
The Lung in Rheumatoid Arthritis—Friend or Enemy?

 , , , and
, , , and
Abstract
:1. Introduction
2. Genetic Predisposition
3. Risk Factors for RA-ILD
3.1. Age
3.2. Patient Gender
3.3. Race
3.4. Smoking
3.5. External Pollutants
3.6. Disease Activity of RA
3.7. Lung Microbiota
4. Immunological Profile in RA-ILD
4.1. ACPA and RF
4.2. Peptidyl Arginine Deiminases Enzymes
4.3. Anticarbamylated Protein Antibodies
4.4. Antibodies against Malondialdehyde
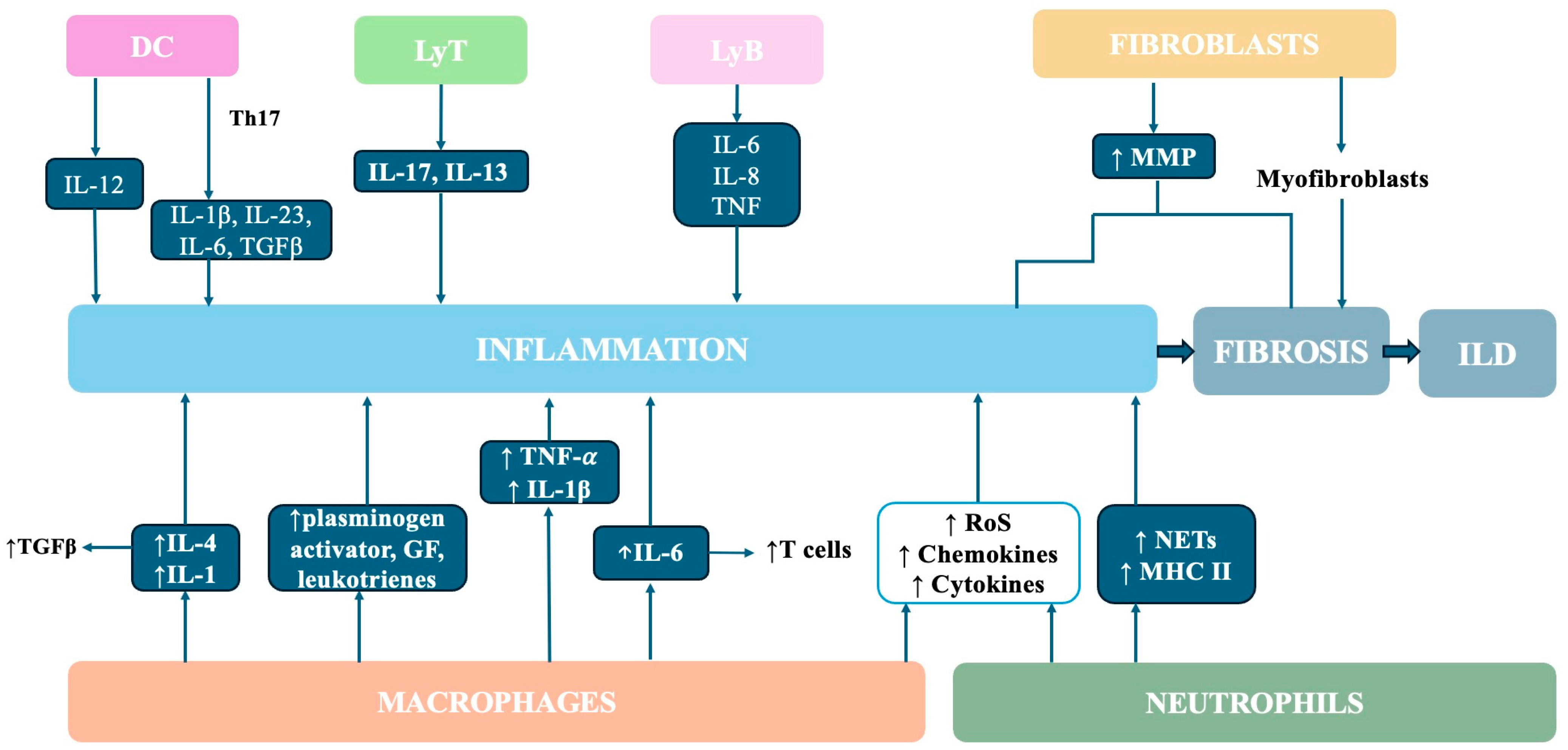
5. Cell Profiles in RA-ILD
5.1. Macrophages
5.2. Neutrophils
5.3. Fibroblasts
5.4. Dendritic Cells
5.5. T and B Cells
6. Cytokine Profile in RA-ILD
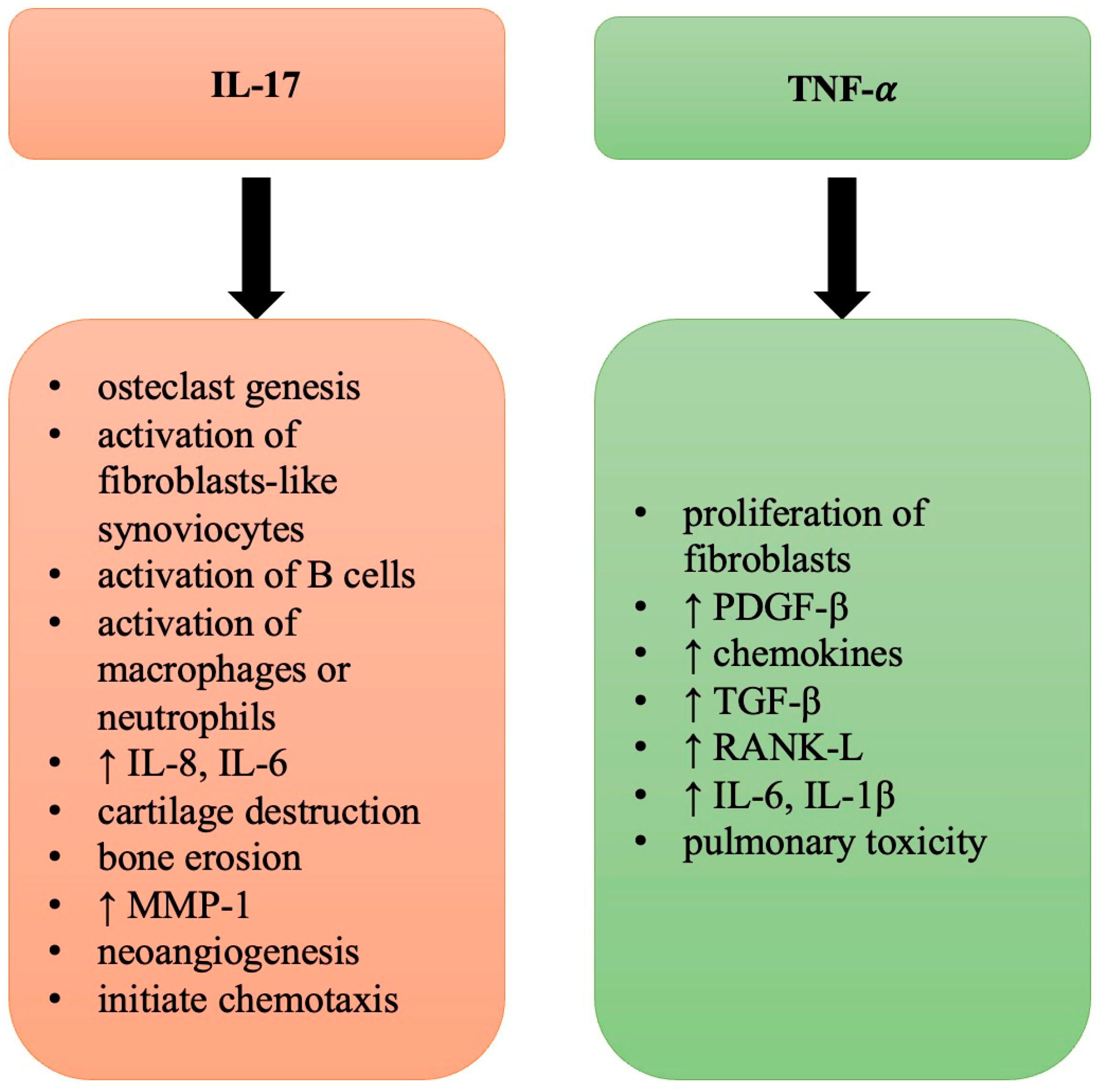
6.1. TNF-α
6.2. IL-17

{kind=link}
{kind=link}
{kind=link}
| Interleukins | References | Pathogenic or Clinical Correlations |
|---|---|---|
| IL-4 | [161] | ↑ RA-ILD |
| IL-18 | [163] | ↑ RA-ILD |
| IL-13 | [159,160,161,162,163] | ↑ RA-ILD; lung fibrosis on HRCT |
| IL-11 | [160,161] | Correlated with RA-ILD activity |
| IL-33 | [160,161] | Correlated with RA-ILD activity |
| IL-23 | [164,165] | Associated with EMT |
| MMP-3 | [173,174,175,176] | Correlated with RA-ILD activity |
| MMP-7 | [173,174,175,176] | Correlated with RA-ILD activity |
| PDGF-β | [90] | Profibrotic/inflammatory role |
| JAK/STAT | [179,180,181] | Associated with lung fibrosis |
| KL-6 | [182,183,184] | ↑ RA-ILD; lung fibrosis on HRCT |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shaw, M.; Collins, B.F.; Ho, L.A.; Raghu, G. Rheumatoid arthritis-associated lung disease. Eur. Respir. Rev. 2015, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Kwon, E.J.; Lee, J.J. Rheumatoid Arthritis: Pathogenic Roles of Diverse Immune Cells. Int. J. Mol. Sci. 2022, 23, 905. [Google Scholar] [CrossRef]
- Mitrović, J.; Hrkač, S.; Tečer, J.; Golob, M.; Ljilja Posavec, A.; Kolar Mitrović, H.; Grgurević, L. Pathogenesis of Extraarticular Manifestations in Rheumatoid Arthritis—A Comprehensive Review. Biomedicines 2023, 11, 1262. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid Arthritis. Nat. Rev. Dis. Prim. 2018, 4, 18001. [Google Scholar] [CrossRef] [PubMed]
- Cerro, C.G.; Parimon, T. Understanding Interstitial Lung Diseases Associated with Connective Tissue Disease (CTD-ILD): Genetics, Cellular Pathophysiology, and Biologic Drivers. Int. J. Mol. Sci. 2023, 24, 2405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yin, J.; Zhang, X. Factors associated with interstitial lung disease in patients with rheumatoid arthritis: A systematic review and meta-analysis. PLoS ONE 2023, 18, e0286191. [Google Scholar] [CrossRef] [PubMed]
- Matson, S.M.; Demoruelle, M.K.; Castro, M. Airway Disease in Rheumatoid Arthritis. Ann. Am. Thorac. Soc. 2022, 19, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Lee, J.S.; Sverzellati, N.; Rossi, G.; Cottin, V. The Lung in Rheumatoid Arthritis: Focus on Interstitial Lung Disease. Arthritis Rheumatol. 2018, 70, 1544–1554. [Google Scholar] [CrossRef]
- Yunt, Z.X.; Solomon, J.J. Lung Disease in Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2015, 41, 225–236. [Google Scholar] [CrossRef]
- McDermott, G.C.; Doyle, T.J.; Sparks, J.A. Interstitial lung disease throughout the rheumatoid arthritis disease course. Curr. Opin. Rheumatol. 2021, 33, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, J.; Lau, J.; Wang, S.; Taneja, V.; Matteson, E.L.; Vassallo, R. Mechanisms of lung disease development in rheumatoid arthritis. Nat. Rev. Rheumatol. 2019, 15, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Olivas-Flores, E.M.; Bonilla-Lara, D.; Gamez-Nava, J.I.; Rocha-Muñoz, A.D.; Gonzalez-Lopez, L. Interstitial lung disease in rheumatoid arthritis: Current concepts in pathogenesis, diagnosis and therapeutics. World J. Rheumatol. 2015, 12, 1–22. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, L.; Rocha-Muñoz, A.D.; Ponce-Guarneros, M.; Flores-Chavez, A.; Salazar-Paramo, M.; Nava, A.; Cardona-Muñoz, E.G.; Fajardo-Robledo, N.S.; Zavaleta-Muñiz, S.A.; Garcia-Cobian, T.; et al. Anti-cyclic citrullinated peptide (anti-CCP) and anti-mutated citrullinated vimentin (anti-MCV) relation with extra-articular manifestations in rheumatoid arthritis. J. Immunol. Res. 2014, 2014, 536050. [Google Scholar] [CrossRef] [PubMed]
- Matson, S.; Lee, J.; Eickelberg, O. Two sides of the same coin? A review of the similarities and differences between idiopathic pulmonary fibrosis and rheumatoid arthritis-associated interstitial lung disease. Eur. Respir. J. 2021, 57, 2002533. [Google Scholar] [CrossRef]
- De Zorzi, E.; Spagnolo, P.; Cocconcelli, E.; Balestro, E.; Iaccarino, L.; Gatto, M.; Benvenuti, F.; Bernardinello, N.; Doria, A.; Maher, T.M.; et al. Thoracic involvement in systemic autoimmune rheumatic diseases: Pathogenesis and management. Clin. Rev. Allergy Immunol. 2022, 63, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Cavagna, L.; Monti, S.; Grosso, V.; Boffini, N.; Scorletti, E.; Crepaldi, G. The multifaceted aspects of interstitial lung disease in rheumatoid arthritis. BioMed Res. Int. 2013, 2013, 759760. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.J.; Patel, A.S.; Hatabu, H.; Nishino, M.; Wu, G.; Osorio, J.C.; Golzarri, M.F.; Traslosheros, A.; Chu, S.G.; Frits, M.L.; et al. Detection of rheumatoid arthritis-interstitial lung disease is enhanced by serum biomarkers. Am. J. Respir. Crit. Care Med. 2015, 191, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, A.J.; Junker, P.; Houen, G.; Kyvik, K.O.; Nielsen, C.; Skytthe, A.; Holst, R. Incidence of Chronic Persistent Rheumatoid Arthritis and the Impact of Smoking: A Historical Twin Cohort Study. Arthritis Care Res. 2017, 69, 616–624. [Google Scholar] [CrossRef]
- Kuwana, M.; Gil-Vila, A.; Selva-O’Callaghan, A. Role of autoantibodies in the diagnosis and prognosis of interstitial lung disease in autoimmune rheumatic disorders. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211032457. [Google Scholar] [CrossRef]
- Paulin, F.; Babini, A.; Mamani, M.; Mercado, J.; Caro, F. Practical Approach to the Evaluation and Management of Rheumatoid Arthritis-Interstitial Lung Disease Based on its Proven and Hypothetical Mechanisms. Rev. Investig. Clin. 2017, 69, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Paulin, F.; Doyle, T.J.; Fletcher, E.A.; Ascherman, D.P.; Rosas, I.O. Rheumatoid Arthritis-Associated Interstitial Lung Disease and Idiopathic Pulmonary Fibrosis: Shared Mechanistic and Phenotypic Traits Suggest Overlapping Disease Mechanisms. Rev. Investig. Clin. 2015, 67, 280–286. [Google Scholar]
- Quirke, A.M.; Lugli, E.B.; Wegner, N.; Hamilton, B.C.; Charles, P.; Chowdhury, M.; Ytterberg, A.J.; Zubarev, R.A.; Potempa, J.; Culshaw, S.; et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: A potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 263–269. [Google Scholar] [CrossRef]
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid Arthritis and the Mucosal Origins Hypothesis: Protection Turns to Destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. The immunology of rheumatoid arthritis. Nat. Immunol. 2021, 22, 10–18. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet Lond. Engl. 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Manfredi, A.; Cassone, G.; Luppi, F.; Atienza-Mateo, B.; Cavazza, A.; Sverzellati, N.; González-Gay, M.A.; Salvarani, C.; Sebastiani, M. Rheumatoid arthritis related interstitial lung disease. Expert Rev. Clin. Immunol. 2021, 17, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef]
- Al-Baldawi, S.; Zúñiga Salazar, G.; Zúñiga, D.; Balasubramanian, S.; Mehmood, K.T. Interstitial Lung Disease in Rheumatoid Arthritis: A Review. Cureus 2024, 16, e53632. [Google Scholar] [CrossRef]
- Littlejohn, E.A.; Monrad, S. Early Diagnosis and Treatment of Rheumatoid Arthritis. Prim. Care Clin. Off. Pract. 2018, 45, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Arleevskaya, M.I.; Kravtsova, O.A.; Lemerle, J.; Renaudineau, Y.; Tsibulkin, A.P. How Rheumatoid Arthritis Can Result from Provocation of the Immune System by Microorganisms and Viruses. Front. Microbiol. 2016, 7, 1296. [Google Scholar] [CrossRef] [PubMed]
- Kadura, S.; Raghu, G. Rheumatoid arthritis-interstitial lung disease: Manifestations and current concepts in pathogenesis and management. Eur. Respir. Rev. 2021, 30, 210011. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.A.; Oldham, J.M.; Ley, B.; Anand, V.; Adegunsoye, A.; Liu, G.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.; et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur. Respir. J. 2019, 53, 1801641. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, S.I.; Kim, H.O. Recent Advances in Basic and Clinical Aspects of Rheumatoid Arthritis-associated Interstitial Lung Diseases. J. Rheum. Dis. 2022, 29, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Koga, Y.; Sugimoto, M. Different risk factors between interstitial lung disease and airway disease in rheumatoid arthri- tis. Respir. Med. 2012, 106, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.J.; Sweatman, M.C.; Markwick, J.R.; Maini, R.N. HLA-B40: A marker for susceptibility to lung disease in rheumatoid arthritis. Dis. Mark. 1991, 9, 97–101. [Google Scholar]
- Sugiyama, Y.; Ohno, S.; Kano, S.; Maeda, H.; Kitamura, S. Diffuse pan-bronchiolitis and rheumatoid arthritis: A possible correlation with HLA-B54. Intern Med. 1994, 33, 612–614. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef]
- Juge, P.-A.; Borie, R.; Kannengiesser, C.; Gazal, S.; Revy, P.; Wemeau-Stervinou, L.; Debray, M.-P.; Ottaviani, S.; Marchand-Adam, S.; Nathan, N. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1602314. [Google Scholar] [CrossRef]
- Kim, Y.; Yang, H.-I.; Kim, K.-S. Etiology and Pathogenesis of Rheumatoid Arthritis-Interstitial Lung Disease. Int. J. Mol. Sci. 2023, 24, 14509. [Google Scholar] [CrossRef] [PubMed]
- Stainer, A.; Tonutti, A.; De Santis, M.; Amati, F.; Ceribelli, A.; Bongiovanni, G.; Torrisi, C.; Iacopino, A.; Mangiameli, G.; Aliberti, S.; et al. Unmet needs and perspectives in rheumatoid arthritis-associated interstitial lung disease: A critical review. Front. Med. 2023, 10, 1129939. [Google Scholar] [CrossRef]
- Jönsson, E.; Ljung, L.; Norrman, E.; Freyhult, E.; Ärlestig, L.; Dahlqvist, J.; Rantapää-Dahlqvist, S. Pulmonary fibrosis in relation to genetic loci in an inception cohort of patients with early rheumatoid arthritis from northern Sweden. Rheumatology 2022, 61, 943–952. [Google Scholar] [CrossRef]
- Akiyama, M.; Kaneko, Y. Pathogenesis, clinical features, and treatment strategy for rheumatoid arthritis-associated interstitial lung disease. Autoimmun. Rev. 2022, 21, 103056. [Google Scholar] [CrossRef]
- Khan, T.; Jose, R.J.; Renzoni, E.A.; Mouyis, M. A Closer Look at the Role of Anti-CCP Antibodies in the Pathogenesis of Rheumatoid Arthritis-Associated Interstitial Lung Disease and Bronchiectasis. Rheumatol. Ther. 2021, 8, 1463–1475. [Google Scholar] [CrossRef]
- Juge, P.A.; Lee, J.S.; Ebstein, E.; Furukawa, H.; Dobrinskikh, E.; Gazal, S.; Kannengiesser, C.; Ottaviani, S.; Oka, S.; Tohma, S.; et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N. Engl. J. Med. 2018, 379, 2209–2219. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Qu, D.; Yu, J.; Yang, J. The Possible Pathogenesis of Idiopathic Pulmonary Fibrosis considering MUC5B. BioMed Res. Int. 2019, 2019, 9712464. [Google Scholar]
- Vermant, M.; Goos, T.; Gogaert, S.; De Cock, D.; Verschueren, P.; Wuyts, W.A. Are genes the missing link to detect and prognosticate RA-ILD? Rheumatol. Adv. Pract. 2023, 7, rkad023. [Google Scholar] [CrossRef] [PubMed]
- Palomäki, A.; Palotie, A.; Koskela, J.; Eklund, K.K.; Pirinen, M.; Ripatti, S.; Laitinen, T.; Mars, N.; Group, F.R.C.E. Lifetime risk of rheumatoid arthritis-associated interstitial lung disease in MUC5B mutation carriers. Ann. Rheum. Dis. 2021, 80, 1530–1536. [Google Scholar] [CrossRef]
- Shirai, Y.; Honda, S.; Ikari, K.; Kanai, M.; Takeda, Y.; Kamatani, Y.; Morisaki, T.; Tanaka, E.; Kumanogoh, A.; Harigai, M. Association of the RPA3-UMAD1 locus with interstitial lung diseases complicated with rheumatoid arthritis in Japanese. Ann. Rheum. Dis. 2020, 79, 1305–1309. [Google Scholar] [CrossRef]
- Higuchi, T.; Oka, S.; Furukawa, H.; Shimada, K.; Tsunoda, S.; Ito, S.; Okamoto, A.; Katayama, M.; Saisho, K.; Shinohara, S. Association of a FAM13A variant with interstitial lung disease in Japanese rheumatoid arthritis. RMD Open 2023, 9, e002828. [Google Scholar] [CrossRef] [PubMed]
- Innala, L.; Berglin, E.; Möller, B.; Ljung, L.; Smedby, T.; Södergren, A.; Magnusson, S.; Rantapää-Dahlqvist, S.; Wållberg-Jonsson, S. Age at onset determines severity and choice of treatment in early rheumatoid arthritis: A prospective study. Arthrit. Res. Ther. 2014, 16, R94. [Google Scholar] [CrossRef] [PubMed]
- Koduri, G.; Norton, S.; Young, A.; Cox, N.; Davies, P.; Devlin, J.; Dixey, J.; Gough, A.; Prouse, P.; Winfield, J. Interstitial lung disease has a poor prognosis in rheumatoid arthritis: Results from an inception cohort. Rheumatology 2010, 49, 1483–1489. [Google Scholar] [CrossRef]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- Murtha, L.A.; Morten, M.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Ngo, D.T.; Sverdlov, A.L.; Knight, D.A. The role of pathological aging in cardiac and pulmonary fibrosis. Aging Dis. 2019, 10, 419–428. [Google Scholar] [CrossRef]
- Assayag, D.; Lubin, M.; Lee, J.S.; King, T.E.; Collard, H.R.; Ryerson, C.J. Predictors of mortality in rheumatoid arthritis-related interstitial lung disease. Respirology 2014, 19, 493–500. [Google Scholar] [CrossRef]
- Saag, K.G.; Cerhan, J.R.; Kolluri, S.; Ohashi, K.; Hunninghake, G.W.; Schwartz, D.A. Cigarette smoking and rheumatoid arthritis severity. Ann. Rheum. Dis. 1997, 56, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.A.; Saravanan, V.; Nisar, M.; Arthanari, S.; Woodhead, F.A.; Price-Forbes, A.N.; Dawson, J.; Sathi, N.; Ahmad, Y.; Koduri, G. Rheumatoid arthritis-related interstitial lung disease: Associations, prognostic factors and physiological and radiological characteristics—A large multicentre UK study. Rheumatology 2014, 53, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, J.F.; Del Rincón, I.; Battafarano, D.F.; Haas, R.W.; Doria, M.; Escalante, A. Clinical and laboratory factors associated with interstitial lung disease in rheumatoid arthritis. Clin. Rheumatol. 2015, 34, 1529–1536. [Google Scholar] [CrossRef]
- Klester, E.; Klester, K.; Shoykhet, Y.; Elykomov, V.; Yarkova, V.; Berdyugina, A.; Mukhtarova, E. Risk factors of interstitial lung diseases in patients with rheumatoid arthritis. Eur. Respir. Soc. 2019, 54, PA1365. [Google Scholar]
- Chen, N.; Diao, C.-Y.; Gao, J.; Zhao, D.-B. Risk factors for the progression of rheumatoid arthritis-related interstitial lung disease: Clinical features, biomarkers, and treatment options. Semin. Arthritis Rheum. 2022, 55, 152004. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, N.; Nguyen, E.; Sathananthan, M. Rheumatoid arthritis and associated interstitial lung disease: Mortality rates and trends. Ann. Am. Thorac. Soc. 2021, 18, 1970–1977. [Google Scholar] [CrossRef] [PubMed]
- Peschken, C.A.; Hitchon, C.A.; Robinson, D.B.; Smolik, I.; Barnabe, C.R.; Prematilake, S.; El-Gabalawy, H.S. Rheumatoid arthritis in a north american native population: Longitudinal followup and comparison with a white population. J. Rheumatol. 2010, 37, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.E.; Corte, T.J. Nintedanib for idiopathic pulmonary fibrosis in the Japanese population. Respirology 2017, 22, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Tekaya, A.B.; Mokaddem, S.; Athimini, S.; Kamoun, H.; Mahmoud, I.; Abdelmoula, L. Risk factors for rheumatoid arthritis-associated interstitial lung disease: A retrospective study. Multidiscip. Respir. Med. 2022, 17, 877. [Google Scholar] [PubMed]
- Klareskog, L.; Rönnelid, J.; Saevarsdottir, S.; Padyukov, L.; Alfredsson, L. The importance of differences; On environment and its interactions with genes and immunity in the causation of rheumatoid arthritis. J. Intern. Med. 2020, 287, 514–533. [Google Scholar] [CrossRef] [PubMed]
- Hedstroem, A.K.; Stawiarz, L.; Klareskog, L.; Alfredsson, L. Smoking and susceptibility to rheumatoid arthritis in a Swedish population-based case control study. Eur. J. Epidemiol. 2018, 33, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human Inflammatory Dendritic Cells Induce Th17 Cell Differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef]
- Lee, J.; Luria, A.; Rhodes, C.; Raghu, H.; Lingampalli, N.; Sharpe, O.; Rada, B.; Sohn, D.H.; Robinson, W.H.; Sokolove, J. Nicotine Drives Neutrophil Extracellular Traps Formation and Accelerates Collagen-Induced Arthritis. Rheumatology 2017, 56, 644–653. [Google Scholar] [CrossRef]
- Lee, K.H.; Kronbichler, A.; Park, D.D.Y.; Park, Y.; Moon, H.; Kim, H.; Choi, J.H.; Choi, Y.; Shim, S.; Lyu, I.S.; et al. Neutrophil Extracellular Traps (NETs) in Autoimmune Diseases: A Comprehensive Review. Autoimmun. Rev. 2017, 16, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Hedstroem, A.K.; Roennelid, J.; Klareskog, L.; Alfredsson, L. Complex relationships of smoking, HLA-DRB1 Genes, and serologic profiles in patients with early rheumatoid arthritis: Update from a swedish population-based case-control study. Arthritis Rheumatol. 2019, 71, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Zafar, I.; Mariappan, N.; Husain, M.; Wei, C.-C.; Vetal, N.; Eltoum, I.A.; Ahmad, A. Acute pulmonary effects of aerosolized nicotine. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L94–L104. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Sasaki, S.; Yokoyama, T.; Chida, K.; Azuma, A.; Suda, T.; Kudoh, S.; Sakamoto, N.; Okamoto, K.; Kobashi, G.; et al. Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Ann. Occup. Hyg. 2005, 49, 259–265. [Google Scholar] [PubMed]
- Saag, K.G.; Kolluri, S.; Koehnke, R.K.; Georgou, T.A.; Rachow, J.W.; Hunninghake, G.W.; Schwartz, D.A. Rheumatoid arthritis lung disease. Determinants of radiographic and physiologic abnormalities. Arthritis Rheum. 1996, 39, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Yahya, A.; Bengtsson, C.; Larsson, P.; Too, C.L.; Mustafa, A.N.; Abdullah, N.A.; Muhamad, N.A.; Klareskog, L.; Murad, S.; Alfredsson, L. Silica exposure is associated with an increased risk of developing ACPA-positive rheumatoid arthritis in an Asian population: Evidence from the Malaysian MyEIRA case–control study. Mod. Rheumatol. 2014, 24, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Yahya, A.; Bengtsson, C.; Källberg, H.; Rönnelid, J.; Lundberg, I.; Klareskog, L.; Alfredsson, L.; Group, E.S. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Källberg, H.; Lundberg, I.; Sjögren, B.; Klareskog, L.; Alfredsson, L. Silica exposure is associated with increased risk of developing rheumatoid arthritis: Results from the Swedish EIRA study. Ann. Rheum. Dis. 2005, 64, 582–586. [Google Scholar] [CrossRef]
- Zhao, N.; Al-Aly, Z.; Zheng, B.; van Donkelaar, A.; Martin, R.V.; Pineau, C.A.; Bernatsky, S. Fine particulate matter components and interstitial lung disease in rheumatoid arthritis. Eur. Respir. J. 2022, 60, 2102149. [Google Scholar] [CrossRef]
- Liu, B.; Sun, G.; Liu, Y.; Hou, Y. Observational studies: Ambient air pollution and hospitalization for RA-ILD in a heavily polluted city in China. Medicine 2022, 101, e29309. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Smolen, J.S. Diagnosis and management of rheumatoid arthritis: A review. JAMA 2018, 320, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.; Reed, G.; Kremer, J.; Curtis, J.; Farkouh, M.; Harrold, L.; Hochberg, M.; Tsao, P.; Greenberg, J. Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheumatol. 2015, 67, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.A.; He, X.; Huang, J.; Fletcher, E.A.; Zaccardelli, A.; Friedlander, H.M.; Gill, R.R.; Hatabu, H.; Nishino, M.; Murphy, D.J. Rheumatoid arthritis disease activity predicting incident clinically apparent rheumatoid arthritis–associated interstitial lung disease: A prospective cohort study. Arthritis Rheumatol. 2019, 71, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Paulin, F.; Doyle, T.J.; Mercado, J.F.; Fassola, L.; Fernández, M.; Caro, F.; Alberti, M.L.; Espíndola, M.E.C.; Buschiazzo, E. Development of a risk indicator score for the identification of interstitial lung disease in patients with rheumatoid arthritis. Reumatol. Clin. 2021, 17, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.; Lama, S.; Knapp, K.; Gutierrez, C.; Lovett, K.; Thai, S.; Craig, G.L. Epidemiology and clinical characteristics of interstitial lung disease in patients with rheumatoid arthritis from the JointMan database. Sci. Rep. 2023, 13, 11678. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zheng, J.; Yuan, M.; Yuan, L.; Jia, X.; Liu, H. Differentially expressed lncRNAs in peripheral blood mononuclear cells from middle-aged female patients with rheumatoid arthritis–associated interstitial lung disease. Clin. Rheumatol. 2020, 39, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Severo, C.R.; Chomiski, C.; Valle, M.B.d.; Escuissato, D.L.; Paiva, E.d.S.; Storrer, K.M. Assessment of risk factors in patients with rheumatoid arthritis-associated interstitial lung disease. J. Bras. Pneumol. 2022, 48, e20220145. [Google Scholar] [PubMed]
- Rojas-Serrano, J.; Mejía, M.; Rivera-Matias, P.A.; Herrera-Bringas, D.; Pérez-Román, D.I.; Pérez-Dorame, R.; Mateos-Toledo, H. Rheumatoid arthritis-related interstitial lung disease (RA-ILD): A possible association between disease activity and prognosis. Clin. Rheumatol. 2022, 41, 1741–1747. [Google Scholar] [CrossRef]
- Florescu, A.; Gherghina, F.L.; Mușetescu, A.E.; Pădureanu, V.; Roșu, A.; Florescu, M.M.; Criveanu, C.; Florescu, L.-M.; Bobircă, A. Novel Biomarkers, Diagnostic and Therapeutic Approach in Rheumatoid Arthritis Interstitial Lung Disease—A Narrative Review. Biomedicines 2022, 10, 1367. [Google Scholar] [CrossRef]
- Demoruelle, M.K.; Solomon, J.J.; Fischer, A.; Deane, K.D. The lung may play a role in the pathogenesis of rheumatoid arthritis. Int. J. Clin. Rheumatol. 2014, 9, 295. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Joshua, V.; Artacho, A.; Abdollahi-Roodsaz, S.; Öckinger, J.; Kullberg, S.; Sköld, M.; Eklund, A.; Grunewald, J.; Clemente, J.C.; et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome 2016, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R.; Payne, J.B.; Deane, K.D.; Thiele, G.M. Autoimmunity of the lung and oral mucosa in a multisystem inflammatory disease: The spark that lights the fire in rheumatoid arthritis? J. Allergy Clin. Immunol. 2016, 137, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Drakopanagiotakis, F.; Stavropoulou, E.; Tsigalou, C.; Nena, E.; Steiropoulos, P. The Role of the Microbiome in Connective-Tissue-Associated Interstitial Lung Disease and PulmonaryVasculitis. Biomedicines 2022, 10, 3195. [Google Scholar] [CrossRef] [PubMed]
- Kronzer, V.L.; Davis, J.M. Etiologies of rheumatoid arthritis: Update on mucosal, genetic, and cellular pathogenesis. Curr. Rheumatol. 2021, 23, 21. [Google Scholar] [CrossRef] [PubMed]
- Demoruelle, M.K.; Weisman, M.H.; Simonian, P.L.; Lynch, D.A.; Sachs, P.B.; Pedraza, I.F.; Harrington, A.R.; Kolfenbach, J.R.; Striebich, C.C.; Pham, Q.N.; et al. Brief report: Airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: Early injury or initiating site of autoimmunity? Arthritis Rheum. 2012, 64, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Solomon, J.J.; Du Bois, R.M.; Deane, K.D.; Olson, A.L.; Fernandez-Perez, E.R.; Huie, T.J.; Stevens, A.D.; Gill, M.B.; Rabinovitch, A.M.; et al. Lung disease with anti-CCP antibodies but not rheumatoid arthritis or connective tissue disease. Respir. Med. 2012, 106, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Proal, A.D.; Albert, P.J.; Marshall, T.G. The human microbiome and autoimmunity. Curr. Opin. Rhumatol. 2013, 25, 234–240. [Google Scholar] [CrossRef]
- Yeoh, N.; Burton, J.P.; Suppiah, P.; Reid, G.; Stebbings, S. The role of the microbiome in rheumatic diseases. Curr. Rheumatol. Rep. 2013, 15, 314. [Google Scholar] [CrossRef]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.M.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to porphyromonas gingivalis indicate interaction between oral infection, smoking, and risk genes in rheumatoid arthritis etiology. Arthritis Rheumatol. 2016, 68, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, D.; Platzer, A.; Studenic, P.; Alasti, F.; Grundhuber, M.; Swiniarski, S.; Horn, T.; Haslacher, H.; Blüml, S.; Smolen, J.; et al. Determination of autoantibody isotypes increases the sensitivity of serodiag- nostics in rheumatoid arthritis. Front. Immunol. 2018, 9, 876. [Google Scholar] [CrossRef] [PubMed]
- Van Delft, M.A.M.; Huizinga, T.W.J. An Overview of Autoantibodies in Rheumatoid Arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef] [PubMed]
- Kissel, T.; Reijm, S.; Slot, L.M.; Cavallari, M.; Wortel, C.M.; Vergroesen, R.D.; Stoeken-Rijsbergen, G.; Kwekkeboom, J.C.; Kampstra, A.S.B.; Levarht, E.W.N.; et al. Antibodies and B cells recognising citrullinated proteins display a broad cross-reactivity towards other post-translational modifications. Ann. Rheum. Dis. 2020, 79, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Coutant, F. Pathogenic effects of anti-citrullinated protein antibodies in rheumatoid arthritis—Role for glycosylation. Jt. Bone Spine 2019, 86, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2015, 75, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Van Kalsbeek, D.; Brooks, R.; Shaver, D.; Ebel, A.; Hershberger, D.; Schmidt, C.; Poole, J.A.; Ascherman, D.P.; Thiele, G.M.; Mikuls, T.R.; et al. Peripheral Blood Biomarkers for Rheumatoid Arthritis–Associated Interstitial Lung Disease: A Systematic Review. ACR Open Rheumatol. 2023, 5, 201–226. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Holmdahl, R. The structure, specificity and function of anti-citrullinated protein antibodies. Nat. Rev. Rheumatol. 2019, 15, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Muñoz, A.D.; Ponce-Guarneros, M.; Gamez-Nava, J.I.; Olivas-Flores, E.M.; Mejía, M.; Juárez-Contreras, P.; Martínez-García, E.A.; Corona-Sánchez, E.G.; Rodríguez-Hernández, T.M.; Vázquez-del Mercado, M.; et al. Anti-cyclic citrullinated peptide antibodies and severity of interstitial lung disease in women with rheumatoid arthritis. J. Immunol. Res. 2015, 2015, 151626. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, W.; Yu, Y.; Hu, S. Rheumatoid arthritis–associated interstitial lung disease: An overview of epidemiology, pathogenesis and management. Clin. Rheumatol. 2021, 40, 1211–1220. [Google Scholar] [CrossRef]
- Correia, C.S.; Briones, M.R.; Guo, R.; Ostrowski, R.A. Elevated anti-cyclic citrullinated peptide antibody titer is associated with increased risk for interstitial lung disease. Clin. Rheumatol. 2019, 38, 1201–1206. [Google Scholar] [CrossRef]
- Giles, J.T.; Danoff, S.K.; Sokolove, J.; Wagner, C.A.; Winchester, R.; Pappas, D.A.; Siegelman, S.; Connors, G.; Robinson, W.H.; Bathon, J.M. Association of fine specificity and repertoire expansion of anticitrullinated peptide antibodies with rheumatoid arthritis associated interstitial lung disease. Ann. Rheum. Dis. 2014, 73, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhou, Y.; Chen, X.; Li, J. A metaanalysis of the increased risk of rheumatoid arthritis-related pulmonary disease as a result of serum anticitrullinated protein antibody positivity. J. Rheumatol. 2014, 41, 1282–1289. [Google Scholar] [CrossRef]
- Aubart, F.; Crestani, B.; Nicaise-Roland, P.; Tubach, F.; Bollet, C.; Dawidowicz, K.; Quintin, E.; Hayem, G.; Palazzo, E.; Meyer, O. High levels of anti-cyclic citrullinated peptide autoantibodies are associated with co-occurrence of pulmonary diseases with rheumatoid arthritis. J. Rheumatol. 2011, 38, 979–982. [Google Scholar] [CrossRef]
- Kronzer, V.L.; Huang, W.; Dellaripa, P.F.; Huang, S.; Feathers, V.; Lu, B.; Iannaccone, C.K.; Gill, R.R.; Hatabu, H.; Nishino, M. Lifestyle and clinical risk factors for incident rheumatoid arthritis-associated interstitial lung disease. J. Rheumatol. 2021, 48, 656–663. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Suzuki, A.; Maher, T.M. Interstitial lung diseases. Lancet 2022, 400, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Kelmenson, L.B.; Demoruelle, M.K.; Deane, K.D. The complex role of the lung in the pathogenesis and clinical outcomes of rheumatoid arthritis. Curr. Rheumatol. Rep. 2016, 18, 69. [Google Scholar] [CrossRef]
- Darrah, E.; Giles, J.T.; Davis, R.L.; Naik, P.; Wang, H.; Konig, M.F.; Cappelli, L.C.; Bingham, C.O., 3rd; Danoff, S.K.; Andrade, F. Autoantibodies to peptidylarginine Deiminase 2 are associated with less severe disease in rheumatoid arthritis. Front. Immunol. 2018, 9, 2696. [Google Scholar] [CrossRef] [PubMed]
- Giles, J.T.; Darrah, E.; Danoff, S.; Johnson, C.; Andrade, F.; Rosen, A.; Bathon, J.M. Association of cross-reactive antibodies targeting peptidyl-arginine deiminase 3 and 4 with rheumatoid arthritis-associated interstitial lung disease. PLoS ONE 2014, 9, e98794. [Google Scholar] [CrossRef]
- Kolarz, B.; Ciesla, M.; Rosenthal, A.K.; Dryglewska, M.; Majdan, M. The value of anti-car P and anti-PAD4 as markers of rheumatoid arthritis in ACPA/RF negative rheumatoid arthritis patients. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X2198986. [Google Scholar] [CrossRef]
- Nava-Quiroz, K.J.; Rojas-Serrano, J.; Pérez-Rubio, G.; Buendia-Roldan, I.; Mejía, M.; Fernández-López, J.C.; Rodríguez-Henríquez, P.; Ayala-Alcantar, N.; Ramos-Martínez, E.; López-Flore, L.A.; et al. Molecular factors in PAD2 (PADI2) and PAD4 (PADI4) are associated with interstitial lung disease susceptibility in rheumatoid arthritis patients. Cells 2023, 12, 2235. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Moreira, R.; Rodríguez-García, S.C.; Gomara, M.J.; Ruiz-Esquide, V.; Cuervo, A.; Casafont-Solé, I.; Ramírez, J.; Holgado, S.; Gómez-Puerta, J.A.; Cañete, J.D.; et al. Anti-carbamylated proteins antibody repertoire in rheumatoid arthritis: Evidence of a new autoantibody linked to interstitial lung disease. Ann. Rheum. Dis. 2020, 79, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.M.; de Smit, M.J.; Brouwer, E.; de Kok, F.A.; Kraan, J.; Altenburg, J.; Verheul, M.K.; Trouw, L.A.; van Winkelhoff, A.J.; Vissink, A.; et al. Rheumatoid arthritis–associated autoantibodies in non–rheumatoid arthritis patients with mucosal inflammation: A case–control study. Arthritis Res. Ther. 2015, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Verheul, M.K.; van Erp, S.J.H.; van der Woude, D.; Levarht, E.W.; Mallat, M.J.; Verspaget, H.W.; Stolk, J.; Toes, R.E.; van der Meulen-de Jong, A.E.; Hiemstra, P.S.; et al. Anti-carbamylated protein antibodies: A specific hallmark for rheumatoid arthritis. Comparison to conditions known for enhanced carbamylation; renal failure, smoking and chronic inflammation. Ann. Rheum. Dis. 2016, 75, 1575–1576. [Google Scholar] [CrossRef] [PubMed]
- England, B.R.; Duryee, M.J.; Roul, P.; Mahajan, T.D.; Singh, N.; Poole, J.A.; Ascherman, D.P.; Caplan, L.; Demoruelle, M.K.; Deane, K.D.; et al. Malondialdehyde-acetaldehyde adducts and antibody responses in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. 2019, 71, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Enein, A.; Abdelsalam, N.; Balata, M.; Abdellatif, S.; Rizk, E.; Fathy, A. Utility of Anti-Carbamylated Protein Antibodies in the Diagnosis of Early Rheumatoid Arthritis. Indian J. Rheumatol. 2019, 14, 37–41. [Google Scholar] [CrossRef]
- Nakabo, S. Clinical and Etiological Meaning of Anti-Carbamylated Protein Antibodies in Rheumatoid Arthritis. Immunol. Med. 2018, 41, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Yang, H.Y.; Luo, S.F.; Lai, J.H. From Rheumatoid Factor to Anti-Citrullinated Protein Antibodies and Anti-Carbamylated Protein Antibodies for Diagnosis and Prognosis Prediction in Patients with Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 686. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.C.; Sung, Y.W.; Chen, K.Y.; Wang, P.Y.; Yen, C.Y.; Sung, W.Y.; Wu, C.C.; Ou, T.T.; Tsai, W.C.; Liao, W.T.; et al. The Role of Macrophages in Connective Tissue Disease-Associated Interstitial Lung Disease: Focusing on Molecular Mechanisms and Potential Treatment Strategies. Int. J. Mol. Sci. 2023, 24, 11995. [Google Scholar] [CrossRef]
- Kulshrestha, R.; Dhanda, H.; Pandey, A.; Singh, A.; Kumar, R. Immunopathogenesis and therapeutic potential of macrophage influx in diffuse parenchymal lung diseases. Expert Rev. Respir. Med. 2020, 14, 917–928. [Google Scholar] [CrossRef]
- Tanguy, J.; Pommerolle, L.; Garrido, C.; Kolb, M.; Bonniaud, P.; Goirand, F.; Bellaye, P.S. Extracellular heat shock proteins as therapeutic targets and biomarkers in fibrosing interstitial lung diseases. Int. J. Mol. Sci. 2021, 22, 9316. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kotani, T.; Kuwabara, H.; Suzuka, T.; Kiboshi, T.; Wada, Y.; Ishida, T.; Fujiki, Y.; Shiba, H.; Hata, K.; et al. Association of M2 macrophages, Th2, and B cells with pathomechanism in microscopic polyangiitis complicated by interstitial lung disease. J. Rheumatol. 2022, 49, 913–921. [Google Scholar] [CrossRef] [PubMed]
- McCubbrey, A.L.; Barthel, L.; Mohning, M.P.; Redente, E.F.; Mould, K.J.; Thomas, S.M.; Leach, S.M.; Danhorn, T.; Gibbings, S.L.; Jakubzick, C.V.; et al. Deletion of c-FLIP from CD11b(hi) Macrophages Prevents Development of Bleomycin-induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Bozec, A.; Ramming, A.; Schett, G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 2018, 15, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Iwasaki, T.; Kitano, S.; Satake, A.; Nomura, S.; Furukawa, T.; Matsui, K.; Sano, H. IL-2-Anti-IL-2 Mono-clonal antibody immune complexes inhibit collagen induced arthritis by augmenting regulatory T cell functions. J. Immunol. 2018, 201, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Hong, W.; Guo, Y.; Zhang, P.; Fang, Y.; Wang, X.; Chen, X.; Lu, S.; Wei, W. Ontogeny of synovial macrophages and the roles of synovial macrophages from different origins in arthritis. Front. Immunol. 2019, 10, 1146. [Google Scholar] [CrossRef]
- Gabbiani, G.; Ryan, G.; Majno, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Hansel, C.; Jendrossek, V.; Klein, D. Cellular senescence in the lung: The central role of senescent epithelial cells. Int. J. Mol. Sci. 2020, 21, 3279. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Marmai, C.; Sutherland, R.E.; Kim, K.K.; Dolganov, G.M.; Fang, X.; Kim, S.S.; Jiang, S.; Golden, J.A.; Hoopes, C.W.; Matthay, M.A. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L71–L78. [Google Scholar] [CrossRef] [PubMed]
- Coutant, F.; Miossec, P. Altered dendritic cell functions in autoimmune diseases: Distinct and overlapping profiles. Nat. Rev. Rheumatol. 2016, 12, 703–715. [Google Scholar] [CrossRef]
- Johnson, C. Recent Advances in the Pathogenesis, Prediction, and Management of Rheumatoid Arthritis-Associated Interstitial Lung Disease. Curr. Opin. Rheumatol. 2017, 29, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Pruijn, G.J.M. Citrullination and Carbamylation in the Pathophysiology of Rheumatoid Arthritis. Front. Immunol. 2015, 6, 192. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Minhas, R.; Shankar, S.; Taha, O. Rheumatoid arthritis-associated interstitial lung disease. QJM 2019, 112, 815–816. [Google Scholar] [CrossRef] [PubMed]
- Douglas, D.; Keating, L.; Strykowski, R.; Lee, C.T.; Garcia, N.; Selvan, K.; Kaushik, N.; Bauer Ventura, I.; Jablonski, R.; Vij, R. Tobacco smoking is associated with combined pulmonary fibrosis and emphysema and worse outcomes in interstitial lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2023, 325, L233–L243. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.U.; Denton, C.P. Interstitial lung disease in connective tissue disease--mechanisms and management. Nat. Rev. Rheumatol. 2014, 10, 728–739. [Google Scholar] [CrossRef]
- Kondo, N.; Kuroda, T.; Kobayashi, D. Cytokine networks in the pathogenesis of rheumatoid arthritis. Int. J. Mol. Sci. 2021, 22, 10922. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, D.A.; Poenariu, I.S.; Crînguș, L.I.; Vreju, F.A.; Turcu-Stiolica, A.; Tica, A.A.; Padureanu, V.; Dumitrascu, R.M.; Banicioiu-Covei, S.; Dinescu, S.C.; et al. JAK/STAT pathway in pathology of rheumatoid arthritis. Exp. Ther. Med. 2020, 20, 498–3503. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Jantan, I.; Bukhari, S.N.A. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed. Pharmacother. 2017, 92, 615–633. [Google Scholar] [CrossRef] [PubMed]
- Györfi, A.H.; Matei, A.-E.; Distler, J.H. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lin, W.; Chen, Z.; Wang, Y.; Huang, Y.; Tu, S. Effect of tumor necrosis factor inhibitors on interstitial lung disease in rheumatoid arthritis: Angel or demon? Drug Des. Dev. Ther. 2019, 1, 2111–2125. [Google Scholar] [CrossRef] [PubMed]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.-R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [PubMed]
- Mena-Vázquez, N.; Godoy-Navarrete, F.J.; Lisbona-Montañez, J.M.; Redondo-Rodriguez, R.; Manrique-Arija, S.; Rioja, J.; Mucientes, A.; Ruiz-Limón, P.; Garcia-Studer, A.; Ortiz-Márquez, F.; et al. Inflammatory Biomarkers in the Diagnosis and Prognosis of Rheumatoid Arthritis–Associated Interstitial Lung Disease. Int. J. Mol. Sci. 2023, 24, 6800. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Xia, L.; Lu, J. Interleukin-4 in rheumatoid arthritis patients with interstitial lung disease: A pilot study. Indian J. Med. Res. 2013, 138, 919–921. [Google Scholar]
- Wang, X.; Zhu, G.; Ren, Q.; Wu, J.; Gu, B.; Su, D.; Shen, M. Increased interleukin-11 associated with disease activity and development of interstitial lung disease in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2022, 40, 135–141. [Google Scholar] [CrossRef]
- Xiangyang, Z.; Lutian, Y.; Lin, Z.; Liping, X.; Hui, S.; Jing, L. Increased levels of interleukin-33 associated with bone erosion and interstitial lung diseases in patients with rheumatoid arthritis. Cytokine 2012, 58, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.S.; El-Barbary, A.M.; Nada, D.W.; Gaber, R.A.; Elkolaly, R.M.; Aboelhawa, M.A. Identification of serum interleukin-13 and interleukin-13 receptor subunit expressions: Rheumatoid arthritis–associated interstitial lung disease. Int. J. Rheum. Dis. 2021, 24, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Hashimoto, M.; Ito, I.; Kubo, T.; Uozumi, R.; Furu, M.; Ito, H.; Fujii, T.; Tanaka, M.; Terao, C.; et al. Interleukin-18 is associated with the presence of interstitial lung disease in rheumatoid arthritis: A cross-sectional study. Scand. J. Rheumatol. 2019, 48, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, D.; Wang, L.; Wang, S.; Roden, A.C.; Zhao, H.; Li, X.; Prakash, Y.S.; Matteson, E.L.; Tschumperlin, D.J.; et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L487–L497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, S.; Lau, J.; Roden, A.C.; Matteson, E.L.; Sun, J.; Luo, F.; Tschumperlin, D.J.; Vassallo, R. IL-23 amplifies the epithelial-mesenchymal transition of mechanically conditioned alveolar epithelial cells in rheumatoid arthritis-associated interstitial lung disease through mTOR/S6 signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L1006–L1022. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.; Miossec, P. IL-17 in Rheumatoid Arthritis and Precision Medicine: From Synovitis Expression to Circulating Bioactive Levels. Front. Med. 2019, 5, 364. [Google Scholar] [CrossRef]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; Barbera, L.L.; Ciccia, F.; Guggino, G. Role of IL-23/IL-17 pathway in rheumatic diseases: An overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef] [PubMed]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune mecha- nisms in pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef]
- Przybylo, J.A.; Radisky, D.C. Matrix metalloproteinase-induced epithelial–mesenchymal transition: Tumor progression at Snail’s pace. Int. J. Biochem. Cell Biol. 2007, 39, 1082–1088. [Google Scholar] [CrossRef]
- Yamashita, C.M.; Dolgonos, L.; Zemans, R.L.; Young, S.K.; Robertson, J.; Briones, N.; Suzuki, T.; Campbell, M.N.; Gauldie, J.; Radisky, D.C. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am. J. Pathol. 2011, 179, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Hu, W.; Wu, S.; Wang, J.; Chi, S.; Liu, X. Development of a risk nomogram model for identifying interstitial lung disease in patients with rheumatoid arthritis. Front. Immunol. 2022, 13, 823669. [Google Scholar] [CrossRef]
- Kollar, B.; Uffing, A.; Borges, T.J.; Shubin, A.V.; Aoyama, B.T.; Dagot, C.; Haug, V.; Kauke, M.; Safi, A.-F.; Talbot, S.G. MMP3 is a non-invasive biomarker of rejection in skin-bearing vascularized composite allotransplantation: A multicenter validation study. Front. Immunol. 2019, 10, 2771. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Doyle, T.J.; Liu, Y.; Aggarwal, R.; Wang, X.; Shi, Y.; Ge, S.X.; Huang, H.; Lin, Q.; Liu, W. Biomarkers of rheumatoid arthritis–associated interstitial lung disease. Arthritis Rheumatol. 2015, 67, 28–38. [Google Scholar] [CrossRef]
- Doyle, T.J.; Lee, J.S.; Dellaripa, P.F.; Lederer, J.A.; Matteson, E.L.; Fischer, A.; Ascherman, D.P.; Glassberg, M.K.; Ryu, J.H.; Danoff, S.K. A roadmap to promote clinical and translational research in rheumatoid arthritis-associated interstitial lung disease. Chest 2014, 145, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.; Assassi, S. Biomarkers in Rheumatic Diseases: How Can They Facilitate Diagnosis and Assessment of Disease Activity? BMJ 2015, 351, h5079. [Google Scholar] [CrossRef]
- Kass, D.J.; Nouraie, M.; Glassberg, M.K.; Ramreddy, N.; Fernandez, K.; Harlow, L.; Zhang, Y.; Chen, J.; Kerr, G.S.; Reimold, A.M.; et al. Comparative Profiling of Serum Protein Biomarkers in Rheumatoid Arthritis–Associated Interstitial Lung Disease and Idiopathic Pulmonary Fibrosis. Arthritis Rheumatol. 2020, 72, 409–419. [Google Scholar] [CrossRef]
- Mahalanobish, S.; Saha, S.; Dutta, S.; Sil, P.C. Matrix Metalloproteinase: An Upcoming Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Pharmacol. Res. 2020, 152, 104591. [Google Scholar] [CrossRef]
- Ma, X.; Chen, R.; Liu, X.; Xie, J.; Si, K.; Duan, L. Effects of matrine on JAK-STAT signaling transduction pathways in bleomycin-induced pulmonary fibrosis. Afr. J. Tradit. Complement. Altern. Med. 2013, 10, 442–448. [Google Scholar] [CrossRef]
- Simon, L.S.; Taylor, P.C.; Choy, E.H.; Sebba, A.; Quebe, A.; Knopp, K.L.; Porreca, F. The Jak/STAT pathway: A focus on pain in rheumatoid arthritis. Semin. Arthritis Rheum. 2021, 51, 278–284. [Google Scholar] [CrossRef]
- Montero, P.; Milara, J.; Roger, I.; Cortijo, J. Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int. J. Mol. Sci. 2021, 22, 6211. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, E.Y.; Ha, Y.J.; Kang, E.H.; Lee, Y.J.; Song, Y.W. Serum Kl-6 Levels Reflect the Severity of Interstitial Lung Disease Associated with Connective Tissue Disease. Arthritis Res. Ther. 2019, 21, 58. [Google Scholar] [CrossRef] [PubMed]
- Oguz, E.O.; Kucuksahin, O.; Turgay, M.; Yildizgoren, M.T.; Ates, A.; Demir, N.; Kumbasar, O.O.; Kinikli, G.; Duzgun, N. Association of Serum KL-6 Levels with Interstitial Lung Disease in Patients with Connective Tissue Disease: A Cross-Sectional Study. Clin. Rheumatol. 2016, 35, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Choi, K.H.; Jacob, J.; Song, J.W. Prognostic role of blood KL-6 in rheumatoid arthritis–associated interstitial lung disease. PLoS ONE 2020, 15, e0229997. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anton, M.-L.; Cardoneanu, A.; Burlui, A.M.; Mihai, I.R.; Richter, P.; Bratoiu, I.; Macovei, L.A.; Rezus, E. The Lung in Rheumatoid Arthritis—Friend or Enemy? Int. J. Mol. Sci. 2024, 25, 6460. https://doi.org/10.3390/ijms25126460
Anton M-L, Cardoneanu A, Burlui AM, Mihai IR, Richter P, Bratoiu I, Macovei LA, Rezus E. The Lung in Rheumatoid Arthritis—Friend or Enemy? International Journal of Molecular Sciences. 2024; 25(12):6460. https://doi.org/10.3390/ijms25126460
Chicago/Turabian StyleAnton, Maria-Luciana, Anca Cardoneanu, Alexandra Maria Burlui, Ioana Ruxandra Mihai, Patricia Richter, Ioana Bratoiu, Luana Andreea Macovei, and Elena Rezus. 2024. "The Lung in Rheumatoid Arthritis—Friend or Enemy?" International Journal of Molecular Sciences 25, no. 12: 6460. https://doi.org/10.3390/ijms25126460
APA StyleAnton, M.-L., Cardoneanu, A., Burlui, A. M., Mihai, I. R., Richter, P., Bratoiu, I., Macovei, L. A., & Rezus, E. (2024). The Lung in Rheumatoid Arthritis—Friend or Enemy? International Journal of Molecular Sciences, 25(12), 6460. https://doi.org/10.3390/ijms25126460