
Modifying the Secretome of Mesenchymal Stem Cells Prolongs the Regenerative Treatment Window for Encephalopathy of Prematurity


 and
and 
Abstract
:1. Introduction
2. Results
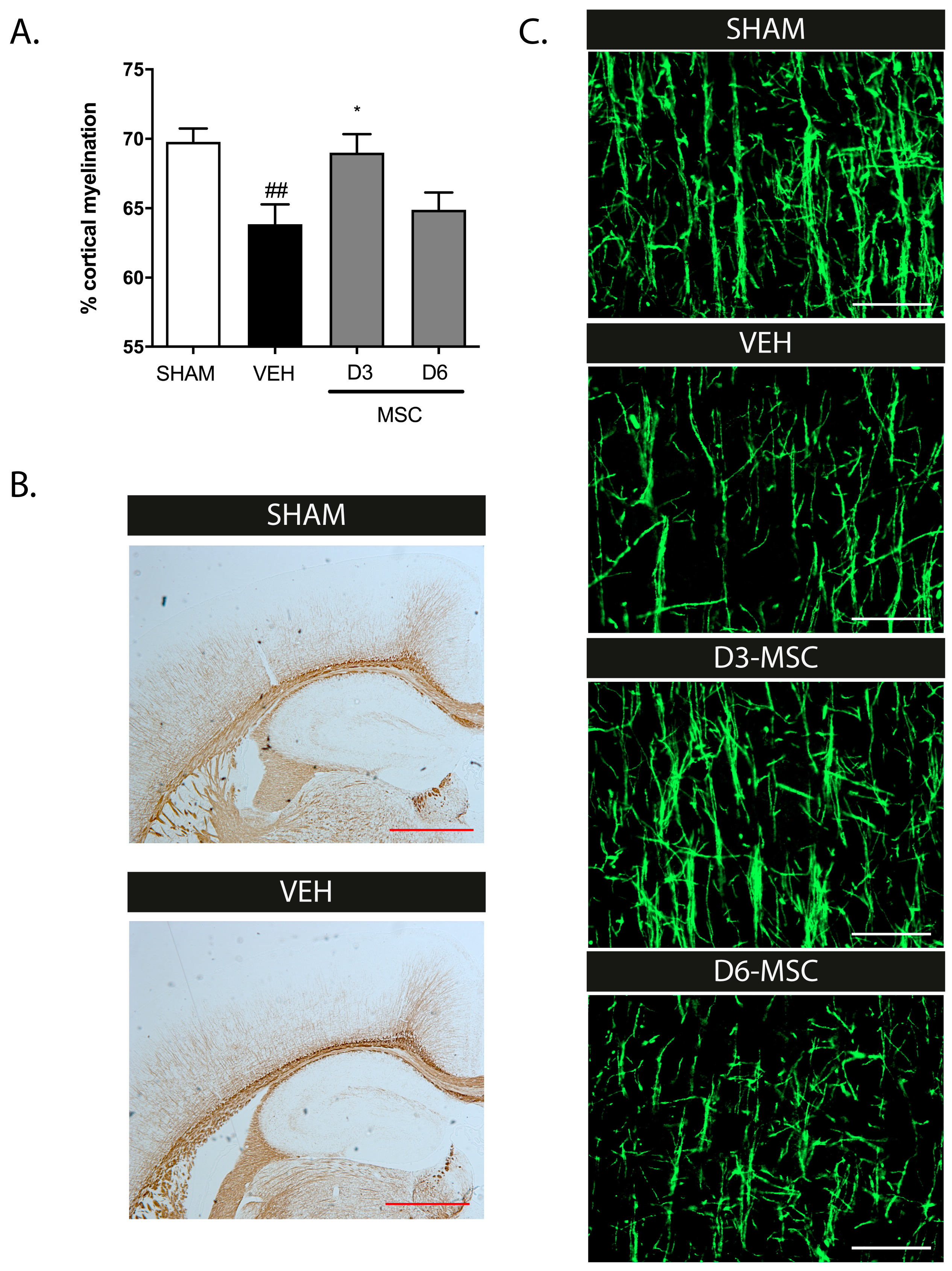
2.1. Intranasal MSC Treatment Efficacy in dWMI: Timing Matters
2.2. Expression of Chemotactic Signals in the Brain following dWMI Induction
2.3. MSCs Change their Secretome In Situ after Treatment Delay
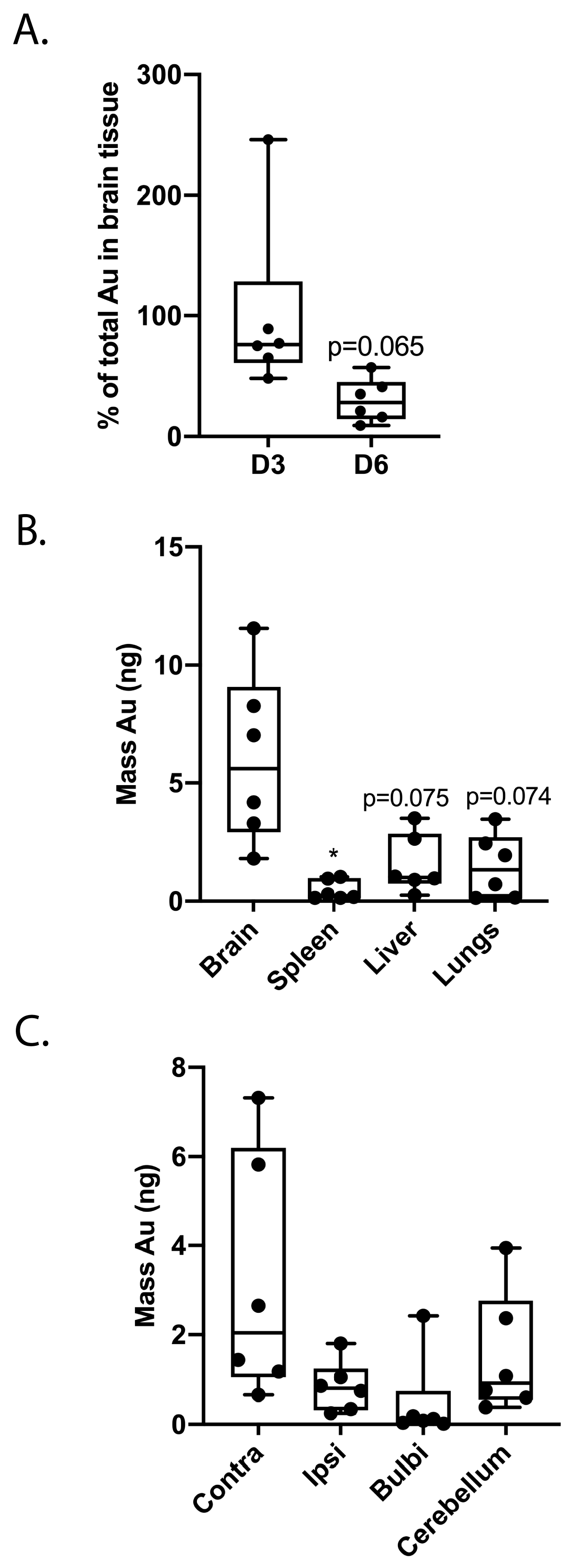
2.4. Treatment Delay Limits MSC Migration after Intranasal Administration
2.5. MSC Modification Leads to Hypersecretion of Selected Factors
2.6. Modification of the MSC Secretome Enhances Myelination and Prolongs the Treatment Window In Vitro
2.7. Modified MSCs Display Superior Anti-Inflammatory Properties on Microglia In Vitro
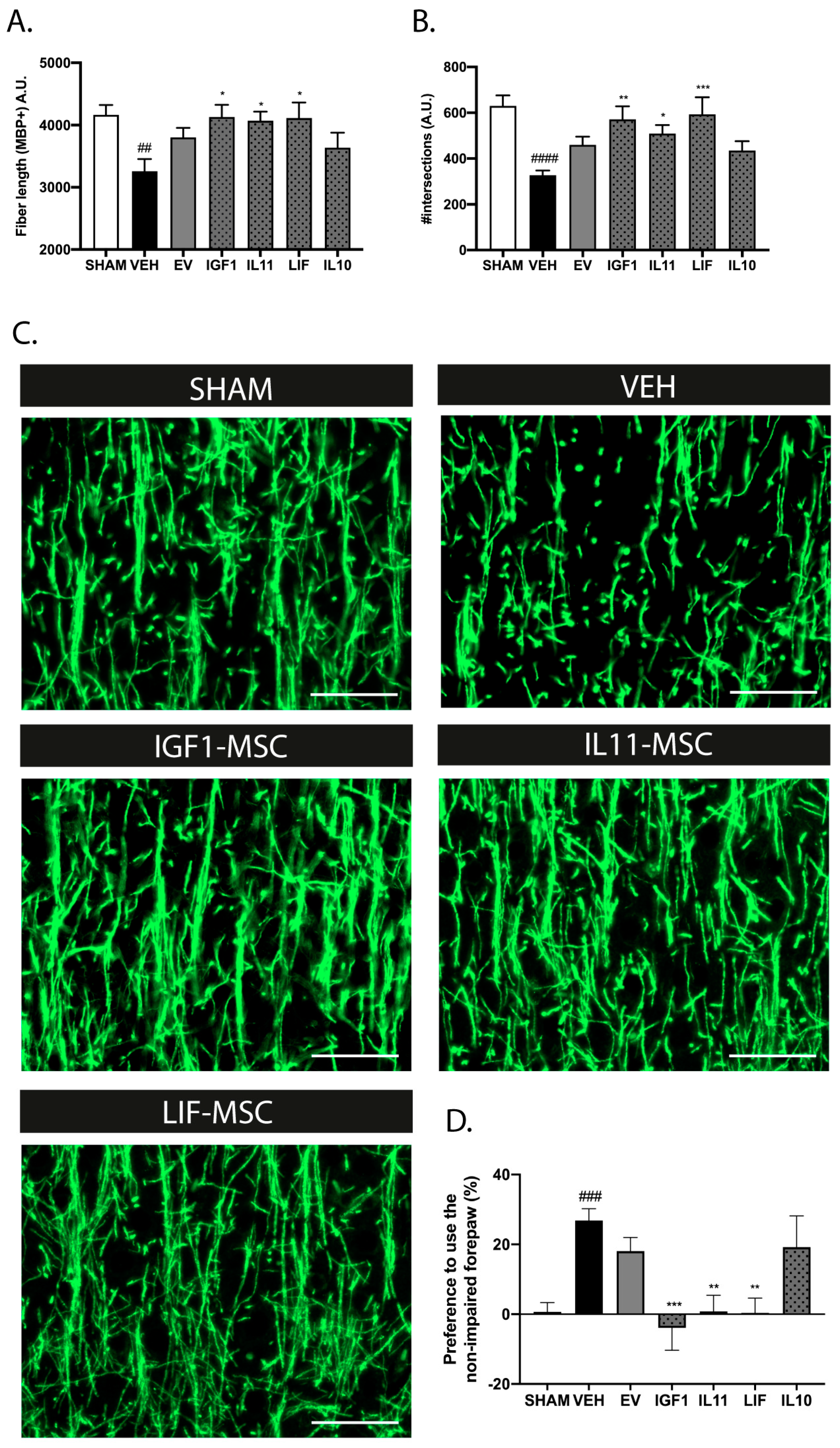
2.8. Intranasal Administration of Modified MSCs Prolongs the Treatment Window after dWMI
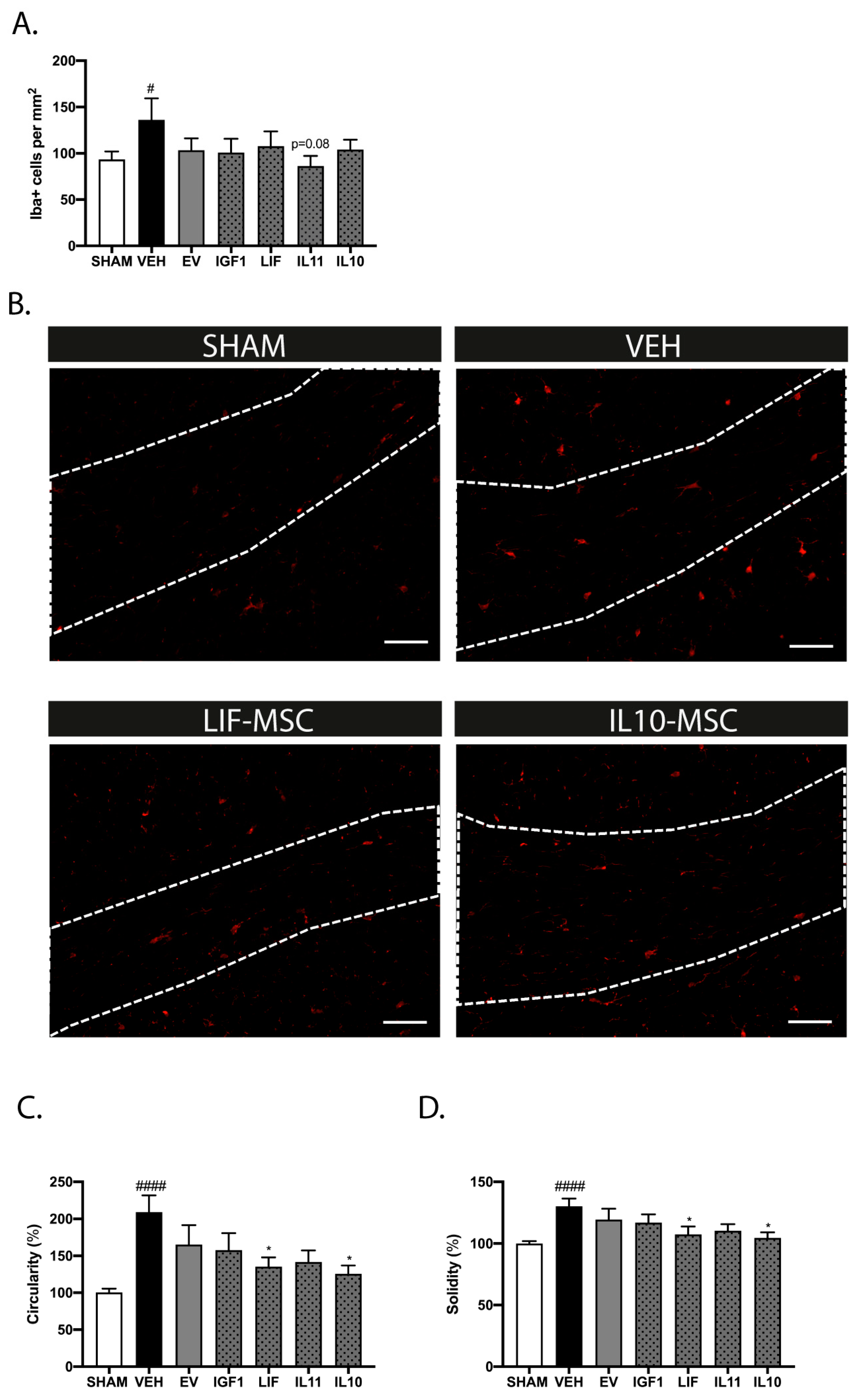
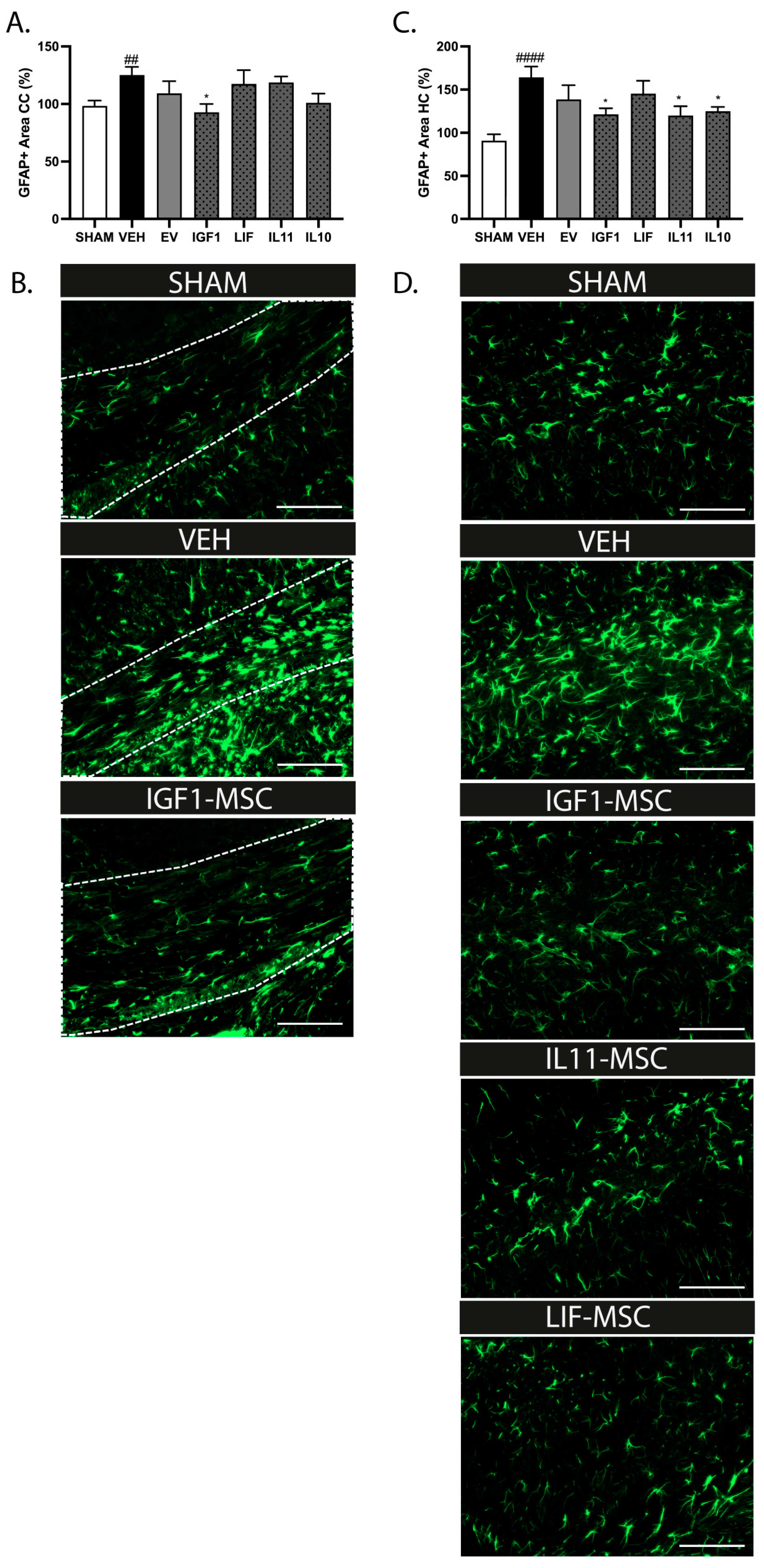
2.9. Modified MSCs Attenuate Neuroinflammation after Delayed Administration following dWMI
3. Discussion
4. Materials and Methods
4.1. MSC Culture
4.2. MSC Transfection
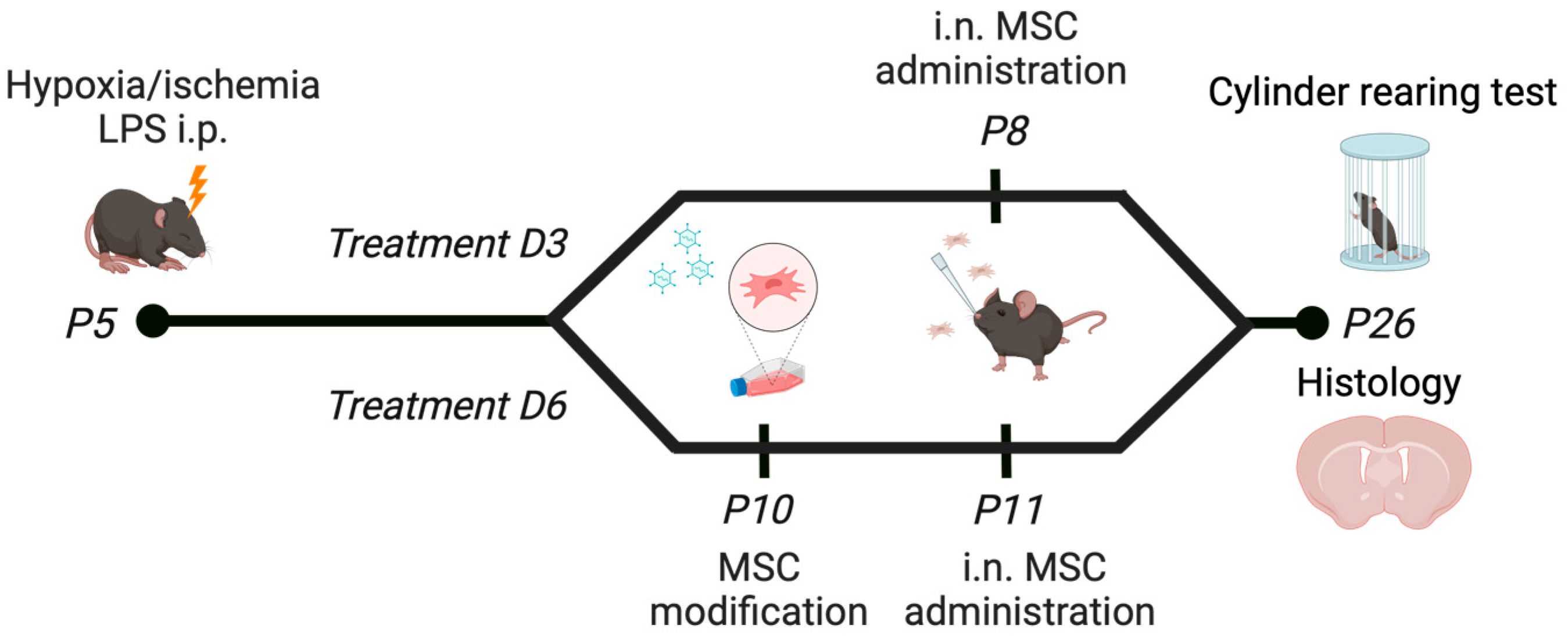
4.3. In Vivo Model of Diffuse White Matter Injury
4.3.1. Cerebral Chemokine Expression Profiles
4.3.2. MSC Gene Expression Profiles after Exposure to Brain Extracts
4.4. MSC Labeling and Cell Tracing
4.5. Immunohistochemistry
4.6. Microscopy and Image Analysis
4.7. Behavioral Assessment
4.8. In Vitro Models of dWMI
4.8.1. Primary Rat Glial Cultures
4.8.2. Primary Mouse Microglia Culture
4.8.3. Non-Contact MSC Glia Cocultures
4.8.4. ELISA
4.8.5. Luminex Assay
4.8.6. Immunocytochemistry of Primary Oligodendrocyte Cultures
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volpe, J.J. The Encephalopathy of Prematurity—Brain injury and impaired brain development inextricably intertwined. Semin. Pediatr. Neurol. 2009, 16, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A. White matter injury in the preterm infant: Pathology and mechanisms. Acta Neuropathol. 2017, 134, 331–349. [Google Scholar] [CrossRef] [PubMed]
- van Tilborg, E.; Heijnen, C.J.; Benders, M.J.; van Bel, F.; Fleiss, B.; Gressens, P.; Nijboer, C.H. Impaired oligodendrocyte maturation in preterm infants: Potential therapeutic targets. Prog. Neurobiol. 2016, 136, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J.; Kinney, H.C.; Jensen, F.E.; Rosenberg, P.A. The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int. J. Dev. Neurosci. 2011, 29, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Vaes, J.E.G.; Vink, M.A.; De Theije, C.G.M.; Hoebeek, F.E.; Benders, M.J.N.L.; Nijboer, C.H.A. The Potential of Stem Cell Therapy to Repair White Matter Injury in Preterm Infants: Lessons Learned From Experimental Models. Front. Physiol. 2019, 10, 540. [Google Scholar] [CrossRef] [PubMed]
- Oppliger, B.; Joerger-Messerli, M.; Mueller, M.; Reinhart, U.; Schneider, P.; Surbek, D.V.; Schoeberlein, A. Intranasal Delivery of Umbilical Cord-Derived Mesenchymal Stem Cells Preserves Myelination in Perinatal Brain Damage. Stem Cells Dev. 2016, 25, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Paton, M.C.B.; Allison, B.J.; Li, J.; Fahey, M.C.; Sutherland, A.E.; Nitsos, I.; Bischof, R.J.; Dean, J.M.; Moss, T.J.M.; Polglase, G.R.; et al. Human Umbilical Cord Blood Therapy Protects Cerebral White Matter from Systemic LPS Exposure in Preterm Fetal Sheep. Dev. Neurosci. 2018, 40, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Vaes, J.E.G.; van Kammen, C.M.; Trayford, C.; van der Toorn, A.; Ruhwedel, T.; Benders, M.J.N.L.; Dijkhuizen, R.M.; Möbius, W.; van Rijt, S.H.; Nijboer, C.H. Intranasal mesenchymal stem cell therapy to boost myelination after Encephalopathy of Prematurity. Glia 2021, 69, 655–680. [Google Scholar] [CrossRef]
- Van Velthoven, C.T.; Kavelaars, A.; Van Bel, F.; Heijnen, C.J. Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav. Immun. 2010, 24, 387–393. [Google Scholar] [CrossRef]
- Liang, X.; Ding, Y.; Zhang, Y.; Tse, H.F.; Lian, Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: Current status and perspectives. Cell Transplant. 2014, 23, 1045–1059. [Google Scholar] [CrossRef]
- Kassis, I.; Vaknin-Dembinsky, A.; Karussis, D. Bone marrow mesenchymal stem cells: Agents of immunomodulation and neuroprotection. Curr. Stem Cell Res. Ther. 2011, 6, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Douglas-Escobar, M.; Weiss, M.D. Biomarkers of brain injury in the premature infant. Front. Neurol. 2012, 3, 185. [Google Scholar] [CrossRef]
- Ment, L.R.; Hirtz, D.; Hüppi, P.S. Imaging biomarkers of outcome in the developing preterm brain. Lancet Neurol. 2009, 8, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Donega, V.; Nijboer, C.H.; Braccioli, L.; Slaper-Cortenbach, I.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Intranasal administration of human MSC for ischemic brain injury in the mouse: In vitro and in vivo neuroregenerative functions. PLoS ONE 2014, 9, e112339. [Google Scholar] [CrossRef]
- Kalwitz, G.; Andreas, K.; Endres, M.; Neumann, K.; Notter, M.; Ringe, J.; Sittinger, M.; Kaps, C. Chemokine Profile of Human Serum from Whole Blood: Migratory Effects of CXCL-10 and CXCL-11 on Human Mesenchymal Stem Cells. Connect. Tissue Res. 2010, 51, 113–122. [Google Scholar] [CrossRef]
- Sordi, V.; Malosio, M.L.; Marchesi, F.; Mercalli, A.; Melzi, R.; Giordano, T.; Belmonte, N.; Ferrari, G.; Leone, B.E.; Bertuzzi, F.; et al. Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood 2005, 106, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Van Tilborg, E.; van Kammen, C.M.; de Theije, C.G.M.; van Meer, M.P.A.; Dijkhuizen, R.M.; Nijboer, C.H. A quantitative method for microstructural analysis of myelinated axons in the injured rodent brain. Sci. Rep. 2017, 7, 16492. [Google Scholar] [CrossRef]
- De Vries, L.S.; Benders, M.J.N.L.; Groenendaal, F. Imaging the premature brain: Ultrasound or MRI? Neuroradiology 2013, 55, 13–22. [Google Scholar] [CrossRef]
- van Velthoven, C.T.; Braccioli, L.; Willemen, H.L.; Kavelaars, A.; Heijnen, C.J. Therapeutic potential of genetically modified mesenchymal stem cells after neonatal hypoxic-ischemic brain damage. Mol. Ther. 2014, 22, 645–654. [Google Scholar] [CrossRef]
- Liu, A.M.; Lu, G.; Tsang, K.S.; Li, G.; Wu, Y.; Huang, Z.S.; Ng, H.K.; Kung, H.F.; Poon, W.S. Umbilical cord-derived mesenchymal stem cells with forced expression of hepatocyte growth factor enhance remyelination and functional recovery in a rat intracerebral hemorrhage model. Neurosurgery 2010, 67, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hu, X.; Zhu, C.; Wang, D.; Zheng, X.; Liu, Q. Overexpression of CNTF in Mesenchymal Stem Cells reduces demyelination and induces clinical recovery in experimental autoimmune encephalomyelitis mice. J. Neuroimmunol. 2009, 206, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, R.; Spohn, G.; Baer, P.C. Mesenchymal Stem/Stromal Cells in Regenerative Medicine: Can Preconditioning Strategies Improve Therapeutic Efficacy. Transfus. Med. Hemotherapy 2016, 43, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Ahn, S.Y.; Sung, S.I.; Ahn, J.-Y.; Chang, Y.S. Strategies to enhance paracrine potency of transplanted mesenchymal stem cells in intractable neonatal disorders. Pediatr. Res. 2018, 83, 214–222. [Google Scholar] [CrossRef]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Mesenchymal stem cell transplantation changes the gene expression profile of the neonatal ischemic brain. Brain Behav. Immun. 2011, 25, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.K.; Kondziolka, D.; Wechsler, L.R.; Lunsford, L.D.; Coburn, M.L.; Billigen, J.B.; Kim, A.S.; Johnson, J.N.; Bates, D.; King, B.; et al. Clinical Outcomes of Transplanted Modified Bone Marrow–Derived Mesenchymal Stem Cells in Stroke: A Phase 1/2a Study. Stroke 2016, 47, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Liu, D.D.; Thakor, A.S. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 2019, 15, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Oppliger, B.; Joerger-Messerli, M.S.; Simillion, C.; Mueller, M.; Surbek, D.V.; Schoeberlein, A. Mesenchymal stromal cells from umbilical cord Wharton’s jelly trigger oligodendroglial differentiation in neural progenitor cells through cell-to-cell contact. Cytotherapy 2017, 19, 829–838. [Google Scholar] [CrossRef]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J. Biomed. Sci. 2018, 25, 31. [Google Scholar] [CrossRef]
- Chamberlain, G.; Wright, K.; Rot, A.; Ashton, B.; Middleton, J. Murine Mesenchymal Stem Cells Exhibit a Restricted Repertoire of Functional Chemokine Receptors: Comparison with Human. PLoS ONE 2008, 3, e2934. [Google Scholar] [CrossRef]
- Moretti, R.; Pansiot, J.; Bettati, D.; Strazielle, N.; Ghersi-Egea, J.-F.; Damante, G.; Fleiss, B.; Titomanlio, L.; Gressens, P. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 2015, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Yap, V.; Perlman, J.M. Mechanisms of brain injury in newborn infants associated with the fetal inflammatory response syndrome. Semin. Fetal Neonatal Med. 2020, 25, 101110. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, L.; Schäfer, R.; von Ameln-Mayerhofer, A.; Buadze, M.; Geisler, J.; Klopfer, T.; Burkhardt, U.; Proksch, B.; Verleysdonk, S.; Ayturan, M.; et al. Intranasal delivery of cells to the brain. Eur. J. Cell Biol. 2009, 88, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, J. Regulatory factors of mesenchymal stem cell migration into injured tissues and their signal transduction mechanisms. Front. Med. 2011, 5, 33–39. [Google Scholar] [CrossRef] [PubMed]
- De Becker, A.; Van Riet, I. Homing and migration of mesenchymal stromal cells: How to improve the efficacy of cell therapy? World J. Stem Cells 2016, 8, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Sung, S.I.; Ahn, S.Y.; Sung, D.K.; Im, G.H.; Yoo, H.S.; Choi, S.J.; Chang, Y.S. Optimal Timing of Mesenchymal Stem Cell Therapy for Neonatal Intraventricular Hemorrhage. Cell Transplant. 2016, 25, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Zappia, E.; Casazza, S.; Pedemonte, E.; Benvenuto, F.; Bonanni, I.; Gerdoni, E.; Giunti, D.; Ceravolo, A.; Cazzanti, F.; Frassoni, F.; et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 2005, 106, 1755–1761. [Google Scholar] [CrossRef]
- Ding, X.; Cao, F.; Cui, L.; Ciric, B.; Zhang, G.-X.; Rostami, A. IL-9 signaling affects central nervous system resident cells during inflammatory stimuli. Exp. Mol. Pathol. 2015, 99, 570–574. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Zanier, E.R.; Fumagalli, S.; Perego, C.; Pischiutta, F.; De Simoni, M.-G. Shape descriptors of the “never resting” microglia in three different acute brain injury models in mice. Intensiv. Care Med. Exp. 2015, 3, 39. [Google Scholar] [CrossRef]
- Chen, Y.; Balasubramaniyan, V.; Peng, J.; Hurlock, E.C.; Tallquist, M.; Li, J.; Lu, Q.R. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat. Protoc. 2007, 2, 1044–1051. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | D3 (P8) | dWMI |
|---|---|---|
| dWMI vs. SHAM | D6 (P11) vs. D3 (P8) | |
| Ccl4 | 9.09 | 1.22 |
| Cxcl10 | 6.47 | 5.21 |
| Ccl3 | 3.74 | −2.86 |
| Cxcl3 | 3.62 | 1.51 |
| Cxcl5 | 3.13 | 1.02 |
| Cxcl1 | −3.57 | 16.45 |
| Symbol | D6 vs. D3 |
|---|---|
| Adipoq | 3.54 |
| Bmp2 | 3.20 |
| Bmp7 | 9.13 |
| Ccl19 | 5.07 |
| Ccl22 | 12.58 |
| Ccl24 | 17.64 |
| Ccl3 | 4.41 |
| Ccl4 | 6.04 |
| Csf2 | 3.42 |
| Cxcl13 | 12.79 |
| Cxcl3 | 3.08 |
| Fasl | −6.64 |
| Fgf13 | 4.72 |
| Ffg3 | 5.86 |
| Fgf4 | 3.59 |
| Fgf5 | 3.61 |
| Fgf8 | 3.42 |
| Hc | 3.07 |
| IfnA2 | 6.34 |
| IfnG | 3.86 |
| Il1b | 3.50 |
| Il2 | 13.65 |
| Il3 | 3.92 |
| Il4 | 4.12 |
| Il5 | 3.04 |
| Il9 | 14.53 |
| Il10 | 8.64 |
| Il12b | 13.52 |
| Il17a | 3.97 |
| Il17f | 7.44 |
| Il22 | 4.33 |
| Il23a | 3.33 |
| Il24 | 6.30 |
| Mstn | 7.99 |
| Nodal | 7.20 |
| Ntf3 | 3.94 |
| Osm | 309.49 |
| Tnf | 7.60 |
| Tnfrsf11b | 5.24 |
| Tnfsf10 | 31.85 |
| Tdgf1 | 12.51 |
| Xcl1 | 10.41 |
| Factor | Condition | MOI | Mean Concentration |
|---|---|---|---|
| IGF1 (pg/mL) | Control (no virus) | - | 30.1 |
| EV-MSC | 2000 | 23.9 | |
| IGF1-MSC | 500 | 111.9 | |
| 1000 | 352.1 | ||
| 2000 | 408 | ||
| EGF (pg/mL) | Control (no virus) | - | 0 |
| EV-MSC | 4000 | 0 | |
| EGF-MSC | 2000 | 2.7 | |
| 4000 | 9.5 | ||
| IL11 (ng/mL) | Control (no virus) | - | 0 |
| EV-MSC | 4000 | 79.5 | |
| IL11-MSC | 2000 | 231.9 | |
| 4000 | 414 | ||
| LIF (pg/mL) | Control (no virus) | - | 2.5 |
| EV-MSC | 8000 | 11.5 | |
| LIF-MSC | 4000 | 278.7 | |
| 8000 | 445.6 | ||
| IL10 (pg/mL) | Control (no virus) | - | 0 |
| EV-MSC | 4000 | 420.5 | |
| IL10-MSC | 1000 | 2295 | |
| 2000 | 2634 | ||
| 4000 | 3725 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaes, J.E.G.; Onstwedder, S.M.; Trayford, C.; Gubbins, E.; Maas, M.; van Rijt, S.H.; Nijboer, C.H. Modifying the Secretome of Mesenchymal Stem Cells Prolongs the Regenerative Treatment Window for Encephalopathy of Prematurity. Int. J. Mol. Sci. 2024, 25, 6494. https://doi.org/10.3390/ijms25126494
Vaes JEG, Onstwedder SM, Trayford C, Gubbins E, Maas M, van Rijt SH, Nijboer CH. Modifying the Secretome of Mesenchymal Stem Cells Prolongs the Regenerative Treatment Window for Encephalopathy of Prematurity. International Journal of Molecular Sciences. 2024; 25(12):6494. https://doi.org/10.3390/ijms25126494
Chicago/Turabian StyleVaes, Josine E. G., Suzanne M. Onstwedder, Chloe Trayford, Eva Gubbins, Mirjam Maas, Sabine H. van Rijt, and Cora H. Nijboer. 2024. "Modifying the Secretome of Mesenchymal Stem Cells Prolongs the Regenerative Treatment Window for Encephalopathy of Prematurity" International Journal of Molecular Sciences 25, no. 12: 6494. https://doi.org/10.3390/ijms25126494
APA StyleVaes, J. E. G., Onstwedder, S. M., Trayford, C., Gubbins, E., Maas, M., van Rijt, S. H., & Nijboer, C. H. (2024). Modifying the Secretome of Mesenchymal Stem Cells Prolongs the Regenerative Treatment Window for Encephalopathy of Prematurity. International Journal of Molecular Sciences, 25(12), 6494. https://doi.org/10.3390/ijms25126494