
Support Vector Machine-Based Formula for Detecting Suspected α Thalassemia Carriers: A Path toward Universal Screening


Abstract
:1. Introduction
2. Results
2.1. Comparative Results of the Formulas
2.2. Analysis of Samples Suspected to Have a Diagnosis of Iron Deficiency Anemia
2.3. Comparison of the Results from the Two Common α Globin Mutations Found
3. Discussion
4. Methods
4.1. Red Blood Count Analysis
4.2. Molecular Analysis
4.3. Analysis of the Data Using Mathematical Formulas
4.4. The Support Vector Machine (SVM) Algorithm
4.5. Analysis of the Red Blood Count Indices and HPLC Results in the Two Most Common α Gene Defects
4.6. Data Analysis
4.7. Ethics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higgs, D.R. The Molecular Basis of -Thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011718. [Google Scholar] [CrossRef] [PubMed]
- Koren, A. The continuing global challenges of treating patients with beta-thalassemia. Br. J. Haematol. 2023, 201, 183–184. [Google Scholar] [CrossRef]
- Koren, A.; Profeta, L.; Zalman, L.; Palmor, H.; Levin, C.; Zamir, R.B.; Shalev, S.; Blondheim, O. Prevention of Β Thalassemia in Northern Israel—A cost-benefit analysis. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014012. [Google Scholar] [CrossRef]
- Musallam, K.M.; Lombard, L.; Kistler, K.D.; Arregui, M.; Gilroy, K.S.; Chamberlain, C.; Zagadailov, E.; Ruiz, K.; Taher, A.T. Epidemiology of clinically significant forms of alpha- and beta-thalassemia: A global map of evidence and gaps. Am. J. Hematol. 2023, 98, 1436–1451. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Zalman, L.; Palmor, H.; Zamir, R.B.; Levin, C.; Openheim, A.; Daniel-Spiegel, E.; Shalev, S.; Filon, D. Sickle cell anemia in northern Israel: Screening and prevention. Isr. Med. Assoc. J. 2009, 11, 229–234. [Google Scholar]
- Koren, A.; Zalman, L.; Palmor, H.; Ekstein, E.; Schneour, Y.; Schneour, A.; Shalev, S.; Rachmilewitz, E.A.; Filon, D.; Openhaim, A. The prevention programs for beta thalassemia in the Jezreel and Eiron valleys: Results of fifteen years experience. Harefuah 2002, 141, 938–943, 1210. [Google Scholar]
- Piel, F.B.; Weatherall, D.J. The α-Thalassemias. N. Engl. J. Med. 2014, 371, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Goh, L.P.W.; Chong, E.T.J.; Lee, P.-C. Prevalence of Alpha(α)-Thalassemia in Southeast Asia (2010–2020): A Meta-Analysis Involving 83,674 Subjects. Int. J. Environ. Res. Public Health 2020, 17, 7354. [Google Scholar] [CrossRef]
- Galanello, R.; Cao, A. Alpha-thalassemia. Genet. Med. 2011, 13, 83–88. [Google Scholar] [CrossRef]
- Songdej, D.; Fucharoen, S. Alpha-Thalassemia: Diversity of Clinical Phenotypes and Update on the Treatment. Thalass. Rep. 2022, 12, 157–172. [Google Scholar] [CrossRef]
- Dehbozorgian, J.; Moghadam, M.; Daryanoush, S.; Haghpanah, S.; Fard, J.I.; Aramesh, A.; Shahsavani, A.; Karimi, M. Distribution of alpha-thalassemia mutations in Iranian population. Hematology 2015, 20, 359–362. [Google Scholar] [CrossRef]
- Keikhaei, B.; Slehi-Fard, P.; Shariati, G.; Khosravi, A. Genetics of Iranian Alpha-Thalassemia Patients: A Comprehensive Original Study. Biochem. Genet. 2018, 56, 506–521. [Google Scholar] [CrossRef]
- Liebhaber, S.A.; Kan, Y.W. Differentiation of the MRNA Transcripts Originating from the Al- and A2-Globin Loci in Normals and a-Thalassemics. J. Clin. Inwvest. 1981, 68, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Oron, V.; Filon, D.; Oppenheim, A.; Rund, D. Severe thalassaemia intermedia caused by interaction of homozygosity for α-globin gene triplication with heterozygosity for β thalassaemia. Br. J. Haematol. 1994, 86, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Amendolia, S.; Cossu, G.; Ganadu, M.; Golosio, B.; Masala, G.; Mura, G. A comparative study of K-Nearest Neighbour, Support Vector Machine and Multi-Layer Perceptron for Thalassemia screening. Chemom. Intell. Lab. Syst. 2003, 69, 13–20. [Google Scholar] [CrossRef]
- Bordbar, E.; Taghipour, M.; Zucconi, B.E. Reliability of Different RBC Indices and Formulas in Discriminating between β-Thalassemia Minor and other Microcytic Hypochromic Anemia. Mediterr. J. Hematol. Infect. Dis. 2015, 7, e2015022. [Google Scholar] [CrossRef] [PubMed]
- Ehsani, M.; Shahgholi, E.; Rahimineja, M.; Seighali, F.; Rashidi, A. A New Index for Discrimination Between Iron Deficiency Anemia and Beta-Thalassemia Minor: Results in 284 Patients. Pak. J. Biol. Sci. 2009, 12, 473–475. [Google Scholar] [CrossRef] [PubMed]
- England, J.; Fraser, P. Discrimination between iron-deficiency and heterozygous-thalassqmia syndromes in differential diagnosis of microcytosis. Lancet 1979, 313, 145–148. [Google Scholar] [CrossRef] [PubMed]
- England, J.; Fraser, P. Differentiation of iron deficiency from thalassæmia trait by routine blood-count. Lancet 1973, 301, 449–452. [Google Scholar] [CrossRef]
- Rahim, F.; Keikhaei, B. Better Differential Diagnosis of Iron Deficiency Anemia from Beta-Thalassemia Trait. Turk. J. Hematol. 2009, 26, 138–145. [Google Scholar]
- Green, R.; King, R. A new red cell discriminant incorporating volume dispersion for differentiating iron deficiency anemia from thalassemia minor. Blood Cells 1989, 15, 481. [Google Scholar] [PubMed]
- Getta, H.A.; Yasseen, H.A.; Said, H.M. Hi & Ha, Are New Indices in Differentiation between Iron Deficiency Anemia and Beta-Thalassaemia Trait/A Study in Sulaimani City-Kurdistan/Iraq. J. Dent. Med. Sci. 2015, 14, 67–72. [Google Scholar]
- Mentzer, W. Differentiation of iron deficiency from thalassæmia trait. Lancet 1973, 301, 882. [Google Scholar] [CrossRef] [PubMed]
- Ricerca, B.M.; Storti, S.; D’Onofrio, G.; Mancini, S.; Vittori, M.; Campisi, S.; Mango, G.; Bizzi, B. Differentiation of iron deficiency from thalassaemia trait: A new approach. Haematologica 1987, 72, 409–413. [Google Scholar]
- Artaza, J.R.; Carbia, C.D.; Ceballo, M.F.; Díaz, N.B. Red cell distribution width (RDW): Its use in the characterization of microcytic and hypochromic anemias. Medicina 1999, 59, 17–22. [Google Scholar]
- Roth, I.L.; Lachover, B.; Koren, G.; Levin, C.; Zalman, L.; Koren, A. Detection of β thalassemia carriers by red cell parameters obtained from automatic counters using mathematical formulas. Mediterr. J. Hematol. Infect. Dis. 2017, 10, 2018008. [Google Scholar] [CrossRef] [PubMed]
- Schriever, H.; Srivastava, P. Differentiation of thalassæmia minor from iron deficiency. Lancet 1973, 302, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Shine, I.; Lal, S. A strategy to detect β-thalassæmia minor. Lancet 1977, 309, 692–694. [Google Scholar] [CrossRef] [PubMed]
- Sirachainan, N.; Iamsirirak, P.; Charoenkwan, P.; Kadegasem, P.; Wongwerawattanakoon, P.; Sasanakul, W.; Chansatitporn, N.; Chuansumrit, A. New Mathematical Formula for Differentiating Thalassemia Trait and Iron Deficiency Anemia in Thalas-semia Prevalent Area: A Study in Healthy School-Age Children. Southeast Asian J. Trop. Med. Public Health 2014, 45, 174–182. [Google Scholar] [PubMed]
- Sirdah, M.; Tarazi, I.; AL Najjar, E.; AL Haddad, R. Evaluation of the diagnostic reliability of different RBC indices and formulas in the differentiation of the β-thalassaemia minor from iron deficiency in Palestinian population. Int. J. Lab. Hematol. 2008, 30, 324–330. [Google Scholar] [CrossRef]
- Urrechaga, E.; Hoffmann, J.J. Critical appraisal of discriminant formulas for distinguishing thalassemia from iron deficiency in patients with microcytic anemia. Clin. Chem. Lab. Med. 2017, 55, 1582–1591. [Google Scholar] [CrossRef]
- Tang, H.; Yu, R.; Yu, Z.; Xi, H. Comparison of screening indicators for different types of thalassemia carriers in Hunan Province. J. Med. Biochem. 2023, 43, 281–289. [Google Scholar] [CrossRef]
- d’Onofrio, G.; Zini, G.; Ricerca, B.M.; Mancini, S.; Mango, G. Automated Measurement of Red Blood Cell Microcytosis and Hy-pochromia in Iron Deficiency and Beta-Thalassemia Trait. Arch. Pathol. Lab. Med. 1992, 116, 84–89. [Google Scholar] [PubMed]
- Koren, A.; Levin, C.; Zalman, L.; Palmor, H.; Filon, D.; Chubar, E.; Resnitzky, P.; Bennett, M. Hb TAYBE: Clinical and morphological findings IN 43 patients. Eur. J. Haematol. 2016, 97, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.J.; Urrechaga, E.; Aguirre, U. Discriminant indices for distinguishing thalassemia and iron deficiency in patients with microcytic anemia: A meta-analysis. Clin. Chem. Lab. Med. 2015, 53, 1883–1894. [Google Scholar] [CrossRef]
- Phirom, K.; Charoenkwan, P.; Shoombuatong, W.; Charoenkwan, P.; Sirichotiyakul, S.; Tongsong, T. DeepThal: A Deep Learning-Based Framework for the Large-Scale Prediction of the α+-Thalassemia Trait Using Red Blood Cell Parameters. J. Clin. Med. 2022, 11, 6305. [Google Scholar] [CrossRef] [PubMed]
- Hatton, C.; Wilkie, A.; Drysdale, H.; Wood, W.; Vickers, M.; Sharpe, J.; Ayyub, H.; Pretorius, I.; Buckle, V. Higgs Alpha-thalassemia caused by a large (62 kb) deletion upstream of the human alpha globin gene cluster. Blood 1990, 76, 221–227. [Google Scholar] [CrossRef]
- Liebhaber, S.A.; Griese, E.U.; Weiss, I.; Cash, F.E.; Ayyub, H.; Higgs, D.R.; Horst, J. Inactivation of human alpha-globin gene expression by a de novo deletion located upstream of the alpha-globin gene cluster. Proc. Natl. Acad. Sci. USA 1990, 87, 9431–9435. [Google Scholar] [CrossRef]
- Wilkie, A.O.M.; Lamb, J.; Harris, P.C.; Finney, R.D.; Higgs, D.R. A truncated human chromosome 16 associated with α thalassaemia is stabilized by addition of telomeric repeat (TTAGGG)n. Nature 1990, 346, 868–871. [Google Scholar] [CrossRef]
- Ribeiro, D.M.; Sonati, M.F. Regulation of Human Alpha-Globin Gene Expression and Alpha-Thalassemia. Genet. Mol. Res. 2008, 7, 1045–1053. [Google Scholar] [CrossRef]
- De Gobbi, M.; Viprakasit, V.; Hughes, J.R.; Fisher, C.; Buckle, V.J.; Ayyub, H.; Gibbons, R.J.; Vernimmen, D.; Yoshinaga, Y.; de Jong, P.; et al. A Regulatory SNP Causes a Human Genetic Disease by Creating a New Transcriptional Promoter. Science 2006, 312, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Akhavan-Niaki, H.; Youssefi Kamangari, R.; Banihashemi, A.; Kholghi Oskooei, V.; Azizi, M.; Tamaddoni, A.; Sedaghat, S.; Vakili, M.; Mahmoudi Nesheli, H.; Shabani, S. Hematologic Features of Alpha Thalassemia Carriers. Int. J. Mol. Cell Med. 2012, 1, 162–167. [Google Scholar] [PubMed]
- Oron-Karni, V.; Filon, D.; Shifrin, Y.; Fried, E.; Pogrebijsky, G.; Oppenheim, A.; Rund, D. Diversity of alpha-globin mutations and clinical presentation of alpha-thalassemia in Israel. Am. J. Hematol. 2000, 65, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Shaulov, A.; Filon, D.; Rund, D. Haplotype analysis of α-thalassemia chromosomes reveals heterogeneity and multiple founders in Ashkenazi Jews. Eur. J. Med. Genet. 2016, 59, 555–558. [Google Scholar] [CrossRef]
- Kattamis, A.C.; Camaschella, C.; Sivera, P.; Surrey, S.; Fortina, P. Human α-Thalassemia Syndromes: Detection of Molecular Defects. Am. J. Hematol. 1996, 53, 81–91. [Google Scholar] [CrossRef]
- Embury, S.H.; A Miller, J.; Dozy, A.M.; Kan, Y.W.; Chan, V.; Todd, D. Two different molecular organizations account for the single alpha-globin gene of the alpha-thalassemia-2 genotype. J. Clin. Investig. 1980, 66, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Gilad, O.; Steinberg-Shemer, O.; Dgany, O.; Krasnov, T.; Noy-Lotan, S.; Tamary, H.; Yacobovich, J. Alpha-Thalassemia Carrier due to -alpha(3.7) Deletion: Not So Silent. Acta Haematol. 2020, 143, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.T. Variable Clinical Phenotypes of α-Thalassemia Syndromes. Sci. World J. 2009, 9, 615–625. [Google Scholar] [CrossRef]
- Arnon, S.; Tamary, H.; Dgany, O.; Litmanovitz, I.; Regev, R.; Bauer, S.; Dolfin, T.; Yacobovich, J.; Wolach, B.; Jaber, L. Hydrops fetalis associated with homozygosity for hemoglobin Taybe (α 38/39 THR deletion) in newborn triplets. Am. J. Hematol. 2004, 76, 263–266. [Google Scholar] [CrossRef]
- Burges, C.J.C. A Tutorial on Support Vector Machines for Pattern Recognition. Data Min. Knowl. Discov. 1998, 2, 121–167. [Google Scholar] [CrossRef]

{kind=link}
| Diagnosis | Non β Thalassemia Trait Mean ± SD (Range) | β Thalassemia Trait Mean ± SD (Range) | α Thalassemia Trait and/or Suspected α Trait Mean ± SD (Range) | p-Value between α and β Trait |
|---|---|---|---|---|
| Number of individuals (%) | 18,572 (81.3) | 2936 (12.8) | 1334 (5.9) | |
| RBC (×109/dL) | 4.2 ± 0.43 (2.17–7.67) | 5.42 ± 0.55 (3.21–7.81) | 4.87 ± 0.44 (3.48–6.34) | <0.001 |
| Hb (g/dL) | 11.75 ± 1.06 (9.00–19.2) | 10.65 ± 0.95 (9.00–15.4) | 11.51 ± 1.07 (9.0–15.6) | <0.001 |
| MCV (fl) | 85.9 ± 6.84 (34–125.9) | 63.14 ± 5.76 (48–91.5) | 73.56 ± 4.52 (53.3–91.4) | <0.001 |
| MCH (pg) | 28.16 ± 2.7 (16.2–40.7) | 19.71 ± 1.95 (14–31.3) | 23.71 ± 1.92 (16.5–29.9) | <0.001 |
| MCHC (g/dL) | 32.75 ± 1.76 (12.6–45.7) | 31.23 ± 1.73 (17.5–65) | 32.17 ± 1.68 (19.2–36.2) | <0.001 |
| RDW (%) | 14.96 ± 2.02 (10.1–36.4) | 16.39 ± 2.71 (12–22.8) | 15.16 ± 1.94 (12.1–28) | <0.001 |
| Hb F (%) | 0.34 ± 0.61 (0–14) | 2.12 ± 2.67 (0–38) | 0.4 ± 0.29 (0.1–1.9) | <0.001 |
| Hb A2 (%) | 1.12 ± 1.38 (0–3.4) | 5.6 ± 0.8 (3.5–8.8) | 2.6 ± 0.26 (2–3.3) | <0.001 |
| Diagnosis | α Thalassemia Trait + Mutation Mean ± SD (Range) | α Thalassemia Trait Suspected Mean ± SD (Range) | α Thalassemia Trait Normal Sequence Mean ± SD (Range) | p-Value (*) |
|---|---|---|---|---|
| Number of individuals (%) | 291 (21.8%) | 962 (72.12%) | 81 (6.07%) | |
| RBC (×109/dL) | 4.93 ± 0.46 (3.79–6.34) | 4.87 ± 0.43 (3.48–6.27) | 4.67 ± 0.36 (3.7–5.69) | <0.001 |
| Hb (g/dL) | 11.58 ± 0.97 (9.0–14.8) | 11.51 ± 1.1 (9.0–15.6) | 11.39 ± 0.93 (9.1–13.5) | NS |
| MCV (fl) | 72.95 ± 5.17 (53.3–83.9) | 73.55 ± 4.25 (57.8–91.4) | 75.1 ± 4.07 (62.2–85) | NS |
| MCH (pg) | 23.61 ± 2.1 (16.5–27.1) | 23.66 ± 1.84 (16.5–29.9) | 24.43 ± 1.81 (18.3–28.6) | NS |
| MCHC (g/dL) | 32.26 ± 1.72 (19.2–36.10) | 32.15 ± 1.33 (25.9–36.2) | 32.51 ± 1.38 (29.4–35.5) | NS |
| RDW (%) | 14.91 ± 1.8 (12.3–23.4) | 15.19 ± 1.87 (12.1–28.0) | 15.89 ± 2.35 (12.9–26.5) | 0.004 |
| Hb F (%) | 0.5 ± 0.52 (0.1–7.3) | 0.4 ± 0.29 (0.1–1.9) | 0.4 ± 0.34 (0–5.1) | NS |
| Hb A2 (%) | 2.6 ± 0.29 (1.3–3.6) | 2.6 ± 0.26 (2–3.3) | 2.6 ± 0.27 (0.8–3.3) | NS |
| No. | Study (Reference) | Formula | βThal Cut-Off | α Thal PPV | α Thal NPV | α Thal Specificity | α Thal Sensitivity | Percentile 75% | Percentile 95% (Lower Limit) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Srivastava [27] | MCH/RBC | <3.8 | 39.39 | 93.59 | 99.35 | 5.82 | 5.38 | 6.04 (5.97) |
| 2 |
| MCV-RBC-(5-Hb)-K* | <0 | 0.87 | 93.10 | 97.53 | 0.3 | 70.29 | 73.37 (72.96) |
| 3 | Mentzer [23] | MCV/RBC | <13 | 45.72 | 93.94 | 99.01 | 11.57 | 16.45 | 18.24 (18.09) |
| 4 | Shine and Lal [28] | MCV2 × MCH/100 | <1530 | 34.42 | 98.83 | 88.22 | 85.54 | 1466.58 | 1626.52 (1607.81) |
| 5 | Ricerca et al. [24] | RDW/RBC | <3.3 | 13.66 | 96.83 | 68.81 | 68.63 | 3.38 | 4.01 (3.95) |
| 6 | Green and King [21] | MCV2 × RDW/(Hb × 100) | <65 | 58.45 | 95.15 | 98.44 | 30.47 | 78.26 | 92.63 (90.12) |
| 7 | D’Onofrio et al. [33] | MCV/MCH | >0.9 | 6.74 | 0 | 0 | 100 | 3.18 | 3.34 (3.33) |
| 8 | Romero Artaza et al. [25] | RDW × MCV/RBC | <220 | 53.65 | 95.86 | 97.4 | 41.64 | 251.18 | 290.99 (285.58) |
| 9 | Sirdah et al. [30] | MCV-RBC-(3XHb) | <27 | 56.63 | 93.69 | 99.55 | 7.15 | 37.19 | 41.73 (41.11) |
| 10 | Ehsani et al. [17] | MCV-(10 × RBC) | <15 | 45.45 | 93.86 | 99.1 | 10.42 | 29.8 | 35.4 (34.8) |
| 11 | Sirachainan et al. [29] | 1.5 × Hb–0.05 × MCV | <14 | 6.08 | 91.95 | 32.69 | 60.39 | 14.58 | 16.01 (15.85) |
| 12 | Bordbar et al. [16] | [80-MCV]*[27-MCH] | >44.76 | 9.17 | 93.72 | 84.63 | 21.46 | 37.8 | 101.32 (93.44) |
| 13 | [20] | Hb × RDW × 100/RBC2 × MCHC | <21 | 63.44 | 95.2 | 98.72 | 30.89 | 25.14 | 29.3 (28.66) |
| 14 | Hisham Index [22] | MCH × RDW/RBC | <67 | 55.85 | 94.99 | 98.4 | 28.13 | 80.87 | 94.52 (92.59) |
| 15 | Hameed Index [22] | MCH × Hct × RDW/(RBC × Hb)2 | <220 | 6.73 | 0 | 0 | 100 | 4.99 | 6.52 (6.35) |
| 16 | Amendolia et al.—SVM [15] | SVM—(RBC, Hb, Hct, MCV) | 98.75 | 13.59 | 98.99 | 57.99 | 1.65 | 1.81 (1.79) | |
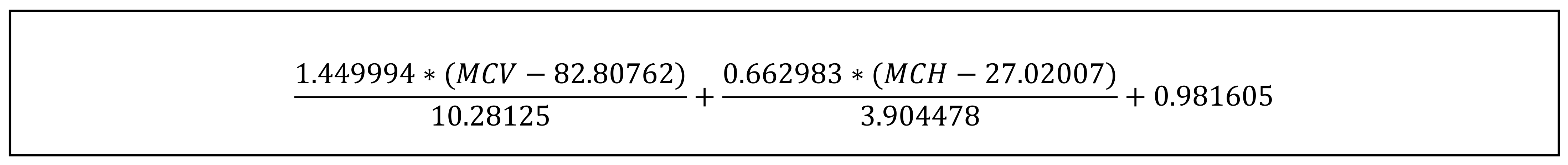
| 17 | SVM [26] | SVM (MCV and MCH) (Figure 1) | <0 | 21.52 | 99.93 | 73.67 | 99.33 | −0.23 | 0.29 (0.22) |
| α Globin Genetics | αα/-α3.7 kb Mean ± STD (Range) | -α3.7 kb/-α3.7 kb Mean ± STD (Range) | p * | αIVS I-1/αα Mean ± STD (Range) | αIVS I-1/αIVS I-1 Mean ± STD (Range) | p ** | αIVS I-1/ -α3.7 kb Mean ± STD (Range) |
|---|---|---|---|---|---|---|---|
| Number of individuals (%) | 134 | 24 | 97 | 8 | 7 | ||
| RBC (×109/dL) | 4.8 ± 0.4 (6.16–3.79) | 5.3 ± 0.49 (4.37–6.21) | <0.001 | 4.9 ± 0.36 (4.1–6.1) | 5.4 ± 0.57 (4.52–5.96) | 0.002 | 5.5 ± 0.56 (4.72–6.14) |
| Hb (g/dL) | 11.6 ± 0.96 (9.1–14.2) | 11.2 ± 1.01 (9.0– 13.3) | NS | 11.8 ± 0.88 (9.4–14.8) | 10.5 ± 1.19 (9.0–11.7) | <0.001 | 11.1 ± 0.66 (10.3–11.8) |
| MCV (fl) | 74.4 ± 4.35 (59–83.9) | 68.7 ±5.6 (58.7–77.1) | <0.001 | 73.7 ± 4.36 (53.3– 82.2) | 64.9 ± 4.66 (59.1–73.6) | <0.001 | 63.3 ± 1.41 (61.8–65.2) |
| MCH (pg) | 24.3 ± 1.75 (18–27.1) | 21.8 ± 2.11 (17.1–24.6) | <0.001 | 23.9 ± 1.63 (16.5– 26.5) | 19.5 ± 2.15 (17.9–24.7) | <0.001 | 20.2 ± 0.96 (19.2–21.9) |
| MCHC (g/dL) | 32.6 ± 1.18 (29.7–36.1) | 31.7 ± 1.52 (29.2–34.4) | <0.001 | 32.4 ± 1.2 (28.9–35.7) | 30.0 ± 2.2 (25.5–33.5) | <0.001 | 28.7 ± 6.66 (19.2–35.4) |
| RDW (%) | 14.8 ± 1.65 (12.5–21.7) | 15.5 ± 2.56 (13.3–23.4) | NS | 14.4 ± 1.29 (12.3– 20.1) | 16.3 ± 2.35 (12.6–20.5) | 0.001 | 18.8 ± 2.64 (16.5–22.6) |
| Hb F (%) | 0.5 ± 0.67 (0.1–7.3) | 0.4 ± 0.38 (0.2–1.6) | NS | 0.5 ± 0.35 (0.1–1.8) | 0.6 ± 0.26 (0.3–1.1) | NS | 0.5 ± 0.46 (0.2–1.1) |
| Hb A2 (%) | 2.7 ± 0.26 (1.7–3.6) | 2.6 ± 0.19 (2.2–2.9) | 0.09 | 2.7 ± 0.3 (1.6–3.1) | 2.3 ± 0.3 (1.7–2.7) | 0.002 | 2.8 ± 0.15 (2.5–2.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachover-Roth, I.; Peretz, S.; Zoabi, H.; Harel, E.; Livshits, L.; Filon, D.; Levin, C.; Koren, A. Support Vector Machine-Based Formula for Detecting Suspected α Thalassemia Carriers: A Path toward Universal Screening. Int. J. Mol. Sci. 2024, 25, 6446. https://doi.org/10.3390/ijms25126446
Lachover-Roth I, Peretz S, Zoabi H, Harel E, Livshits L, Filon D, Levin C, Koren A. Support Vector Machine-Based Formula for Detecting Suspected α Thalassemia Carriers: A Path toward Universal Screening. International Journal of Molecular Sciences. 2024; 25(12):6446. https://doi.org/10.3390/ijms25126446
Chicago/Turabian StyleLachover-Roth, Idit, Sari Peretz, Hiba Zoabi, Eitam Harel, Leonid Livshits, Dvora Filon, Carina Levin, and Ariel Koren. 2024. "Support Vector Machine-Based Formula for Detecting Suspected α Thalassemia Carriers: A Path toward Universal Screening" International Journal of Molecular Sciences 25, no. 12: 6446. https://doi.org/10.3390/ijms25126446
APA StyleLachover-Roth, I., Peretz, S., Zoabi, H., Harel, E., Livshits, L., Filon, D., Levin, C., & Koren, A. (2024). Support Vector Machine-Based Formula for Detecting Suspected α Thalassemia Carriers: A Path toward Universal Screening. International Journal of Molecular Sciences, 25(12), 6446. https://doi.org/10.3390/ijms25126446