphorylation and Activation. EMBO Rep. 2015, 16, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Periquet, M.; Latouche, M.; Lohmann, E.; Rawal, N.; De Michele, G.; Ricard, S.; Teive, H.; Fraix, V.; Vidailhet, M.; Nicholl, D.; et al. Parkin Mutations Are Frequent in Patients with Isolated Early-Onset Parkinsonism. Brain 2003, 126, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Abbas, N.; Lücking, C.B.; Ricard, S.; Dürr, A.; Bonifati, V.; De Michele, G.; Bouley, S.; Vaughan, J.R.; Gasser, T.; Marconi, R.; et al. A Wide Variety of Mutations in the Parkin Gene Are Responsible for Autosomal Recessive Parkinsonism in Europe. Hum. Mol. Genet. 1999, 8, 567–574. [Google Scholar] [CrossRef]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between Early-Onset Parkinson’s Disease and Mutations in the Parkin Gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef]
- Camargos, S.T.; Dornas, L.O.; Momeni, P.; Lees, A.; Hardy, J.; Singleton, A.; Cardoso, F. Familial Parkinsonism and Early Onset Parkinson’s Disease in a Brazilian Movement Disorders Clinic: Phenotypic Characterization and Frequency of SNCA, PRKN, PINK1, and LRRK2 Mutations. Mov. Disord. 2009, 24, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Vinish, M.; Prabhakar, S.; Khullar, M.; Verma, I.; Anand, A. Genetic Screening Reveals High Frequency of PARK2 Mutations and Reduced Parkin Expression Conferring Risk for Parkinsonism in North West India. J. Neurol. Neurosurg. Psychiatry 2010, 81, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Potashkin, J.A.; Blume, S.R.; Runkle, N.K. Limitations of Animal Models of Parkinson’s Disease. Parkinsons. Dis. 2011, 2011, 658083. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Sanchez-Martinez, A.; Zarate, A.M.; Benincá, C.; Mayor, U.; Clague, M.J.; Whitworth, A.J. Basal Mitophagy Is Widespread in Drosophila but Minimally Affected by Loss of Pink1 or Parkin. J. Cell Biol. 2018, 217, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.Y.; Um, J.H.; Yoon, J.H.; Kim, H.; Lee, D.Y.; Lee, Y.J.; Jee, H.J.; Kim, Y.M.; Jang, J.S.; Jang, Y.G.; et al. Assessment of Mitophagy in Mt-Keima Drosophila Revealed an Essential Role of the PINK1-Parkin Pathway in Mitophagy Induction in Vivo. FASEB J. 2019, 33, 9742–9751. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Tong, Y.; Gautier, C.A.; Shen, J. Absence of Nigral Degeneration in Aged Parkin/DJ-1/PINK1 Triple Knockout Mice. J. Neurochem. 2009, 111, 696. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-Deficient Mice Exhibit Nigrostriatal Deficits but Not Loss of Dopaminergic Neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.A.; Palmiter, R.D. Parkin-Deficient Mice Are Not a Robust Model of Parkinsonism. Proc. Natl. Acad. Sci. USA 2005, 102, 2174–2179. [Google Scholar] [PubMed]
- Pickrell, A.M.; Huang, C.H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons Following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.; Karuppagounder, S.S.; Neifert, S.; Kang, B.G.; Wang, H.; Dawson, V.L.; Dawson, T.M. The Absence of Parkin Does Not Promote Dopamine or Mitochondrial Dysfunction in PolgAD257A/D257A Mitochondrial Mutator Mice. J. Neurosci. 2022, 42, 9263–9277. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gldlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature Ageing in Mice Expressing Defective Mitochondrial DNA Polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Kujoth, C.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Medicine: Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar]
- Lim, E.W.; Handzlik, M.K.; Trefts, E.; Gengatharan, J.M.; Pondevida, C.M.; Shaw, R.J.; Metallo, C.M. Progressive Alterations in Amino Acid and Lipid Metabolism Correlate with Peripheral Neuropathy in PolgD257A Mice. Sci. Adv. 2021, 7, eabj4077. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Primiani, C.T.; Langston, R.G.; Kumaran, R.; Cookson, M.R. The Polg Mutator Phenotype Does Not Cause Dopaminergic Neurodegeneration in DJ-1-Deficient Mice. eNeuro 2015, 2, e0075-14.2015. [Google Scholar] [CrossRef]
- Stauch, K.L.; Totusek, S.; Trease, A.J.; Estrella, L.D.; Emanuel, K.; Fangmeier, A.; Fox, H.S. Longitudinal in Vivo Metabolic Labeling Reveals Tissue-Specific Mitochondrial Proteome Turnover Rates and Proteins Selectively Altered by Parkin Deficiency. Sci. Rep. 2023, 13, 1–18. [Google Scholar] [CrossRef]
- Hsieh, E.J.; Shulman, N.J.; Dai, D.F.; Vincow, E.S.; Karunadharma, P.P.; Pallanck, L.; Rabinovitch, P.S.; MacCoss, M.J. Topograph, a Software Platform for Precursor Enrichment Corrected Global Protein Turnover Measurements. Mol. Cell. Proteomics 2012, 11, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An Updated Mitochondrial Proteome Now with Sub-Organelle Localization and Pathway Annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.Y.; Vranas, M.; Krahn, A.I.; Pundlik, S.; Trempe, J.F.; Fon, E.A. Structure-Guided Mutagenesis Reveals a Hierarchical Mechanism of Parkin Activation. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Trempe, J.F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Meńade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of Parkin Reveals Mechanisms for Ubiquitin Ligase Activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin Structure and Function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef]
- Zhao, L. Mitochondrial DNA Degradation: A Quality Control Measure for Mitochondrial Genome Maintenance and Stress Response. Enzymes 2019, 45, 311. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, J.D.; Saneto, R.P.; Copeland, W.C. Clinical and Molecular Features of POLG-Related Mitochondrial Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Caldarazzo Ienco, E.; Mancuso, M.; Siciliano, G. POLG1-Related and Other “Mitochondrial Parkinsonisms”: An Overview. J. Mol. Neurosci. 2011, 44, 17–24. [Google Scholar] [PubMed]
- Luoma, P.T.; Eerola, J.; Ahola, S.; Hakonen, A.H.; Hellström, O.; Kivistö, K.T.; Tienari, P.J.; Suomalainen, A. Mitochondrial DNA Polymerase Gamma Variants in Idiopathic Sporadic Parkinson Disease. Neurology 2007, 69, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Jacome, M.S.; Lei, S.; Hernandez-Franco, P.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Metabolic Disorder Dysfunction in Parkinson’s Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res. Bull. 2017, 133, 12. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Clark, J.; Dai, Y. Do Somatic Mitochondrial DNA Mutations Contribute to Parkinson’s Disease? Parkinsons. Dis. 2011, 2011, 659694. [Google Scholar] [CrossRef] [PubMed]
- Toomey, C.E.; Heywood, W.E.; Evans, J.R.; Lachica, J.; Pressey, S.N.; Foti, S.C.; Al Shahrani, M.; D’Sa, K.; Hargreaves, I.P.; Heales, S.; et al. Mitochondrial Dysfunction Is a Key Pathological Driver of Early Stage Parkinson’s. Acta Neuropathol. Commun. 2022, 10, 1–25. [Google Scholar]
- Gibson, G.E.; Kingsbury, A.E.; Xu, H.; Lindsay, J.G.; Daniel, S.; Foster, O.J.F.; Lees, A.J.; Blass, J.P. Deficits in a Tricarboxylic Acid Cycle Enzyme in Brains from Patients with Parkinson’s Disease. Neurochem. Int. 2003, 43, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Xu, F.; Lai, H.; Yuan, H.; Li, X.Y.; Hu, J.; Li, W.; Liu, L.; Wang, C. ACO2 Deficiency Increases Vulnerability to Parkinson’s Disease via Dysregulating Mitochondrial Function and Histone Acetylation-Mediated Transcription of Autophagy Genes. Commun. Biol. 2023, 6, 1–17. [Google Scholar] [CrossRef]
- Sumi, K.; Hatanaka, Y.; Takahashi, R.; Wada, N.; Ono, C.; Sakamoto, Y.; Sone, H.; Iida, K. Citrate Synthase Insufficiency Leads to Specific Metabolic Adaptations in the Heart and Skeletal Muscles Upon Low-Carbohydrate Diet Feeding in Mice. Front. Nutr. 2022, 9, 925908. [Google Scholar]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199. [Google Scholar] [CrossRef]
- Shimada, E.; Ahsan, F.M.; Nili, M.; Huang, D.; Atamdede, S.; TeSlaa, T.; Case, D.; Yu, X.; Gregory, B.D.; Perrin, B.J.; et al. PNPase Knockout Results in MtDNA Loss and an Altered Metabolic Gene Expression Program. PLoS ONE 2018, 13, e0200925. [Google Scholar] [CrossRef]
- Saleem, A.; Safdar, A.; Kitaoka, Y.; Akhtar, M.; Marquez, O.; Tarnopolsky, M. Polymerase Gamma Mutator (PolG) Mice Rely on Increased Glycolytic Flux for Energy Production. FASEB J. 2015, 29, 947.24. [Google Scholar] [CrossRef]
- Zhou, B.; Caudal, A.; Tang, X.; Chavez, J.D.; McMillen, T.S.; Keller, A.; Villet, O.; Zhao, M.; Liu, Y.; Ritterhoff, J.; et al. Upregulation of Mitochondrial ATPase Inhibitory Factor 1 (ATPIF1) Mediates Increased Glycolysis in Mouse Hearts. J. Clin. Investig. 2022, 132, 155333. [Google Scholar] [CrossRef]
- Yellen, G. Fueling Thought: Management of Glycolysis and Oxidative Phosphorylation in Neuronal Metabolism. J. Cell Biol. 2018, 217, 2235. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial Dysfunction and Oxidative Damage in Parkin-Deficient Mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef]
- Woodall, B.P.; Orogo, A.M.; Najor, R.H.; Cortez, M.Q.; Moreno, E.R.; Wang, H.; Divakaruni, A.S.; Murphy, A.N.; Gustafsson, Å.B. Parkin Does Not Prevent Accelerated Cardiac Aging in Mitochondrial DNA Mutator Mice. JCI Insight 2019, 4, 127713. [Google Scholar] [CrossRef]
- Corsa, C.A.S.; Pearson, G.L.; Renberg, A.; Askar, M.M.; Vozheiko, T.; MacDougald, O.A.; Soleimanpour, S.A. The E3 Ubiquitin Ligase Parkin Is Dispensable for Metabolic Homeostasis in Murine Pancreatic β Cells and Adipocytes. J. Biol. Chem. 2019, 294, 7296–7307. [Google Scholar] [CrossRef] [PubMed]
- Filograna, R.; Gerlach, J.; Choi, H.-N.; Rigoni, G.; Barbaro, M.; Oscarson, M.; Lee, S.; Tiklova, K.; Ringnér, M.; Koolmeister, C.; et al. Parkin Is Not Required to Sustain OXPHOS Function in Adult Mammalian Tissues. bioRxiv 2023, 10, 93. [Google Scholar] [CrossRef]
- Villeneuve, L.M.; Stauch, K.L.; Purnell, P.S.; Fox, H.S. Proteomic and Functional Data Sets on Synaptic Mitochondria from Rats with Genetic Ablation of Parkin. Data Br. 2018, 20, 568. [Google Scholar] [CrossRef]
- Lichter, E.Z.; Trease, A.J.; Cooper, K.; Stauch, K.L.; Fox, H.S. Effects of Parkin on the Mitochondrial Genome in the Heart and Brain of Mitochondrial Mutator Mice. Adv. Biol. 2023, 7, 2300154. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Nikouee, A.; Sun, Y.; Zhang, Q.J.; Liu, Z.P.; Zang, Q.S. Evaluation of Parkin in the Regulation of Myocardial Mitochondria-Associated Membranes and Cardiomyopathy During Endotoxemia. Front. Cell Dev. Biol. 2022, 10, 796061. [Google Scholar] [CrossRef]
- Dhondt, I.; Petyuk, V.A.; Bauer, S.; Brewer, H.M.; Smith, R.D.; Depuydt, G.; Braeckman, B.P. Changes of Protein Turnover in Aging Caenorhabditis Elegans. Mol. Cell. Proteom. 2017, 16, 1621. [Google Scholar] [CrossRef]
- Rattan, S.I.S. Synthesis, Modifications, and Turnover of Proteins during Aging. Exp. Gerontol. 1996, 31, 33–47. [Google Scholar] [CrossRef]
- Ryazanov, A.G.; Nefsky, B.S. Protein Turnover Plays a Key Role in Aging. Mech. Ageing Dev. 2002, 123, 207–213. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef] [PubMed]
- Fornasiero, E.F.; Mandad, S.; Wildhagen, H.; Alevra, M.; Rammner, B.; Keihani, S.; Opazo, F.; Urban, I.; Ischebeck, T.; Sakib, M.S.; et al. Precisely Measured Protein Lifetimes in the Mouse Brain Reveal Differences across Tissues and Subcellular Fractions. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Paß, T.; Wiesner, R.J.; Pla-Martín, D. Selective Neuron Vulnerability in Common and Rare Diseases—Mitochondria in the Focus. Front. Mol. Biosci. 2021, 8, 676187. [Google Scholar] [CrossRef]
- Trease, A.J.; George, J.W.; Roland, N.J.; Lichter, E.Z.; Emanuel, K.; Totusek, S.; Fox, H.S.; Stauch, K.L. Hyperphosphorylated Human Tau Accumulates at the Synapse, Localizing on Synaptic Mitochondrial Outer Membranes and Disrupting Respiration in a Mouse Model of Tauopathy. Front. Mol. Neurosci. 2022, 15, 852368. [Google Scholar] [CrossRef]
- Graham, J.M. Purification of a Crude Mitochondrial Fraction by Density-Gradient Centrifugation. Curr. Protoc. Cell Biol. 1999, 4, 3.4.1–3.4.22. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Jarvie, P.E.; Robinson, P.J. A Rapid Percoll Gradient Procedure for Preparation of Synaptosomes. Nat. Protoc. 2008, 3, 1718–1728. [Google Scholar] [CrossRef]
- Choi, S.W.; Gerencser, A.A.; Nicholls, D.G. Bioenergetic Analysis of Isolated Cerebrocortical Nerve Terminals on a Microgram Scale: Spare Respiratory Capacity and Stochastic Mitochondrial Failure. J. Neurochem. 2009, 109, 1179. [Google Scholar] [CrossRef] [PubMed]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Aging Synaptic Mitochondria Exhibit Dynamic Proteomic Changes While Maintaining Bioenergetic Function. Aging 2014, 6, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.W.; Brand, M.D.; Petrosyan, S.; Ashok, D.; Elorza, A.A.; Ferrick, D.A.; Murphy, A.N. High Throughput Microplate Respiratory Measurements Using Minimal Quantities of Isolated Mitochondria. PLoS ONE 2011, 6, e21746. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Filter-Aided Sample Preparation: The Versatile and Efficient Method for Proteomic Analysis. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 585, pp. 15–27. [Google Scholar]
- Scopes, R.K. Measurement of Protein by Spectrophotometry at 205 Nm. Anal. Biochem. 1974, 59, 277–282. [Google Scholar] [CrossRef]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Quantitative Proteomics of Synaptic and Nonsynaptic Mitochondria: Insights for Synaptic Mitochondrial Vulnerability. J. Proteome Res. 2014, 13, 2620–2636. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.D.; Goode, R.J.A.; Huang, C.; Powell, D.R.; Schittenhelm, R.B. Lfq-Analyst: An Easy-To-Use Interactive Web Platform to Analyze and Visualize Label-Free Proteomics Data Preprocessed with Maxquant. J. Proteome Res. 2019, 19, 204–211. [Google Scholar]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 28 December 2021).
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Hoopmann, M.R.; Finney, G.L.; MacCoss, M.J. High Speed Data Reduction, Feature Detection, and MS/MS Spectrum Quality Assessment of Shotgun Proteomics Datasets Using High Resolution Mass Spectrometry. Anal. Chem. 2007, 79, 5620. [Google Scholar] [CrossRef] [PubMed]
- Hoopmann, M.R.; Maccoss, M.J.; Moritz, R.L. Identification of Peptide Features in Precursor Spectra Using Hardklör and Krönik. Curr. Protoc. Bioinforma. 2012, 37, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, E.J.; Hoopmann, M.R.; MacLean, B.; MacCoss, M.J. Comparison of Database Search Strategies for High Precursor Mass Accuracy MS/MS Data. J. Proteome Res. 2010, 9, 1138. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An Open-Source MS/MS Sequence Database Search Tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-Supervised Learning for Peptide Identification from Shotgun Proteomics Datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Klammer, A.A.; Käli, L.; MacCoss, M.J.; Noble, W.S. Rapid and Accurate Peptide Identification from Tandem Mass Spectra. J. Proteome Res. 2008, 7, 3022. [Google Scholar] [CrossRef] [PubMed]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline Ecosystem: Informatics for Quantitative Mass Spectrometry Proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trease, A.J.; Totusek, S.; Lichter, E.Z.; Stauch, K.L.; Fox, H.S. Mitochondrial DNA Instability Supersedes Parkin Mutations in Driving Mitochondrial Proteomic Alterations and Functional Deficits in Polg Mutator Mice. Int. J. Mol. Sci. 2024, 25, 6441. https://doi.org/10.3390/ijms25126441
Trease AJ, Totusek S, Lichter EZ, Stauch KL, Fox HS. Mitochondrial DNA Instability Supersedes Parkin Mutations in Driving Mitochondrial Proteomic Alterations and Functional Deficits in Polg Mutator Mice. International Journal of Molecular Sciences. 2024; 25(12):6441. https://doi.org/10.3390/ijms25126441
Chicago/Turabian StyleTrease, Andrew J., Steven Totusek, Eliezer Z. Lichter, Kelly L. Stauch, and Howard S. Fox. 2024. "Mitochondrial DNA Instability Supersedes Parkin Mutations in Driving Mitochondrial Proteomic Alterations and Functional Deficits in Polg Mutator Mice" International Journal of Molecular Sciences 25, no. 12: 6441. https://doi.org/10.3390/ijms25126441
APA StyleTrease, A. J., Totusek, S., Lichter, E. Z., Stauch, K. L., & Fox, H. S. (2024). Mitochondrial DNA Instability Supersedes Parkin Mutations in Driving Mitochondrial Proteomic Alterations and Functional Deficits in Polg Mutator Mice. International Journal of Molecular Sciences, 25(12), 6441. https://doi.org/10.3390/ijms25126441