


Maternal Hypermethylated Genes Contribute to Intrauterine Growth Retardation of Piglets in Rongchang Pigs

 ,
,
Abstract
1. Introduction
2. Results
2.1. Statistical Analysis
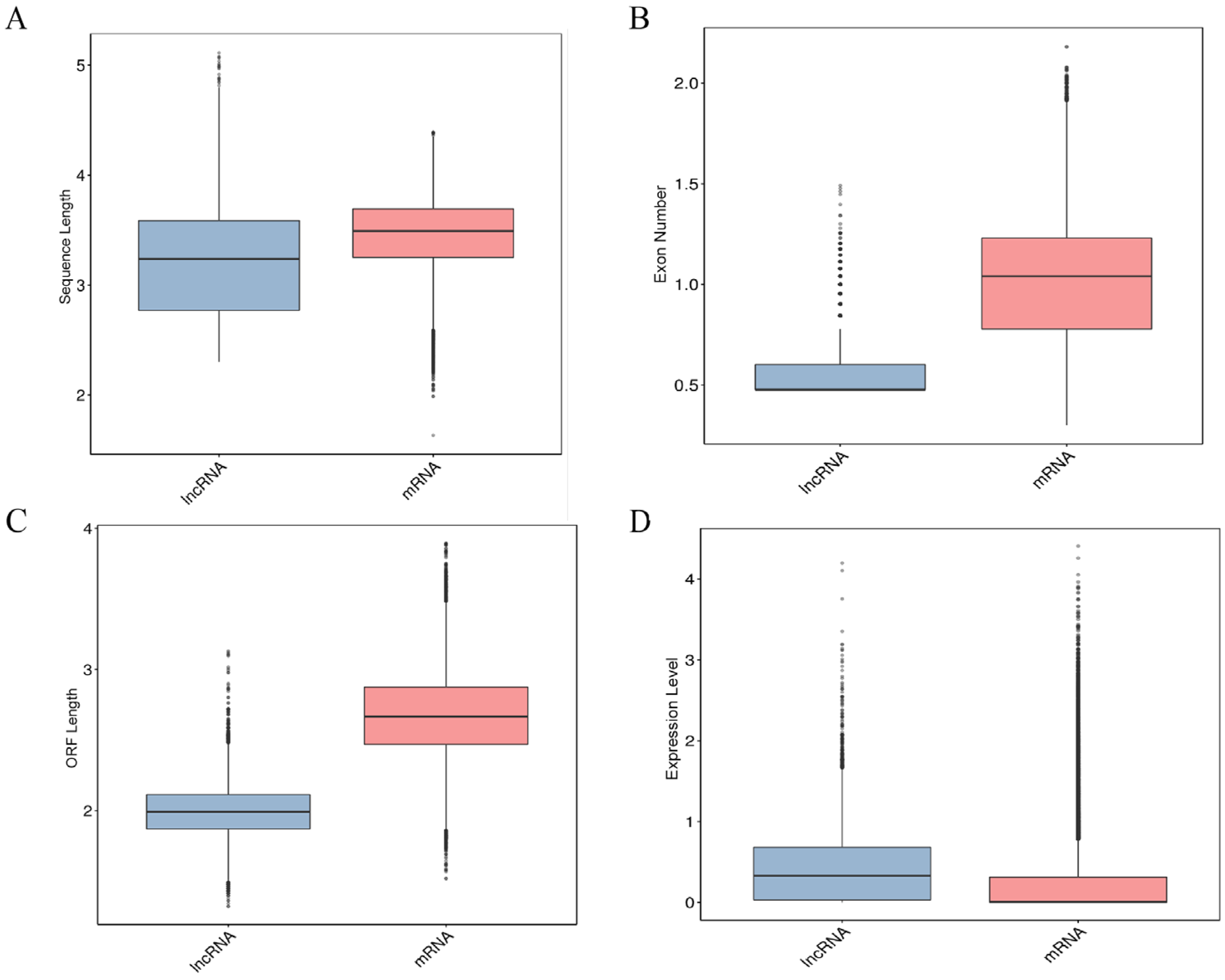
2.2. Comparative Analysis of the Characteristics of lncRNAs and mRNAs
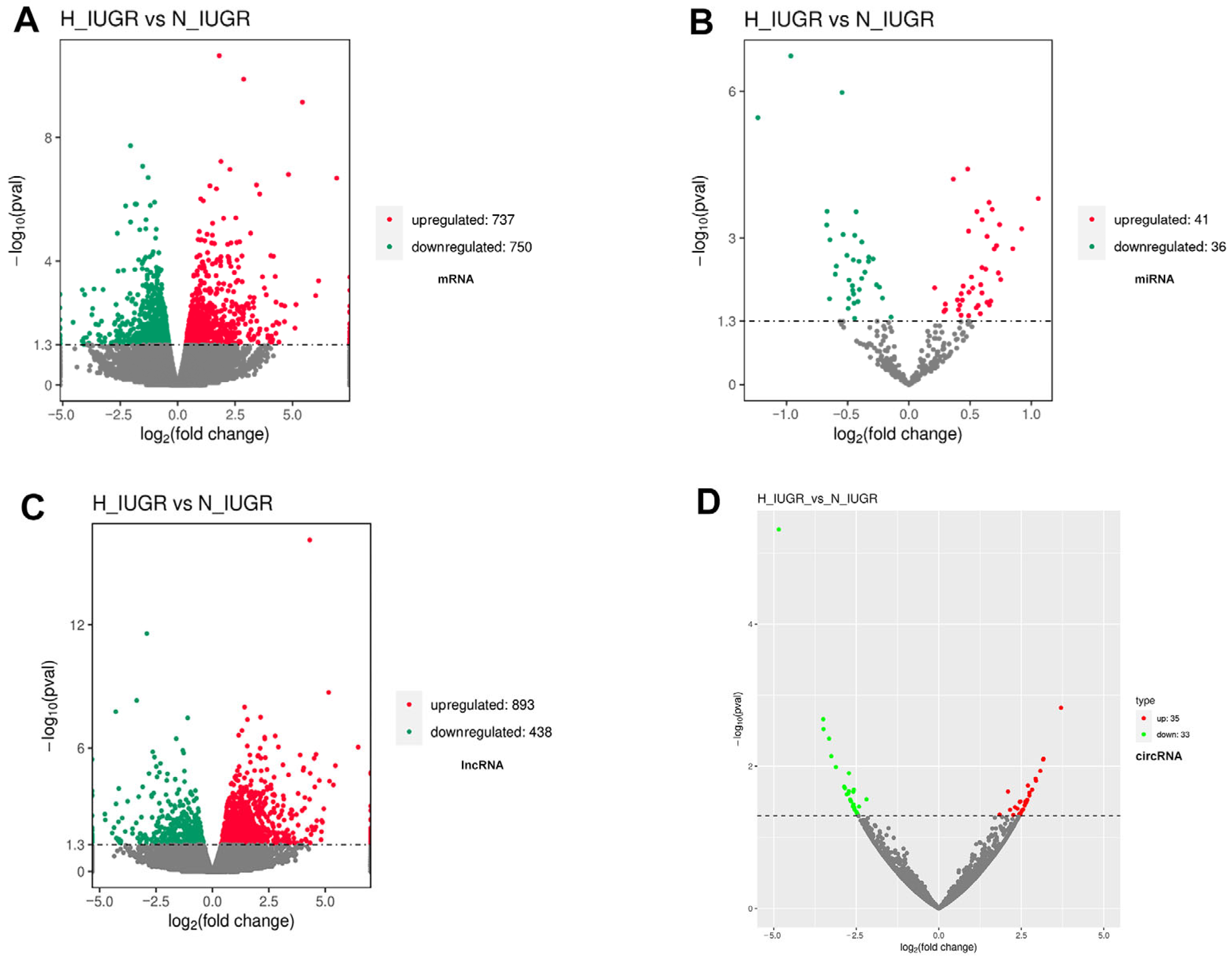
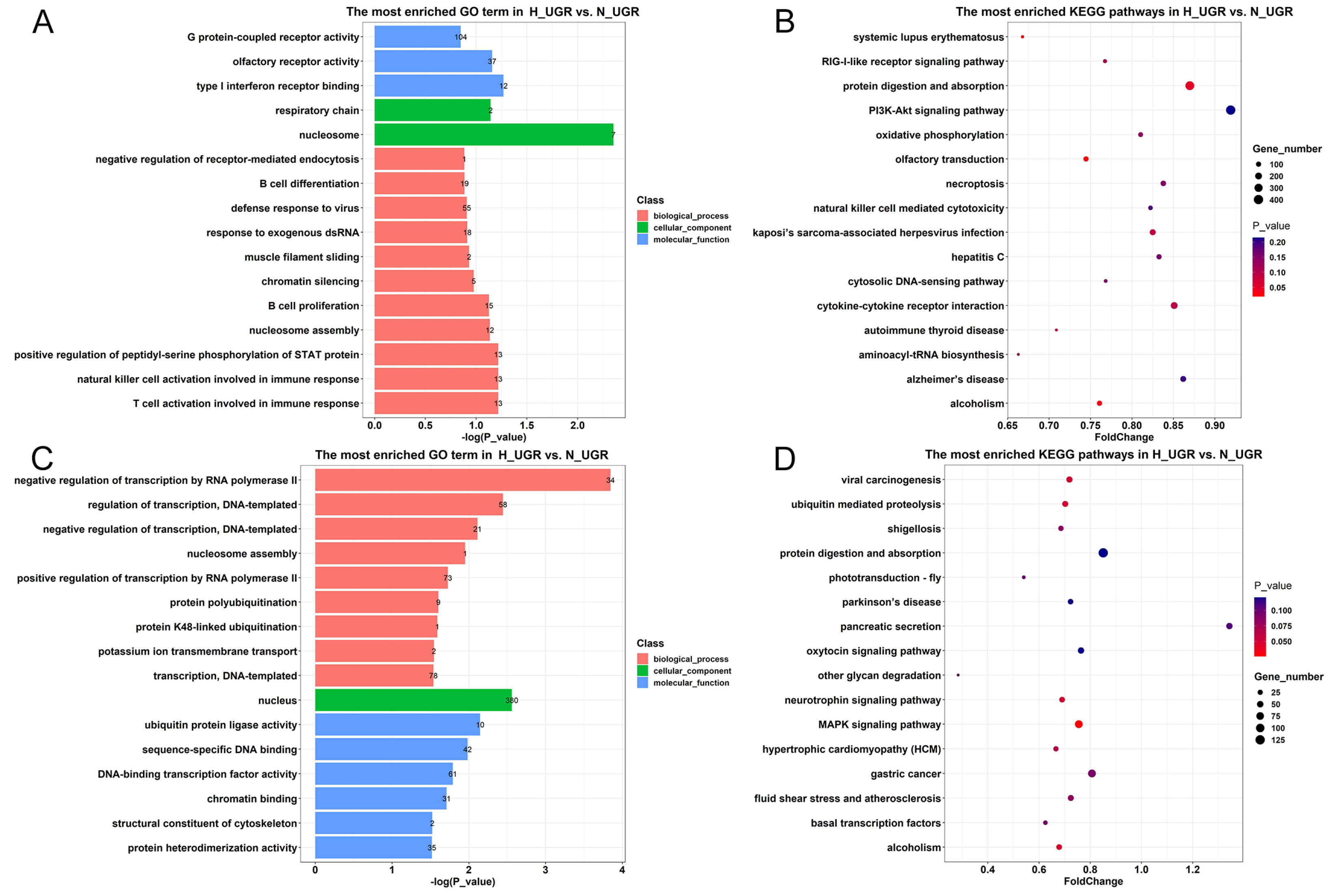
2.3. Detailed Functional Profiles of the DEGs between the H_IUGR and N_IUGR Groups
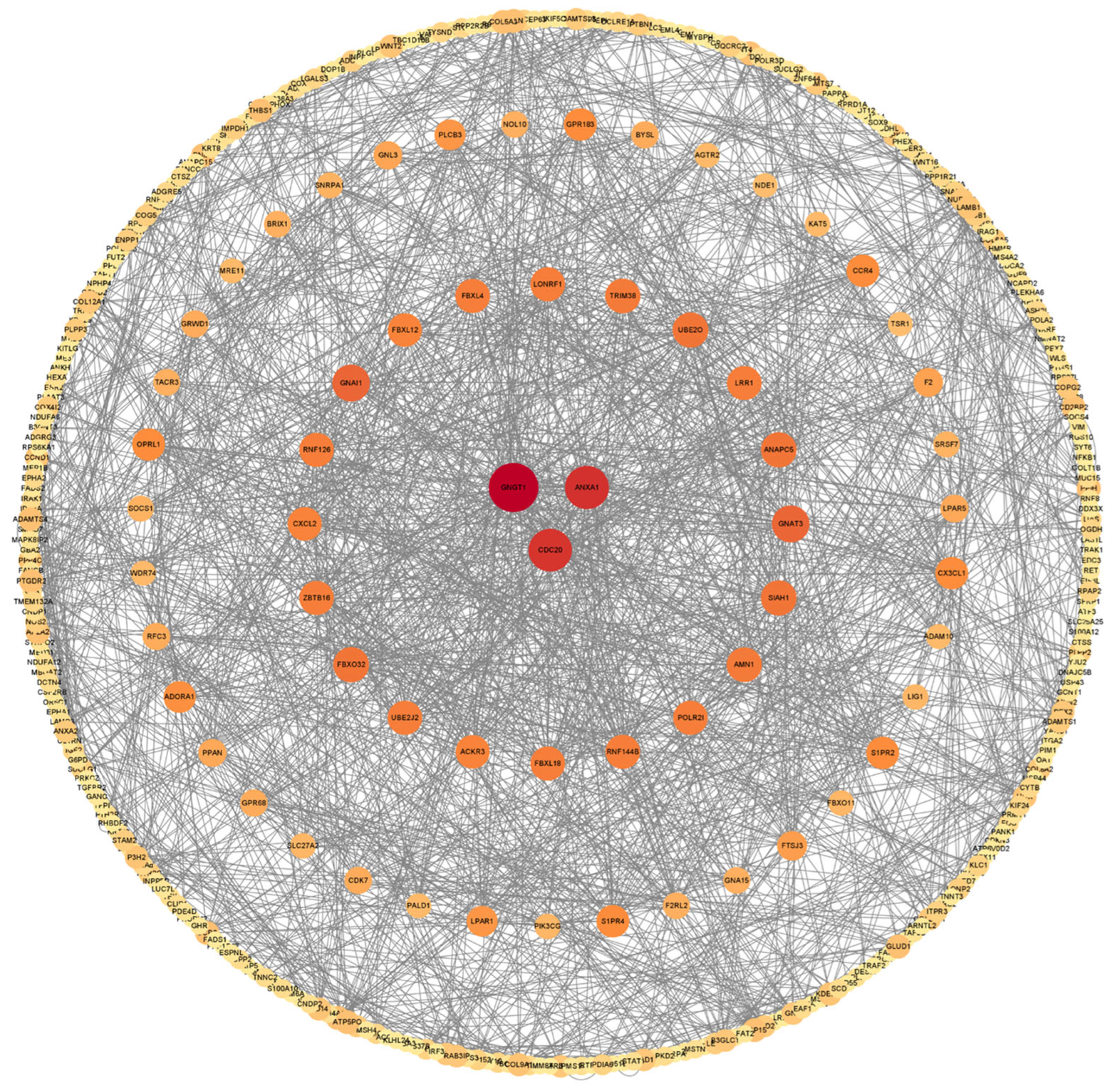
2.4. Protein–Protein Interaction Network of the DEGs
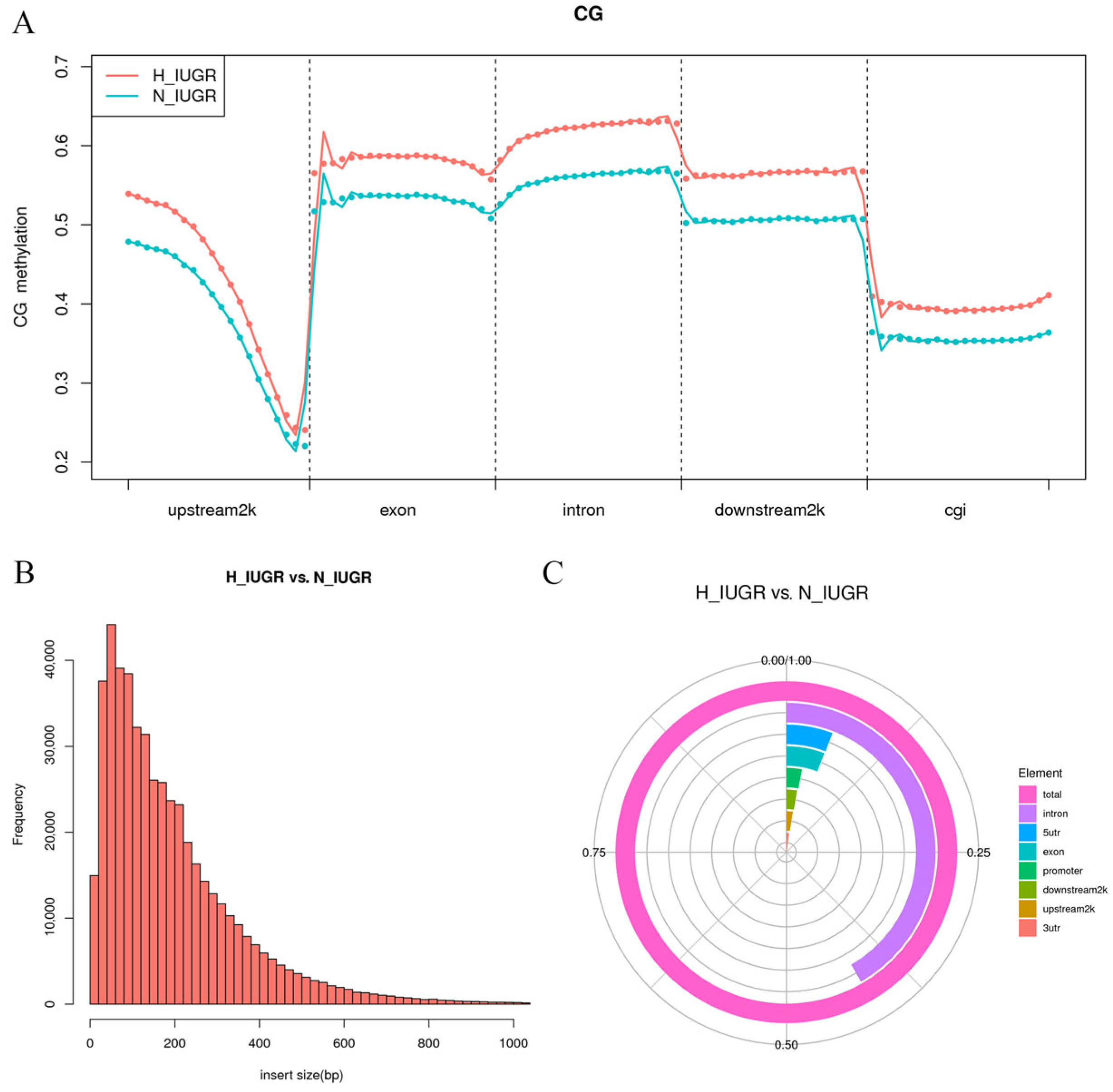
2.5. Genome-Wide DNA Methylation Profiling of Placental Tissues
2.6. Identification of Differentially Methylated Genes and Functional Enrichment Analysis
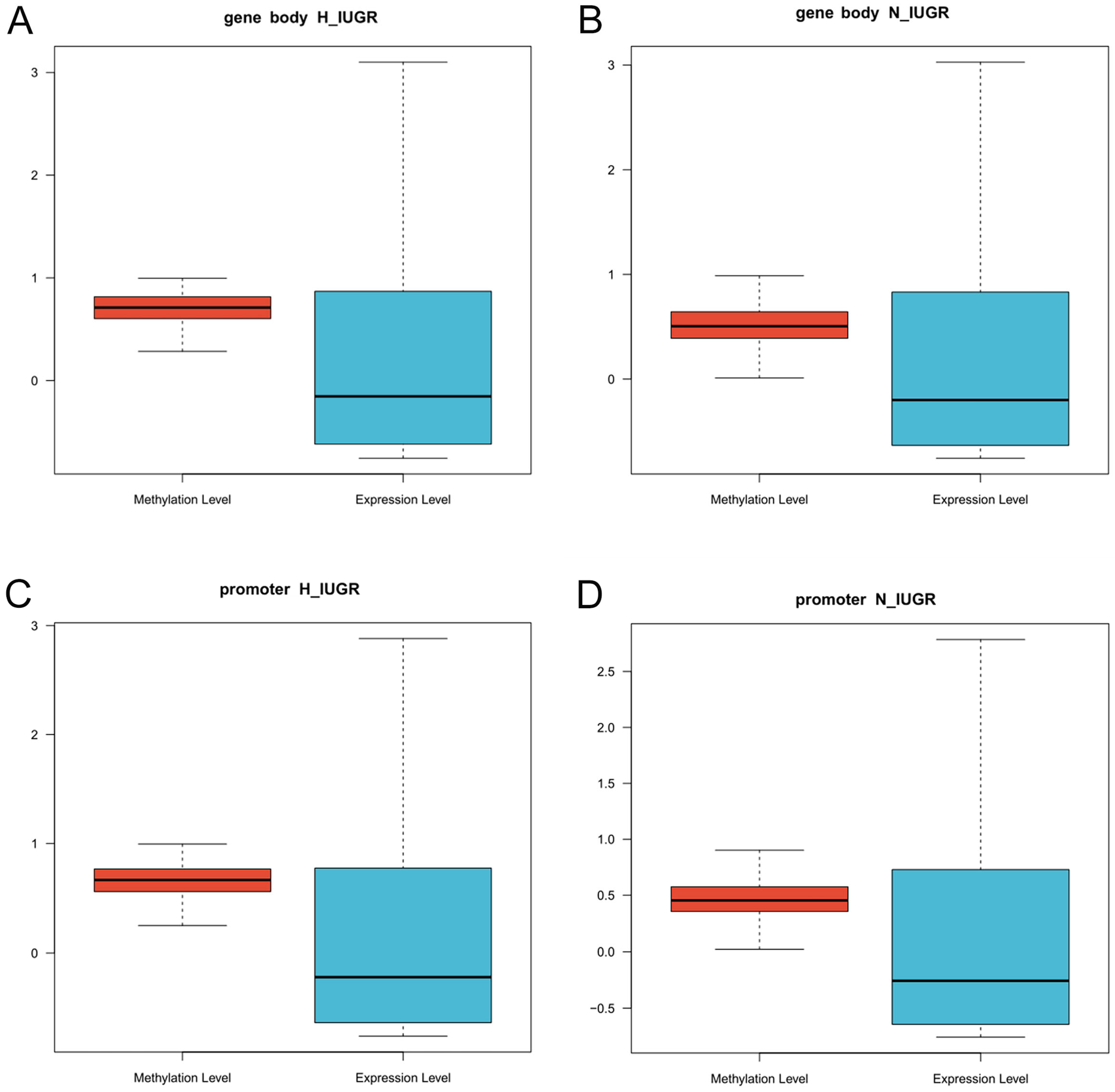
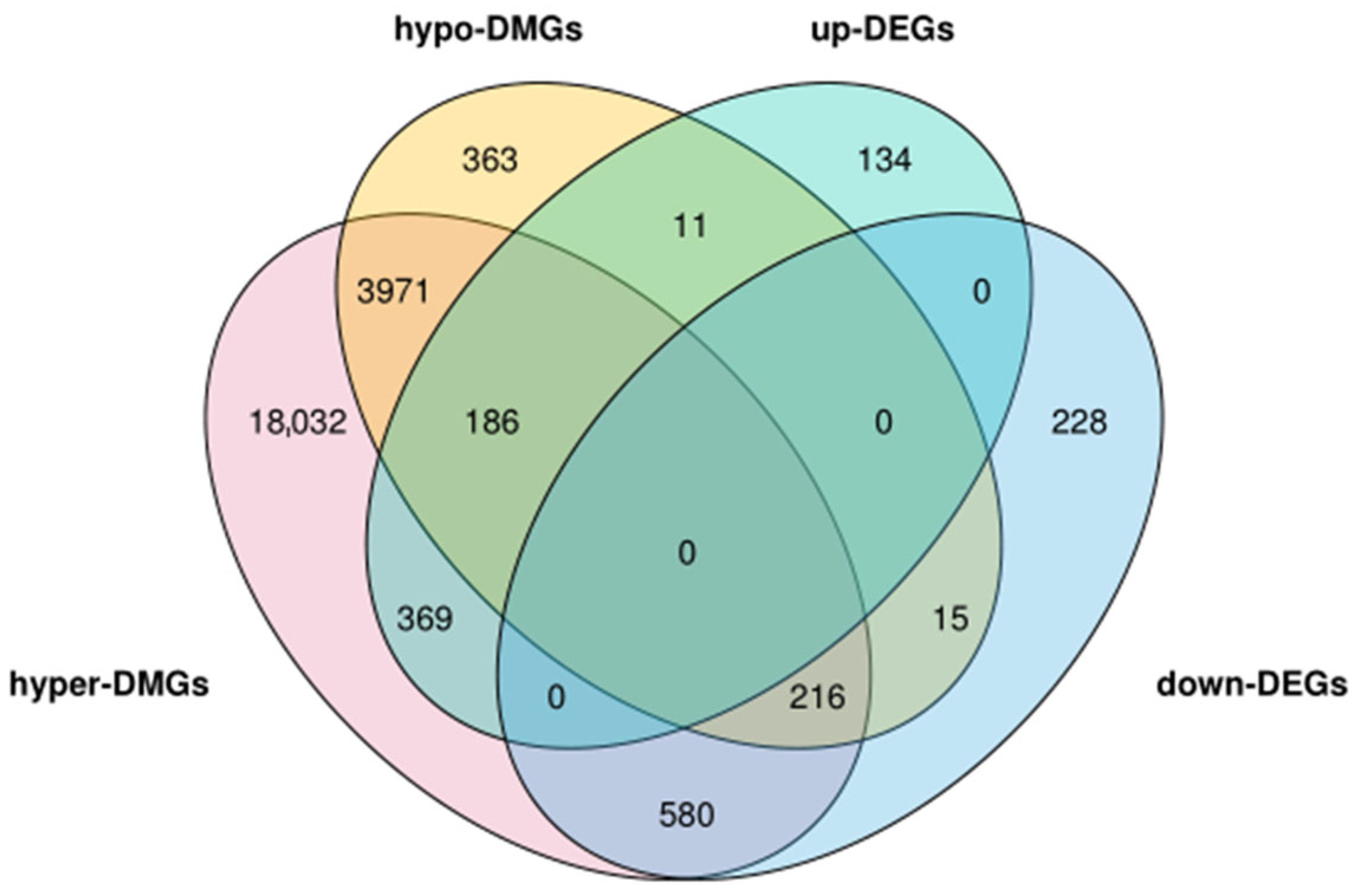
2.7. Combined Analysis of Placental Transcriptome Sequencing and Genome-Wide Methylation Sequencing
2.8. Validation of the Results of RNA-Sequencing Analysis by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Tissue Collection
4.3. Determination of DNA Methylation Degree
4.4. WGBS Analysis
4.5. Annotation of Gene and Genomic Features
4.6. RNA-Seq Analysis
4.7. Identification of DMRs and Enrichment Analysis of DMR-Related Genes
4.8. Quantitative Real-Time PCR (RT-qPCR)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, G.; Bazer, F.W.; Wallace, J.M.; Spencer, T.E. Board-invited review: Intrauterine growth retardation: Implications for the animal sciences. J. Anim. Sci. 2006, 84, 2316–2337. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, Y.; Feng, C.; Lin, G.; Wu, G.; Li, D.; Wang, J. Innate differences and colostrum-induced alterations of jejunal mucosal proteins in piglets with intra-uterine growth restriction. Br. J. Nutr. 2018, 119, 734–747. [Google Scholar] [CrossRef]
- Oksbjerg, N.; Krogh, U.; Jensen, J.A.; Møller, H.S.; Ramaekers, P.; Rasmussen, M.K.; Therkildsen, M.; Theil, P.K. Supplementation of sows with (L)-Arginine during gestating and lactation affects muscle traits of offspring related with postnatal growth and meat quality: From conception to consumption. Meat Sci. 2019, 152, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wu, Z.; Huang, Z.; Hao, X.; Wang, S.; Deng, J.; Yin, Y.; Tan, C. Nox2 impairs VEGF-A-induced angiogenesis in placenta via mitochondrial ROS-STAT3 pathway. Redox Biol. 2021, 45, 102051. [Google Scholar] [CrossRef]
- Sharma, D.; Shastri, S.; Farahbakhsh, N.; Sharma, P. Intrauterine growth restriction—Part 1. J. Matern.-Fetal Neonatal Med. 2016, 29, 3977–3987. [Google Scholar] [CrossRef]
- Salam, R.A.; Das, J.K.; Bhutta, Z.A. Impact of intrauterine growth restriction on long-term health. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Johnson, G.A.; Herring, C.; Seo, H.; Dai, Z.; Wang, J.; Wu, Z.; Wang, X. Functional amino acids in the development of the pig placenta. Mol. Reprod. Dev. 2017, 84, 870–882. [Google Scholar] [CrossRef]
- Robins, J.C.; Marsit, C.J.; Padbury, J.F.; Sharma, S.S. Endocrine disruptors, environmental oxygen, epigenetics and pregnancy. Front. Biosci. 2011, 3, 690–700. [Google Scholar]
- Père, M.C.; Etienne, M. Uterine blood flow in sows: Effects of pregnancy stage and litter size. Reprod. Nutr. Dev. 2000, 40, 369–382. [Google Scholar] [CrossRef]
- Cetin, I.; Antonazzo, P. The role of the placenta in intrauterine growth restriction (IUGR). Z. Geburtshilfe Neonatol. 2009, 213, 84–88. [Google Scholar] [CrossRef]
- Sultana, Z.; Maiti, K.; Dedman, L.; Smith, R. Is there a role for placental senescence in the genesis of obstetric complications and fetal growth restriction? Am. J. Obstet. Gynecol. 2018, 218, S762–S773. [Google Scholar] [CrossRef] [PubMed]
- Guidoni, P.B.; Pasternak, J.A.; Hamonic, G.; MacPhee, D.J.; Harding, J.C.S. Decreased tight junction protein intensity in the placenta of porcine reproductive and respiratory syndrome virus-2 infected fetuses. Placenta 2021, 112, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Cornish, E.F.; McDonnell, T.; Williams, D.J. Chronic Inflammatory Placental Disorders Associated with Recurrent Adverse Pregnancy Outcome. Front. Immunol. 2022, 13, 825075. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ma, W.; Wang, Y.; Wu, C.; Wang, J.; Ma, Z.; Wei, Y.; Cui, C.; Zhang, S.; Guan, W.; et al. Heat Stress-Induced Fetal Intrauterine Growth Restriction Is Associated with Elevated LPS Levels Along the Maternal Intestine-Placenta-Fetus Axis in Pregnant Mice. J. Agric. Food Chem. 2023, 71, 19592–19609. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Busslinger, M.; Hurst, J.; Flavell, R.A. DNA methylation and the regulation of globin gene expression. Cell 1983, 34, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Prather, R.S. Pig genomics for biomedicine. Nat. Biotechnol. 2013, 31, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Wootton, R.; Flecknell, P.A.; Royston, J.P.; John, M. Intrauterine growth retardation detected in several species by non-normal birthweight distributions. J. Reprod. Fertil. 1983, 69, 659–663. [Google Scholar] [CrossRef]
- Terry, E.E.; Zhang, X.; Hoffmann, C.; Hughes, L.D.; Lewis, S.A.; Li, J.; Wallace, M.J.; Riley, L.A.; Douglas, C.M.; Gutierrez-Monreal, M.A.; et al. Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. eLife 2018, 7, e34613. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, S.; Liu, X.; Liu, H.; Hu, T.; Qiu, X.; Zhang, J.; Lei, M. Analyses of Long Non-Coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci. Rep. 2016, 6, 20238. [Google Scholar] [CrossRef]
- Shen, Y.; Mao, H.; Huang, M.; Chen, L.; Chen, J.; Cai, Z.; Wang, Y.; Xu, N. Long Noncoding RNA and mRNA Expression Profiles in the Thyroid Gland of Two Phenotypically Extreme Pig Breeds Using Ribo-Zero RNA Sequencing. Genes 2016, 7, 34. [Google Scholar] [CrossRef]
- Mulligan, T.; Farber, S.A. Central and C-terminal domains of heterotrimeric G protein gamma subunits differentially influence the signaling necessary for primordial germ cell migration. Cell. Signal. 2011, 23, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Lagman, D.; Callado-Pérez, A.; Franzén, I.E.; Larhammar, D.; Abalo, X.M. Transducin duplicates in the zebrafish retina and pineal complex: Differential specialisation after the teleost tetraploidisation. PLoS ONE 2015, 10, e0121330. [Google Scholar] [CrossRef]
- Chen, H.; Leung, T.; Giger, K.E.; Stauffer, A.M.; Humbert, J.E.; Sinha, S.; Horstick, E.J.; Hansen, C.A.; Robishaw, J.D. Expression of the G protein gammaT1 subunit during zebrafish development. Gene Expr. Patterns GEP 2007, 7, 574–583. [Google Scholar] [CrossRef]
- Mendes, M.O.; Rosa, A.I.; Carvalho, A.N.; Nunes, M.J.; Dionísio, P.; Rodrigues, E.; Costa, D.; Duarte-Silva, S.; Maciel, P.; Rodrigues, C.M.P.; et al. Neurotoxic effects of MPTP on mouse cerebral cortex: Modulation of neuroinflammation as a neuroprotective strategy. Mol. Cell. Neurosci. 2019, 96, 1–9. [Google Scholar] [CrossRef]
- Klein, L.; Ophelders, D.; van den Hove, D.; Damoiseaux, M.; Rutten, B.P.F.; Reutelingsperger, C.P.M.; Schurgers, L.J.; Wolfs, T. Prenatal administration of multipotent adult progenitor cells modulates the systemic and cerebral immune response in an ovine model of chorioamnionitis. Brain Behav. Immun.-Health 2022, 23, 100458. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Kim, T.; Oegema, K.; Corbett, K.; Desai, A. A tripartite mechanism catalyzes Mad2-Cdc20 assembly at unattached kinetochores. Science 2021, 371, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Komaravolu, R.K.; Waltmann, M.D.; Konaniah, E.; Jaeschke, A.; Hui, D.Y. ApoER2 (Apolipoprotein E Receptor-2) Deficiency Accelerates Smooth Muscle Cell Senescence via Cytokinesis Impairment and Promotes Fibrotic Neointima After Vascular Injury. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2132–2144. [Google Scholar] [CrossRef]
- Morley, L.C.; Debant, M.; Walker, J.J.; Beech, D.J.; Simpson, N.A.B. Placental blood flow sensing and regulation in fetal growth restriction. Placenta 2021, 113, 23–28. [Google Scholar] [CrossRef]
- Wang, X.; Shi, H.; Li, L.; Yuan, P.; Zhao, Y.; Wei, Y. A study on the correlation of placental characteristics and the onset of selective intrauterine growth restriction. Sci. Prog. 2022, 105, 368504221086358. [Google Scholar] [CrossRef] [PubMed]
- Farsetti, D.; Vasapollo, B.; Pometti, F.; Frantellizzi, R.; Novelli, G.P.; Valensise, H. Maternal hemodynamics for the identification of early fetal growth restriction in normotensive pregnancies. Placenta 2022, 129, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef]
- Lou, S.; Lee, H.M.; Qin, H.; Li, J.W.; Gao, Z.; Liu, X.; Chan, L.L.; Kl Lam, V.; So, W.Y.; Wang, Y.; et al. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 2014, 15, 408. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Coskun, V.; Tao, J.; Xie, W.; Ge, W.; Yoshikawa, K.; Li, E.; Zhang, Y.; Sun, Y.E. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 2010, 329, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef]
- Lou, P.E. Aging-Related DNA Methylomesin Porcine Skeletal Muscles. Ph.D. Thesis, Sichuan Agricultural University, Ya’an, China, 2014. [Google Scholar]
- Hirata, F.; Thibodeau, L.M.; Hirata, A. Ubiquitination and SUMOylation of annexin A1 and helicase activity. Biochim. Biophys. Acta 2010, 1800, 899–905. [Google Scholar] [CrossRef]
- Bełtowski, J.; Jamroz, A. Adrenomedullin--what do we know 10 years since its discovery? Pol. J. Pharmacol. 2004, 56, 5–27. [Google Scholar]
- Wilson, C.; Nikitenko, L.L.; Sargent, I.L.; Rees, M.C. Adrenomedullin: Multiple functions in human pregnancy. Angiogenesis 2004, 7, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Paudel, S.; Flowers, W.L.; Piedrahita, J.A.; Wang, X. Uterine histotroph and conceptus development: III. Adrenomedullin stimulates proliferation, migration and adhesion of porcine trophectoderm cells via AKT-TSC2-MTOR cell signaling pathway. Amino Acids 2023, 55, 743–756. [Google Scholar] [CrossRef]
- Schellenburg, S.; Schulz, A.; Poitz, D.M.; Muders, M.H. Role of neuropilin-2 in the immune system. Mol. Immunol. 2017, 90, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Guo, Y.; Kim, K.; McQueen, P.; Ghaffar, S.; Christ, A.; Lin, C.; Eskander, R.; Zi, X.; Hoang, B.H. Neuropilin-2 expression is inhibited by secreted Wnt antagonists and its down-regulation is associated with reduced tumor growth and metastasis in osteosarcoma. Mol. Cancer 2015, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, G.; von Mering, C.; Basler, K. The hedgehog signaling pathway: Where did it come from? PLoS Biol. 2009, 7, e1000146. [Google Scholar] [CrossRef] [PubMed]
- Salybekov, A.A.; Salybekova, A.K.; Pola, R.; Asahara, T. Sonic Hedgehog Signaling Pathway in Endothelial Progenitor Cell Biology for Vascular Medicine. Int. J. Mol. Sci. 2018, 19, 3040. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sample ID | Birth Weight (kg) | Mean of Litter Weight (kg) | Number of Litter |
|---|---|---|---|---|
| N_IUGR | N_IUGR 1 | 0.76 | 0.68 ± 0.09 | 10 |
| N_IUGR 2 | 0.80 | 0.68 ± 0.10 | 8 | |
| N_IUGR 3 | 0.74 | 0.67 ± 0.09 | 11 | |
| N_IUGR 4 | 0.82 | 0.72 ± 0.09 | 10 | |
| N_IUGR 5 | 0.74 | 0.67 ± 0.10 | 9 | |
| H_IUGR | H_IUGR 1 | 0.44 | 0.67 ± 0.09 | 11 |
| H_IUGR 2 | 0.48 | 0.68 ± 0.10 | 8 | |
| H_IUGR 3 | 0.46 | 0.68 ± 0.08 | 11 | |
| H_IUGR 4 | 0.42 | 0.64 ± 0.08 | 9 | |
| H_IUGR 5 | 0.44 | 0.67 ± 0.09 | 9 |
| Gene | Primer Sequences (5′ to 3′) | TM (°C) | Product Size (bp) |
|---|---|---|---|
| ACTB | F:GATGACGATATTGCTGCGCTC R:TCGATGGGGTACTTGAGGGT | 60 | 121 |
| ANXA1 | F:CTCGATTGCACTGAGGATCA R:GCTGATTCTGGCCACTTCTC | 60 | 116 |
| SPOCK1 | F:GGGCTGGATGTTCAACAAGT R:CTCCAGGTTTCTTGGAAGCAG | 60 | 105 |
| CPS1 | F:AAGTCCTGGGGACCTCAGTT R:AGGACAATGCCTGAGCCTA | 60 | 113 |
| FOSB | F:GTGAAGTTCAAGTCCTCGGC R:TCACAGAGCAAGAAGGGAGG | 60 | 120 |
| IGF2 | F:ATCGTGGAAGAGTGCTGCTT R:CATAGCGGAAGAACTTGCCC | 60 | 114 |
| UCP2 | F:GCCAACAGACGTGGTAAAGG R:TGGCATTACGAGCGACATTG | 60 | 96 |
| ITGA8 | F:CTTTCACATACCTGCGGCTC R:TCAAAGGGTATCTGCCTGCA | 60 | 154 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, P.; Wang, J.; Ji, X.; Chai, J.; Chen, L.; Zhang, T.; Long, X.; Tu, Z.; Chen, S.; Zhang, L.; et al. Maternal Hypermethylated Genes Contribute to Intrauterine Growth Retardation of Piglets in Rongchang Pigs. Int. J. Mol. Sci. 2024, 25, 6462. https://doi.org/10.3390/ijms25126462
Wu P, Wang J, Ji X, Chai J, Chen L, Zhang T, Long X, Tu Z, Chen S, Zhang L, et al. Maternal Hypermethylated Genes Contribute to Intrauterine Growth Retardation of Piglets in Rongchang Pigs. International Journal of Molecular Sciences. 2024; 25(12):6462. https://doi.org/10.3390/ijms25126462
Chicago/Turabian StyleWu, Pingxian, Junge Wang, Xiang Ji, Jie Chai, Li Chen, Tinghuan Zhang, Xi Long, Zhi Tu, Siqing Chen, Lijuan Zhang, and et al. 2024. "Maternal Hypermethylated Genes Contribute to Intrauterine Growth Retardation of Piglets in Rongchang Pigs" International Journal of Molecular Sciences 25, no. 12: 6462. https://doi.org/10.3390/ijms25126462
APA StyleWu, P., Wang, J., Ji, X., Chai, J., Chen, L., Zhang, T., Long, X., Tu, Z., Chen, S., Zhang, L., Wang, K., Zhang, L., Guo, Z., & Wang, J. (2024). Maternal Hypermethylated Genes Contribute to Intrauterine Growth Retardation of Piglets in Rongchang Pigs. International Journal of Molecular Sciences, 25(12), 6462. https://doi.org/10.3390/ijms25126462