
Testing the Simplified Molecular Dynamics Approach to Improve the Reproduction of ECD Spectra and Monitor Aggregation †
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
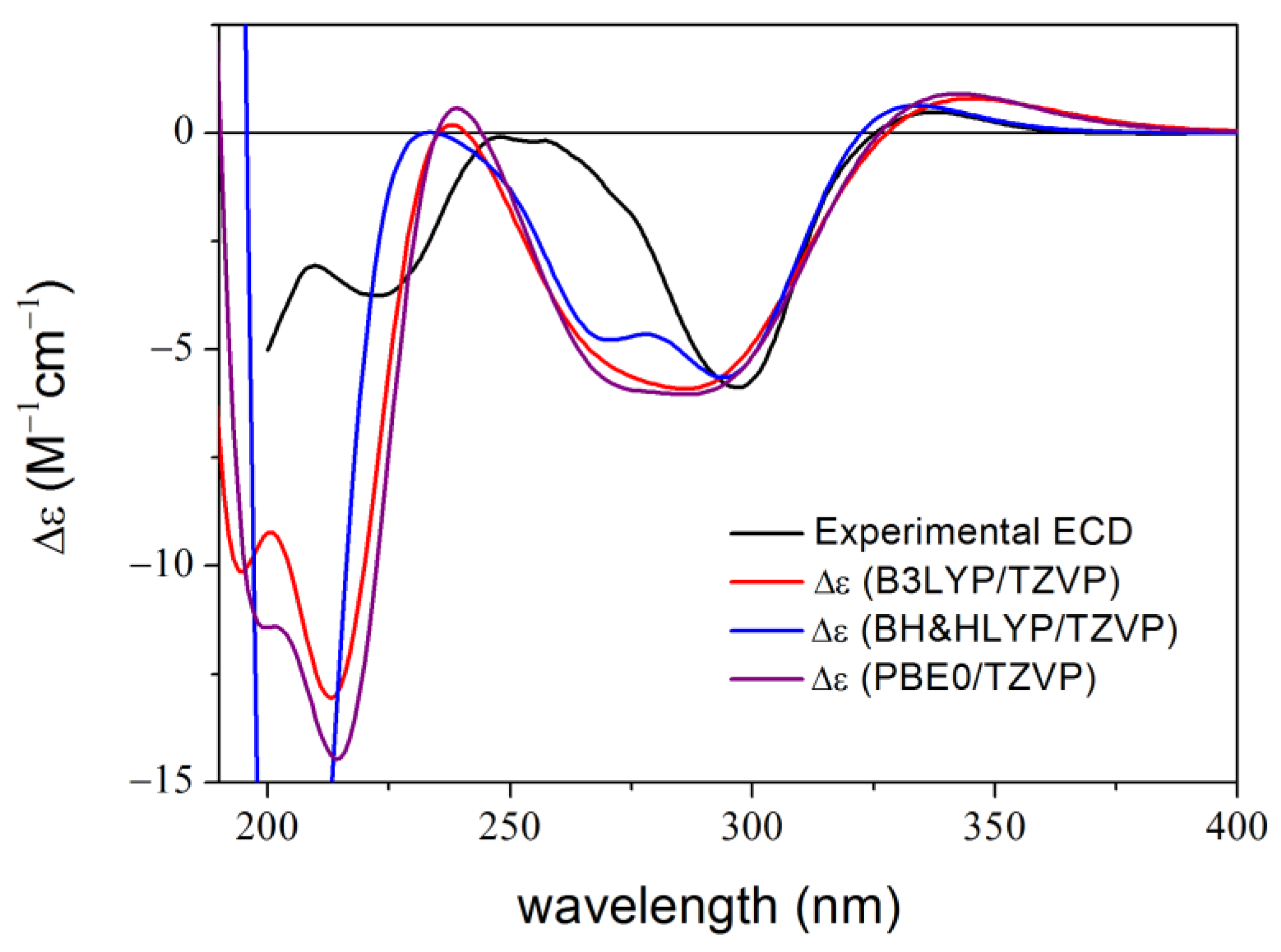
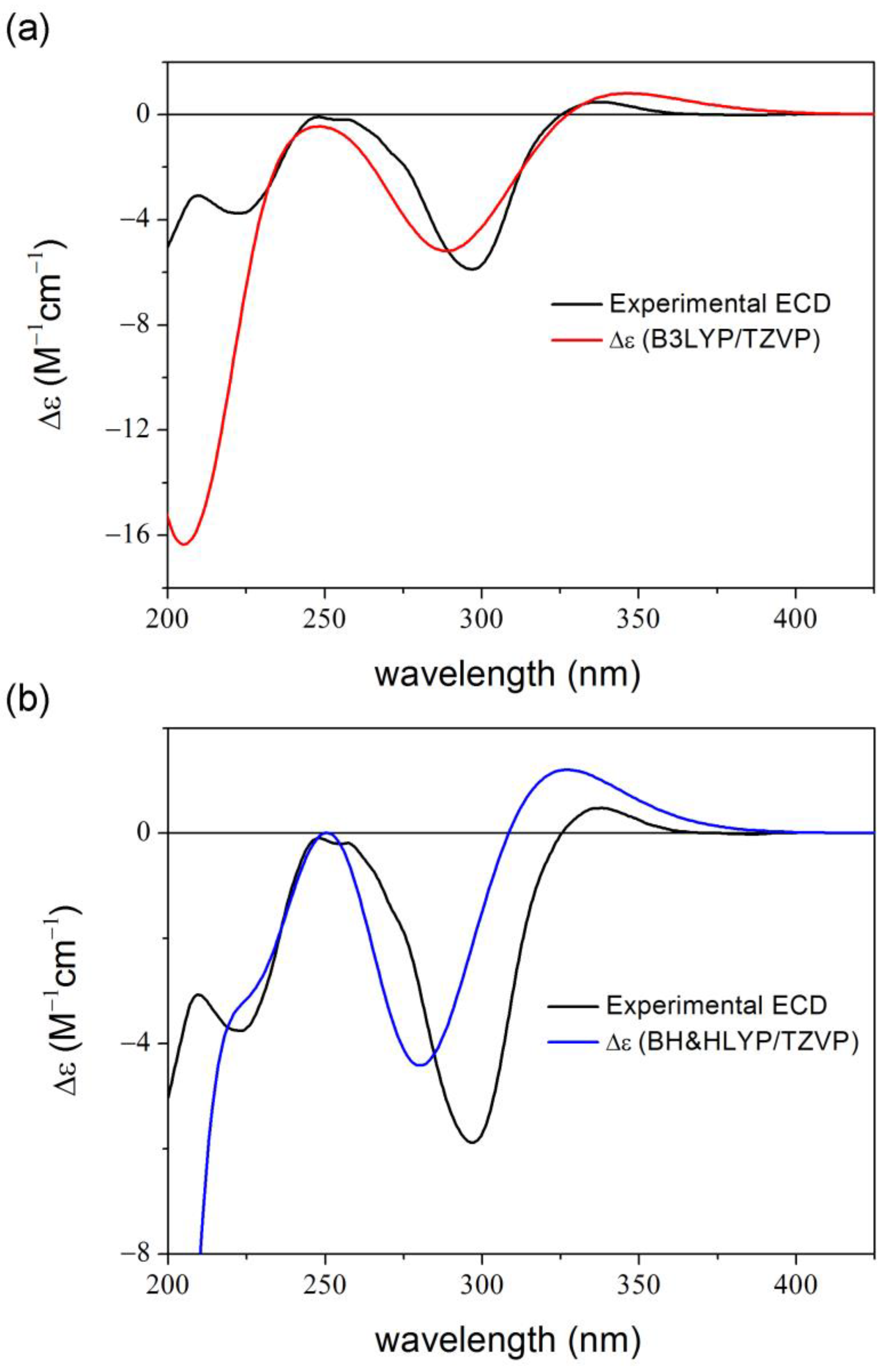
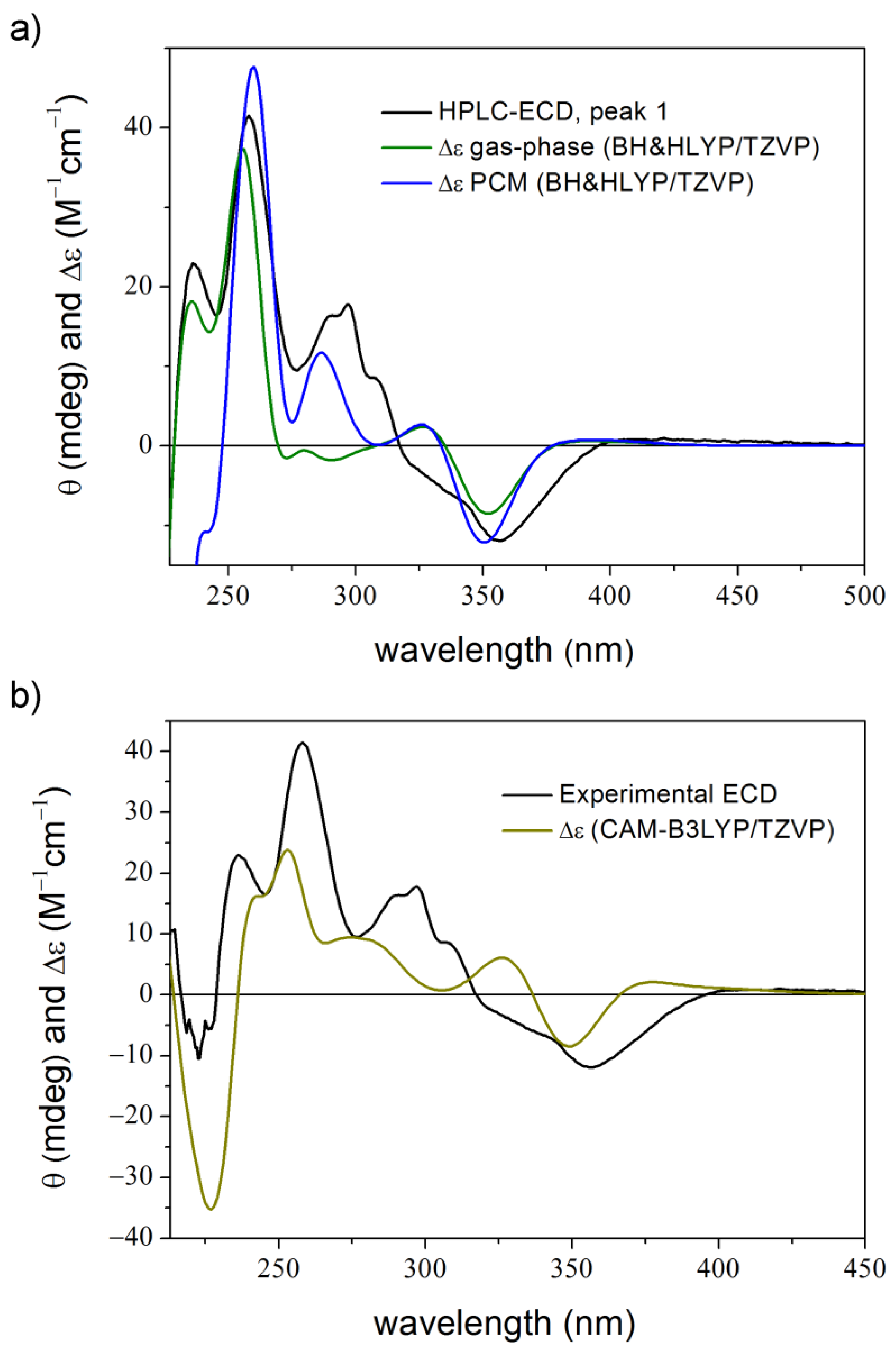
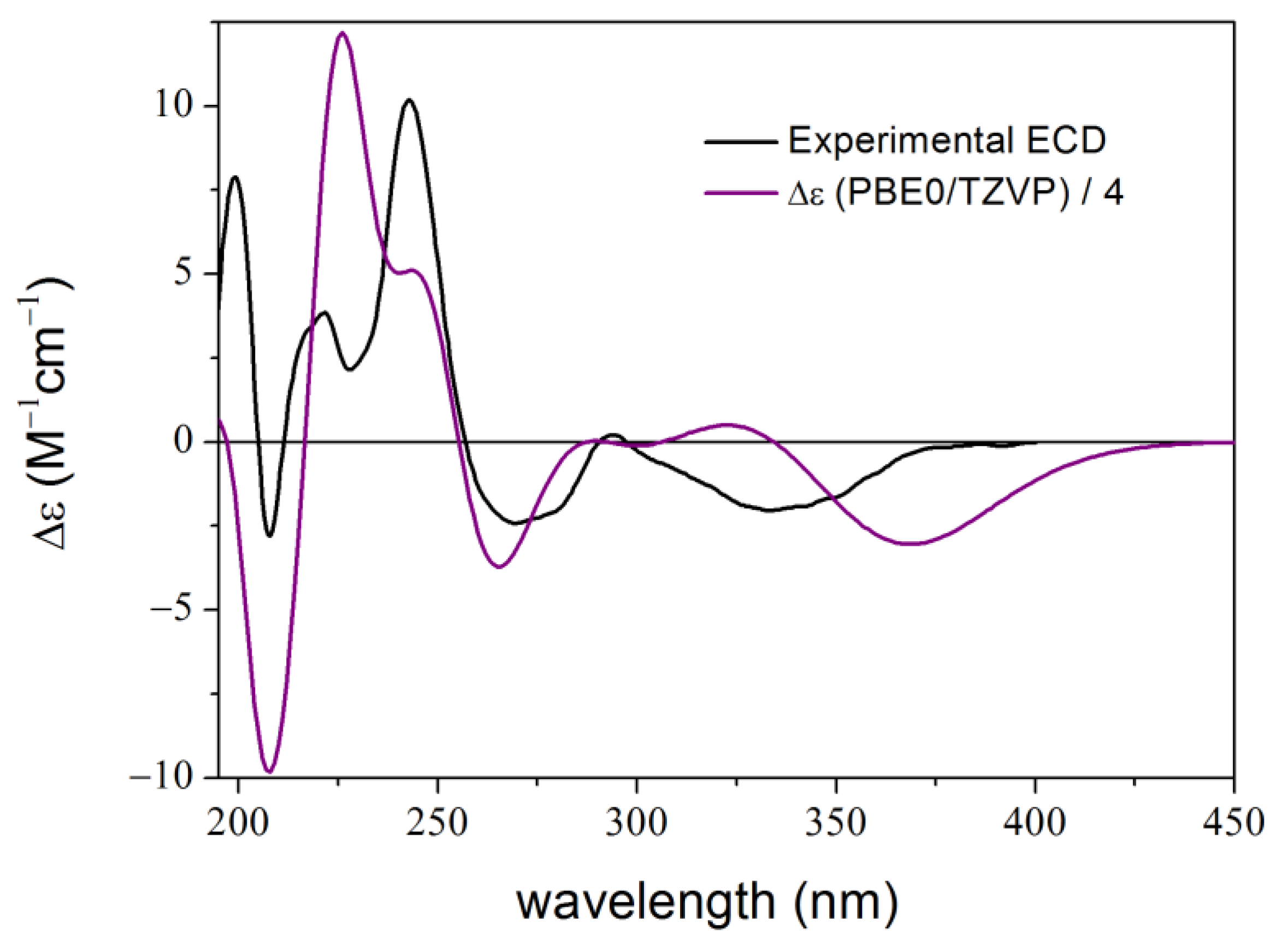
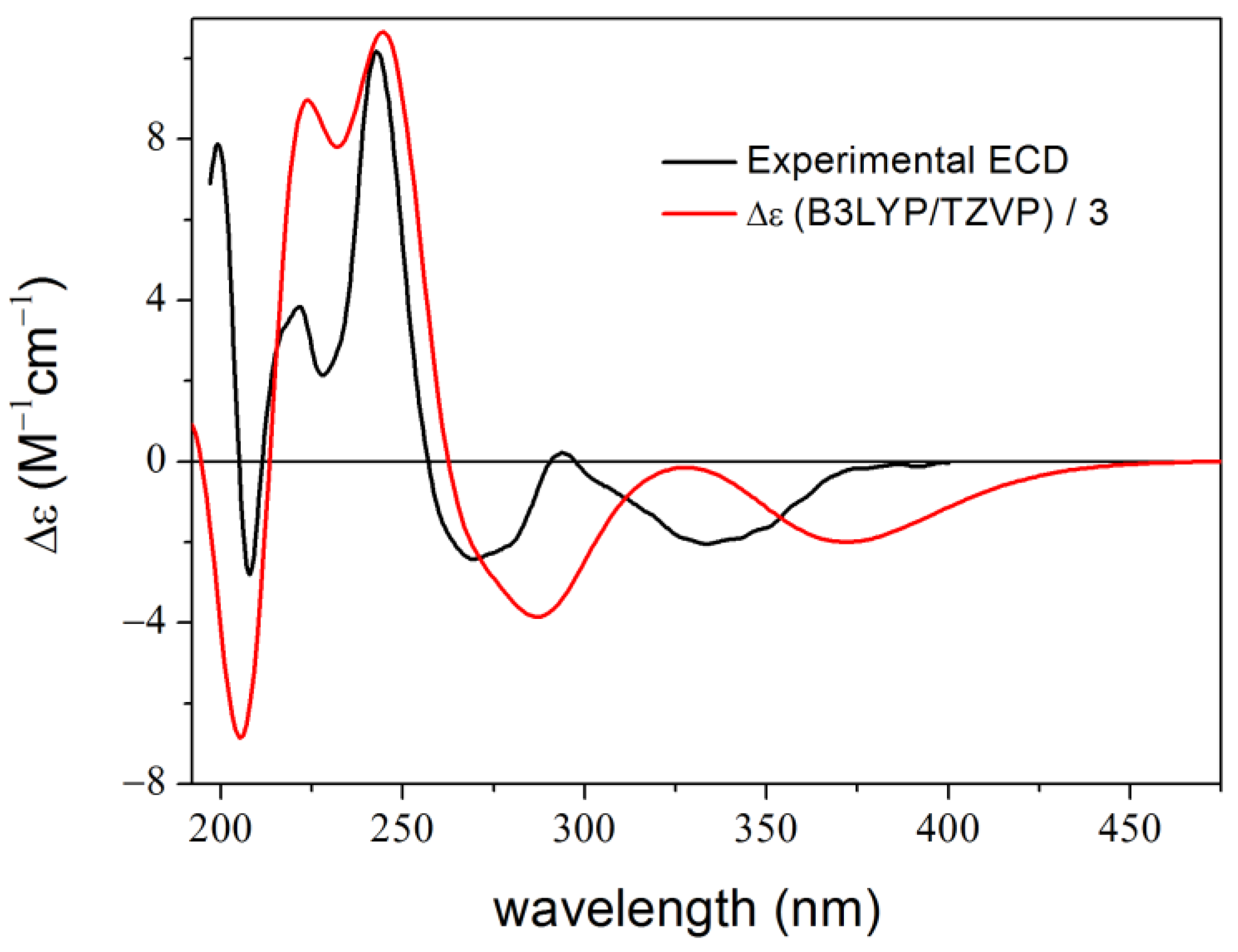
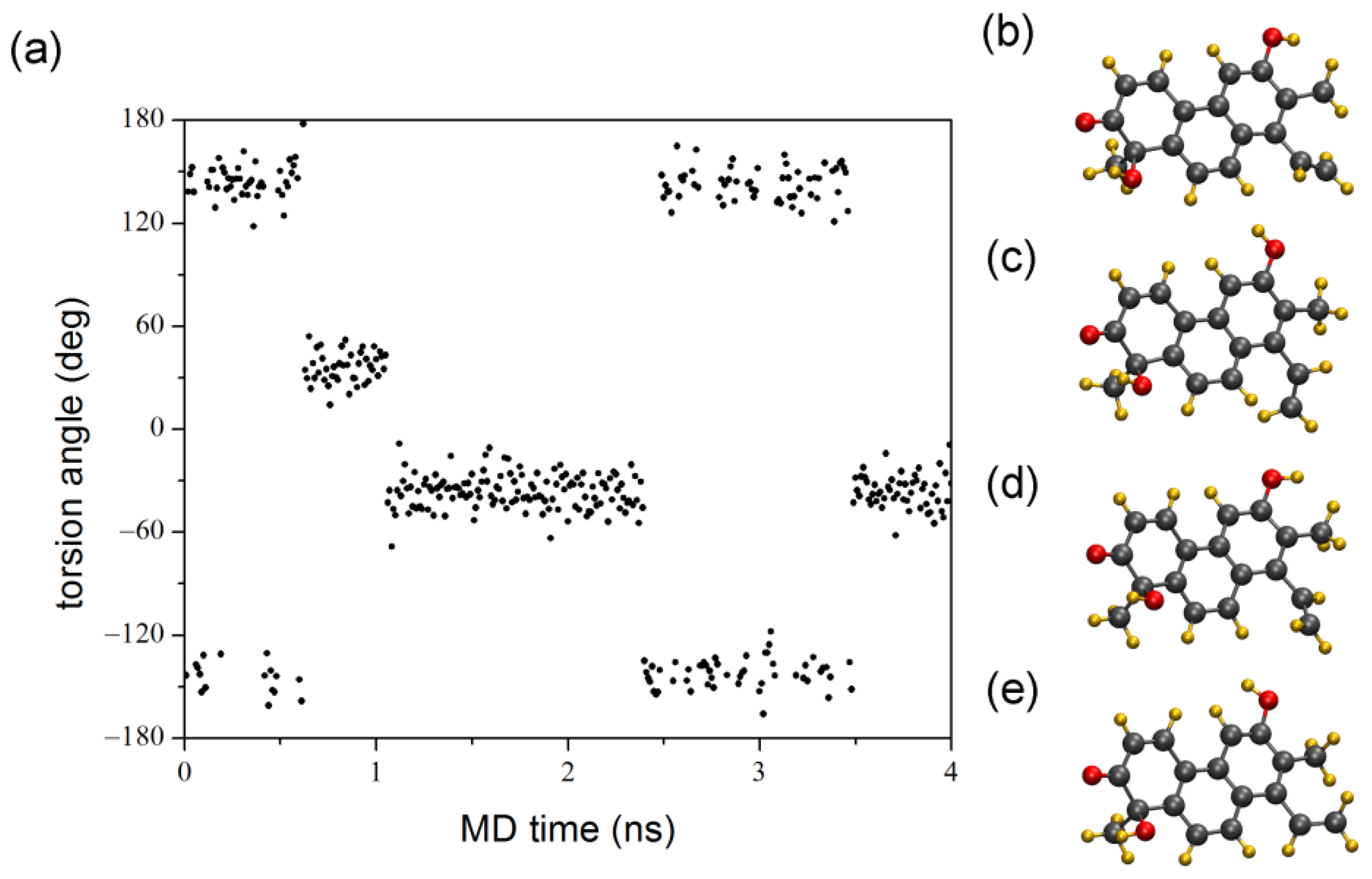
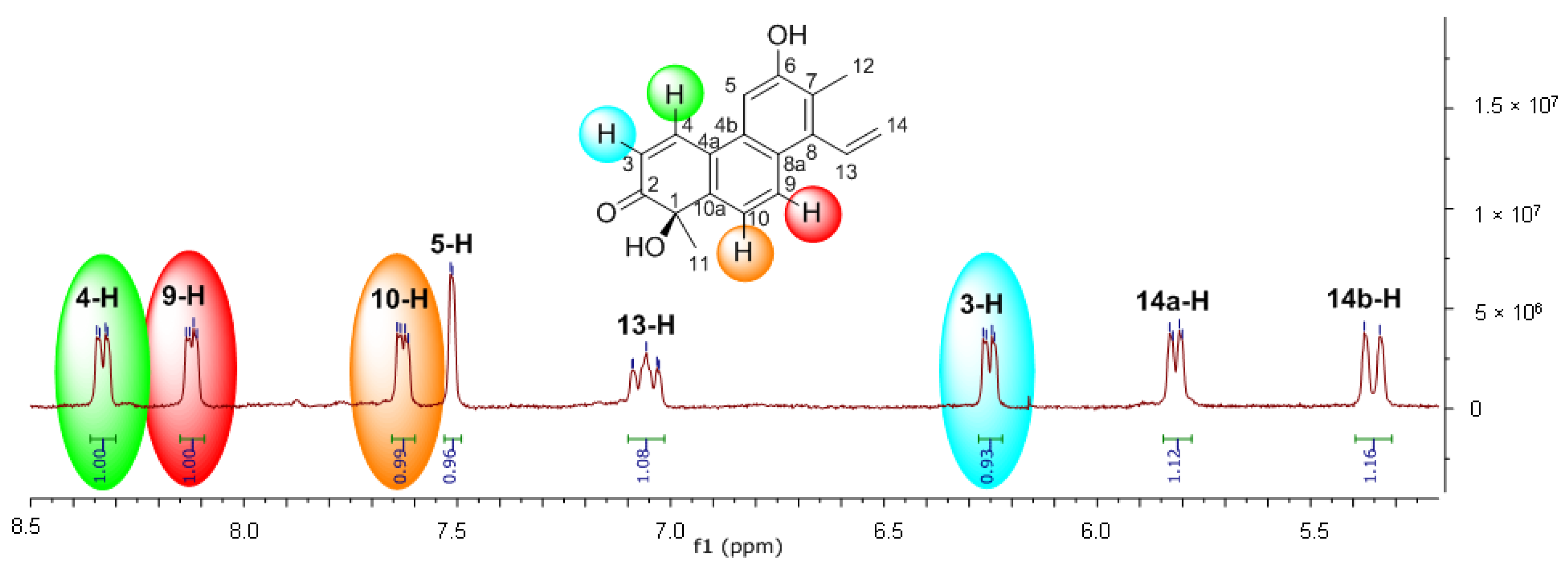
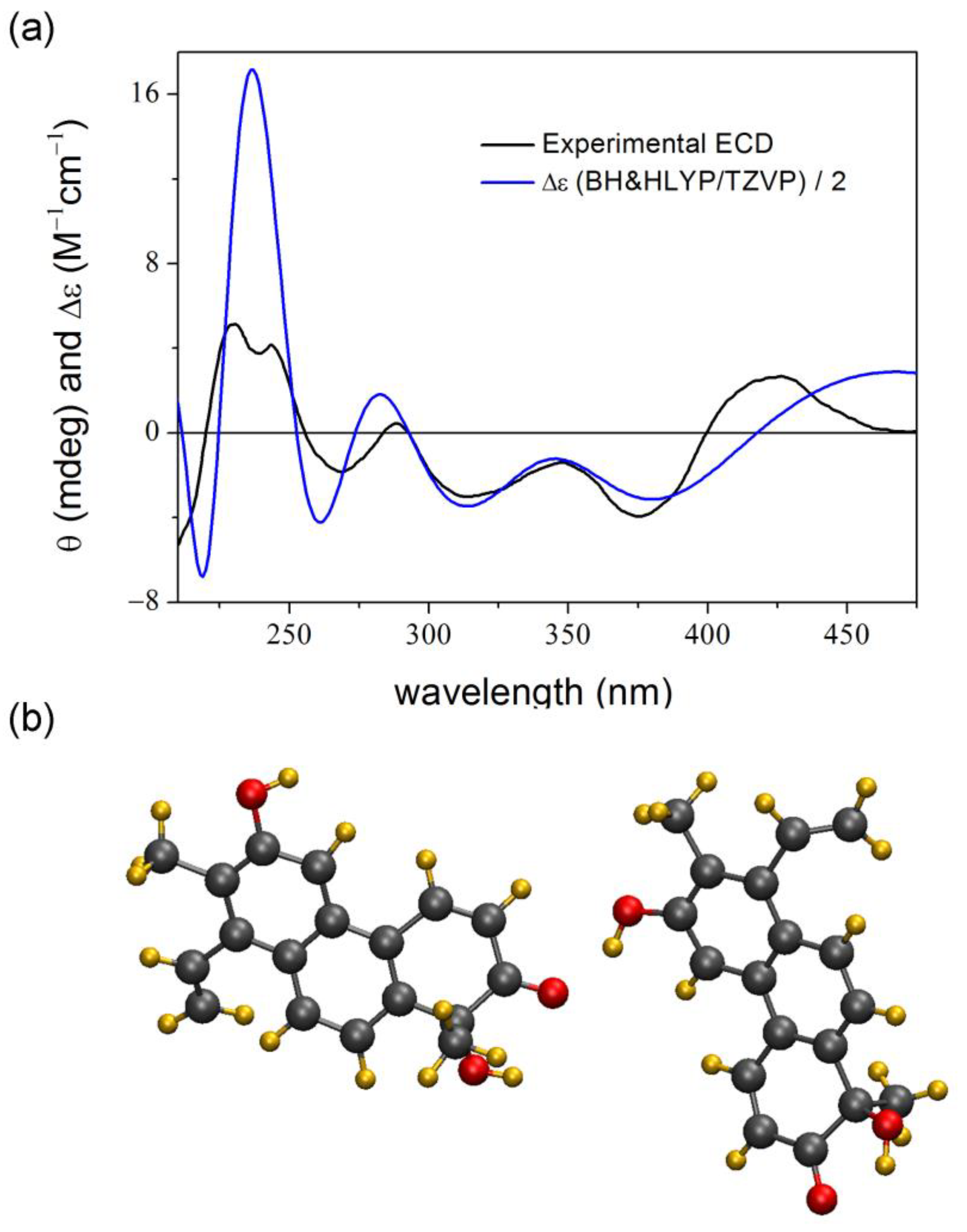
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Computational Section
3.2.1. MD Simulations
3.2.2. Analysis
3.2.3. ECD Calculations
3.2.4. Docking and Solute–Solvent Complex Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Superchi, S.; Scafato, P.; Górecki, M.; Pescitelli, G. Configuration Determination by Quantum Mechanical Calculation of Chiroptical Spectra: Basics and Applications to Fungal Metabolites. Curr. Med. Chem. 2018, 25, 287–320. [Google Scholar] [CrossRef]
- Mándi, A.; Kurtán, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef] [PubMed]
- Grauso, L.; Teta, R.; Esposito, G.; Menna, M.; Mangoni, A. Computational prediction of chiroptical properties in structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 1005–1030. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xu, D.X.; Mándi, A.; Kurtán, T.; Li, T.J.; Schulz, B.; Zhang, W. Structure, Absolute Configuration, and Conformational Study of 12-Membered Macrolides from the Fungus Dendrodochium sp. Associated with the Sea Cucumber Holothuria nobilis Selenka. J. Org. Chem. 2013, 78, 7030–7047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, L.; Wu, C.; Kurtán, T.; Mándi, A.; Liu, Y.; Gu, Q.; Zhu, T.; Guo, P.; Li, D. Penipyridones A−F, Pyridone Alkaloids from Penicillium funiculosum. J. Nat. Prod. 2016, 79, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Mándi, A.; Wu, J.; Kurtán, T. TDDFT-ECD and DFT-NMR studies of thaigranatins A–E and granatumin L isolated from Xylocarpus granatum. RSC Adv. 2020, 10, 32216–32224. [Google Scholar] [CrossRef]
- MacroModel. Schrödinger LLC. 2015. Available online: https://newsite.schrodinger.com/platform/products/macromodel/ (accessed on 7 April 2024).
- Goto, H.; Osawa, E. An efficient algorithm for searching low-energy conformers of cyclic and acyclic molecules. J. Chem. Soc. Perkin Trans. 2 1993, 2, 187–198. [Google Scholar] [CrossRef]
- Spartan, Wavefunction Inc. Irvine, CA, USA. Available online: www.wavefun.com/products/spartan.html (accessed on 7 April 2024).
- Agrafiotis, D.K.; Gibbs, A.C.; Zhu, F.; Izrailev, S.; Martin, E. Conformational Sampling of Bioactive Molecules: A Comparative Study. J. Chem. Inf. Model. 2007, 47, 1067–1086. [Google Scholar] [CrossRef]
- Kolossváry, I.; Guida, W.C. Low-mode conformational search elucidated: Application to C39H80 and flexible docking of 9-deazaguanine inhibitors into PNP. J. Comput. Chem. 1999, 20, 1671–1684. [Google Scholar] [CrossRef]
- Esposito, G.; Bourguet-Kondracki, M.L.; Mai, L.H.; Longeon, A.; Teta, R.; Meijer, L.; van Soest, R.; Mangoni, A.; Costantino, V. Chloromethylhalicyclamine B, a Marine-Derived Protein Kinase CK1δ/ε Inhibitor. J. Nat. Prod. 2016, 79, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Eszenyi, D.; Mándi, A.; Herczeg, M.; Bényei, A.; Komáromi, I.; Borbás, A. Synthesis of C-2- and C-3-Sulfonatomethyl O- and S-Glycosides by Horner–Wadsworth–Emmons Olefination. Eur. J. Org. Chem. 2016, 2016, 3884–3893. [Google Scholar] [CrossRef]
- Matsuo, K.; Namatame, H.; Taniguchi, M.; Gekko, K. Vacuum-Ultraviolet Electronic Circular Dichroism Study of Methyl α-D-Glucopyranoside in Aqueous Solution by Time-Dependent Density Functional Theory. J. Phys. Chem. A 2012, 116, 9996–10003. [Google Scholar] [CrossRef]
- Furuta, M.; Fujisawa, T.; Urago, H.; Eguchi, T.; Shingae, T.; Takahashi, S.; Blanch, E.W.; Unno, M. Raman optical activity of tetra-alanine in the poly(L-proline) II type peptide conformation. Phys. Chem. Chem. Phys. 2017, 19, 2078–2086. [Google Scholar] [CrossRef]
- Bartok, A.; Fehér, K.; Bodor, A.; Rákosi, K.; Tóth, G.K.; Kövér, K.E.; Panyi, G.; Varga, Z. An engineered scorpion toxin analogue with improved Kv1.3 selectivity displays reduced conformational flexibility. Sci. Rep. 2016, 5, 18397. [Google Scholar] [CrossRef]
- Rao, J.; Ashraf, S.; Tan, W.; van der Ven, A.T.; Gee, H.Y.; Braun, D.A.; Fehér, K.; George, S.P.; Esmaeilniakooshkghazi, A.; Choi, W.I.; et al. Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J. Clin. Investig. 2017, 127, 4257–4269. [Google Scholar] [CrossRef] [PubMed]
- Buyst, D.; Gheerardijn, V.; Fehér, K.; van Gasse, B.; van den Begin, J.; Martins, J.C.; Madder, A. Identification of a pKa-regulating motif stabilizing imidazole-modified double-stranded DNA. Nucleic Acids Res. 2015, 43, 51–62. [Google Scholar] [CrossRef]
- Kaltner, H.; Szabó, T.; Fehér, K.; André, S.; Balla, S.; Manning, J.C.; Szilágyi, L.; Gabius, H.J. Bivalent O-glycoside mimetics with S/disulfide/Se substitutions and aromatic core: Synthesis, molecular modeling and inhibitory activity on biomedically relevant lectins in assays of increasing physiological relevance. Bioorg. Med. Chem. 2017, 25, 3158–3170. [Google Scholar] [CrossRef] [PubMed]
- Fehér, K.; Matthews, R.P.; Kövér, K.E.; Naidoo, K.J.; Szilágyi, L. Conformational preferences in diglycosyl disulfides: NMR and molecular modeling studies. Carbohydr. Res. 2011, 346, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, S.F.; Zhang, Y.; Gao, Y.N. Simulation and Application of Molecular Dynamics in Materials Science. Adv. Mater. Res. 2012, 572, 232–236. [Google Scholar] [CrossRef]
- Pristovsek, P.; Fehér, K.; Szilágyi, L.; Kidric, J. Structure of a synthetic fragment of the LALF protein when bound to lipopolysaccharide. J. Med. Chem. 2005, 48, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Fehér, K.; Pristovsek, P.; Szilágyi, L.; Ljevaković, D.; Tomasić, J. Modified glycopeptides related to cell wall peptidoglycan: Conformational studies by NMR and molecular modelling. Bioorg. Med. Chem. 2003, 11, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Wereszczynski, J.; McCammon, J.A. Statistical mechanics and molecular dynamics in evaluating thermodynamic properties of biomolecular recognition. Q. Rev. Biophys. 2012, 45, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Noé, F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Laidig, K.E.; Daggett, V. Conformational search using a molecular dynamics–minimization procedure: Applications to clusters of coulombic charges, Lennard–Jones particles, and waters. Comput. Chem. 1998, 19, 60–70. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Balogh, G.; Gyöngyösi, T.; Timári, I.; Herczeg, M.; Borbás, A.; Fehér, K.; Kövér, K.E. Comparison of Carbohydrate Force Fields Using Gaussian Accelerated Molecular Dynamics Simulations and Development of Force Field Parameters for Heparin-Analogue Pentasaccharides. J. Chem. Inf. Model. 2019, 59, 4855–4867. [Google Scholar] [CrossRef] [PubMed]
- Mándi, A.; Swamy, M.M.M.; Taniguchi, T.; Anetai, M.; Monde, K. Reducing Molecular Flexibility by Cyclization for Elucidation of Absolute Configuration by CD Calculations: Daurichromenic Acid. Chirality 2016, 28, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Yu, Q.; Li, J.; Riccio, R.; Lauro, G.; Bifulco, G.; Kurtán, T.; Mándi, A.; Tang, H.; Li, T.J.; et al. Bissubvilides A and B, Cembrane–Capnosane Heterodimers from the Soft Coral Sarcophyton subviride. J. Nat. Prod. 2016, 79, 2552–2558. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Riccio, R.; Lauro, G.; Bifulco, G.; Li, T.J.; Tang, H.; Zhuang, C.L.; Ma, H.; Sun, P.; et al. Chemistry and Selective Tumor Cell Growth Inhibitory Activity of Polyketides from the South China Sea Sponge Plakortis sp. Mar. Drugs 2017, 15, 129. [Google Scholar] [CrossRef]
- Covington, C.L.; Junior, F.M.S.; Silva, J.H.S.; Kuster, R.M.; de Amorim, M.B.; Polavarapu, P.L. Atropoisomerism in Biflavones: The Absolute Configuration of (−)-Agathisflavone via Chiroptical Spectroscopy. J. Nat. Prod. 2016, 79, 2530–2537. [Google Scholar] [CrossRef] [PubMed]
- Štěpánek, P.; Bouř, P. Multi-scale modeling of electronic spectra of three aromatic amino acids: Importance of conformational averaging and explicit solute–solvent interactions. Phys. Chem. Chem. Phys. 2014, 16, 20639–20649. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Namatame, H.; Taniguchi, M.; Gekko, K. Solution structures of methyl aldopyranosides revealed by vacuum-ultraviolet electronic circular-dichroism spectroscopy. Biomed. Spectrosc. Imaging 2015, 4, 269–282. [Google Scholar] [CrossRef]
- Nicu, V.P.; Neugebauer, J.; Baerends, E.J. Effects of complex formation on vibrational circular dichroism spectra. J. Phys. Chem. A 2008, 112, 6978–6991. [Google Scholar] [CrossRef] [PubMed]
- Nicu, V.P.; Baerends, E.J.; Polavarapu, P.L. Understanding solvent effects in vibrational circular dichroism spectra: [1,1′-binaphthalene]-2,2’-diol in dichloromethane, acetonitrile, and dimethyl sulfoxide solvents. J. Phys. Chem. A 2012, 116, 8366–8373. [Google Scholar] [CrossRef] [PubMed]
- Kaminský, J.; Kubelka, J.; Bouř, P. Theoretical Modeling of Peptide α-Helical Circular Dichroism in Aqueous Solution. J. Phys. Chem. A 2011, 115, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Mustan, F.; Ivanova, A.; Madjarova, G.; Tcholakova, S.; Denkov, N.D. Molecular Dynamics Simulation of the Aggregation Patterns in Aqueous Solutions of Bile Salts at Physiological Conditions. J. Phys. Chem. B 2015, 119, 15631–15643. [Google Scholar] [CrossRef] [PubMed]
- Mannoor, M.; Kang, S.; Suh, Y.K. Molecular Dynamics Simulation of Aggregates in the Dodecane/span80 System and Their Behaviour in an Electric Field. Adv. Cond. Matter Phys. 2015, 2015, 739458. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, X.; Kwok, R.T.K.; Wang, J.; Leung, N.L.C.; Shi, L.; Sun, J.Z.; Tang, Z.; Lam, J.W.Y.; Qin, A.; et al. In situ monitoring of molecular aggregation using circular dichroism. Nat. Commun. 2018, 9, 4961. [Google Scholar] [CrossRef]
- He, L.; Wang, P.; He, L.; Qu, Z.; Luo, J.; Peng, B.; Tang, X.; Pei, Y. Molecular dynamics simulations of the self-organization of side-chain decorated polyaromatic conjugation molecules: Phase separated lamellar and columnar structures and dispersion behaviors in toluene solvent. RSC Adv. 2018, 8, 11134. [Google Scholar] [CrossRef]
- Kang, H.; Zhang, Y.; Yang, M.; Li, L. Molecular Dynamics Simulation on Effect of Nanoparticle Aggregation on Transport Properties of a Nanofluid. J. Nanotechnol. Eng. Med. 2012, 3, 021001. [Google Scholar] [CrossRef]
- Fogolari, F.; Corazza, A.; Viglino, P.; Zuccato, P.; Pieri, L.; Faccioli, P.; Bellotti, V.; Esposito, G. Molecular Dynamics Simulation Suggests Possible Interaction Patterns at Early Steps of b2-Microglobulin Aggregation. Biophys. J. 2007, 92, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.G.; Okumura, H. Promotion and Inhibition of Amyloid-β Peptide Aggregation: Molecular Dynamics Studies. Int. J. Mol. Sci. 2021, 22, 1859. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Seibert, J.; Grimme, S. Electronic Circular Dichroism of [16] Helicene With Simplified TD-DFT: Beyond the Single Structure Approach. Chirality 2016, 28, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Tóth, L.; Mándi, A.; Váradi, D.; Kovács, T.; Szabados, A.; Kiss-Szikszai, A.; Gong, Q.; Zhang, H.; Mátyus, P.; Antus, S.; et al. HPLC-ECD and TDDFT-ECD study of hexahydropyrrolo [1,2-a]quinoline derivatives. Chirality 2018, 30, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Ji, S.; Szalóki, D.; Kovács, T.; Mándi, A.; Antus, S.; Ding, X.; Kurtán, T.; Zhang, H. An optically active isochroman-2H-chromene conjugate potently suppresses neuronal oxidative injuries associated with the PI3K/Akt and MAPK signaling pathways. Acta Pharmacol. Sin. 2021, 42, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.; Chang, F.R.; Hwang, T.L.; Szappanos, Á.; Mándi, A.; Hunyadi, A.; Kurtán, T.; Jakab, G.; Hohmann, J.; Vasas, A. Screening of Luzula species native to the Carpathian Basin for anti-inflammatory activity and bioactivity-guided isolation of compounds from Luzula luzuloides (Lam.) Dandy & Wilmott. Fitoterapia 2017, 116, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.M.M.; Mándi, A.; Anetai, M.; Monde, K. Stereochemistry of a Rhododaurichromanic Acid Derivative. Nat. Prod. Commun. 2016, 11, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Nugroho, A.E.; Morita, H. Circular dichroism calculation for natural products. J. Nat. Med. 2014, 68, 1–10. [Google Scholar] [CrossRef]
- Tao, L.; Yu, W.; Liu, Z.; Zhao, D.; Lin, S.; Szalóki, D.; Kicsák, M.; Kurtán, T.; Zhang, H. JE-133 Suppresses LPS-Induced Neuroinflammation Associated with the Regulation of JAK/STAT and Nrf2 Signaling Pathways. ACS Chem. Neurosci. 2024, 15, 258–267. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474. [Google Scholar] [CrossRef]
- Zajac, G.; Lasota, J.; Dudek, M.; Kaczor, A.; Baranska, M. Pre-resonance enhancement of exceptional intensity in Aggregation-Induced Raman Optical Activity (AIROA) spectra of lutein derivatives. Spectrochim. Acta A 2017, 173, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Zsila, F.; Bikádi, Z.; Keresztes, Z.; Deli, J.; Simonyi, M. Investigation of the Self-Organization of Lutein and Lutein Diacetate by Electronic Absorption, Circular Dichroism Spectroscopy, and Atomic Force Microscopy. J. Phys. Chem. B 2001, 105, 9413–9421. [Google Scholar] [CrossRef]
- Zsila, F.; Deli, J.; Simonyi, M. Color and chirality: Carotenoid self-assemblies in flower petals. Planta 2001, 213, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Zajac, G.; Machalska, E.; Kaczor, A.; Kessler, J.; Bour, P.; Baranska, M. Structure of supramolecular astaxanthin aggregates revealed by molecular dynamics and electronic circular dichroism spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 18038–18046. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Grabuleda, X.; Jaime, C.; Kollman, P.A. Molecular dynamics simulation studies of liquid acetonitrile: New six-site model. J. Comput. Chem. 2000, 21, 901–908. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cisneros, V.W.D.; et al. AMBER 2023; University of California: San Francisco, CA, USA, 2023. [Google Scholar]
- le Grand, S.; Gotz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 1.2r3pre; Schrödinger Inc., LLC.: New York, NY, USA, 2011.
- Varetto, U. MOLEKEL 5.4; Swiss National Supercomputing Centre: Manno, Switzerland, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Ghidinelli, S.; Abbate, S.; Koshoubu, J.; Araki, Y.; Wada, T.; Longhi, G. Solvent Effects and Aggregation Phenomena Studied by Vibrational Optical Activity and Molecular Dynamics: The Case of Pantolactone. J. Phys. Chem. B 2020, 124, 4512–4526. [Google Scholar] [CrossRef]
- Longhi, G.; Ghidinelli, S.; Abbate, S.; Mazzeo, G.; Fusè, M.; Boiadjiev, S.E.; Lightner, D.A. Insights into the Structures of Bilirubin and Biliverdin from Vibrational and Electronic Circular Dichroism: History and Perspectives. Molecules 2023, 28, 2564. [Google Scholar] [CrossRef]
- Ghidinelli, S.; Longhi, G.; Abbate, S.; Boiadjiev, S.E.; Lightner, D.A. Bilirubin and its congeners: Conformational analysis and chirality from metadynamics and related computational methods. Monatsh. Chem. 2019, 150, 801–812. [Google Scholar] [CrossRef]
- Ghidinelli, S.; Longhi, G.; Mazzeo, G.; Abbate, S.; Boiadjiev, S.E.; Lightner, D.A. On the aggregation of bilirubinoids in solution as evidenced by VCD and ECD spectroscopy and DFT calculations. Chirality 2018, 30, 19–28. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mándi, A.; Rimóczi, A.; Vasas, A.; Hohmann, J.; Swamy, M.M.M.; Monde, K.; Barta, R.A.; Kicsák, M.; Komáromi, I.; Fehér, K.; et al. Testing the Simplified Molecular Dynamics Approach to Improve the Reproduction of ECD Spectra and Monitor Aggregation. Int. J. Mol. Sci. 2024, 25, 6453. https://doi.org/10.3390/ijms25126453
Mándi A, Rimóczi A, Vasas A, Hohmann J, Swamy MMM, Monde K, Barta RA, Kicsák M, Komáromi I, Fehér K, et al. Testing the Simplified Molecular Dynamics Approach to Improve the Reproduction of ECD Spectra and Monitor Aggregation. International Journal of Molecular Sciences. 2024; 25(12):6453. https://doi.org/10.3390/ijms25126453
Chicago/Turabian StyleMándi, Attila, Aliz Rimóczi, Andrea Vasas, Judit Hohmann, Mahadeva M. M. Swamy, Kenji Monde, Roland A. Barta, Máté Kicsák, István Komáromi, Krisztina Fehér, and et al. 2024. "Testing the Simplified Molecular Dynamics Approach to Improve the Reproduction of ECD Spectra and Monitor Aggregation" International Journal of Molecular Sciences 25, no. 12: 6453. https://doi.org/10.3390/ijms25126453
APA StyleMándi, A., Rimóczi, A., Vasas, A., Hohmann, J., Swamy, M. M. M., Monde, K., Barta, R. A., Kicsák, M., Komáromi, I., Fehér, K., & Kurtán, T. (2024). Testing the Simplified Molecular Dynamics Approach to Improve the Reproduction of ECD Spectra and Monitor Aggregation. International Journal of Molecular Sciences, 25(12), 6453. https://doi.org/10.3390/ijms25126453