Abstract
Lysosomes are highly dynamic organelles that maintain cellular homeostasis and regulate fundamental cellular processes by integrating multiple metabolic pathways. Lysosomal ion channels such as TRPML1-3, TPC1/2, ClC6/7, CLN7, and TMEM175 mediate the flux of Ca2+, Cl−, Na+, H+, and K+ across lysosomal membranes in response to osmotic stimulus, nutrient-dependent signals, and cellular stresses. These ion channels serve as the crucial transducers of cell signals and are essential for the regulation of lysosomal biogenesis, motility, membrane contact site formation, and lysosomal homeostasis. In terms of pathophysiology, genetic variations in these channel genes have been associated with the development of lysosomal storage diseases, neurodegenerative diseases, inflammation, and cancer. This review aims to discuss the current understanding of the role of these ion channels in the central nervous system and to assess their potential as drug targets.
1. Introduction
Intracellular organelles engage in the mutual exchange of their internal contents through various mechanisms, including vesicular and non-vesicular pathways [1]. The lysosome, an acidic organelle, serves as a degradation center and signaling hub in the cell, playing a crucial role in cellular homeostasis, development, and aging [2]. As a dynamic and heterogeneous organelle with a single limiting membrane, its morphology, size, enzyme content, and substrates vary [3]. The unique low pH characteristic of lysosomes promotes the activity of luminal hydrolytic enzymes, laying the foundation for the breakdown of macromolecules, and leading to the production of amino acids, monosaccharides, and free fatty acids [4]. Lysosomes establish membrane contact sites with the endoplasmic reticulum (ER) and mitochondria, facilitating the bidirectional transfer of metabolites and ions [5]. This interaction influences lysosomal functions, such as mobility, membrane remodeling, and repair.
Lysosomes contain over 60 soluble acidic hydrolases and 200 integral and peripheral membrane proteins [6]. The acidic hydrolases, predominantly located within the lysosomal lumen, carry out the degradation functions of the lysosome and exhibit specificity towards various substrates [7]. The lysosomal membrane proteins, encompassing a variety of transport proteins and ion channels, such as transient receptor potential mucolipin1-3 (TRPML1-3), two-pore channels 1/2 (TPC1/2), and transmembrane protein 175 (TMEM175), not only serve as a natural barrier between the acidic lysosome and the weakly basic cytoplasm, but are also necessary for lysosomal function and balanced physiological environment. These channels function in lysosomal exocytosis, membrane potential, protein degradation, autophagy, phagocytosis, and cellular signal transduction through regulating the influx and efflux of ions such as H+, Na+, K+, Ca2+, and Cl− [8,9,10,11,12,13,14,15,16]. The disruption or dysfunction of lysosomal ion channels can lead to morphological abnormalities, vesicular transport defects, inflammation, and neurodegenerative diseases [17]. This review summarized the expression and functions of lysosomal channels in the central nervous system (CNS).
2. Lysosome Function
Lysosomes, ubiquitous organelles found in nearly all eukaryotic cells, are formed through the fusion of endosomes and vesicles budded from the trans-Golgi network [18]. Originally known as a degradation hub, lysosomes have been found responsible for metabolism, nutrient sensing, and neurologic and immune function. The diversity of lysosomal enzymes facilitates the breakdown of diverse macromolecules like nuclei acids, proteins, polysaccharides, lipids, and other substances [14].
Lysosomal hydrolases are synthesized in the rough ER and specifically transported through the Golgi, and then directed to lysosomes from the trans-Golgi network through a mannose-6-phosphate (M6P)-dependent transport system [19]. M6P receptors (M6PRs) aid in packaging hydrolases into clathrin-coated vesicles, delivering their contents to early endosomes, which eventually mature into late endosomes and lysosomes [20]. These enzymes are exclusively active within the acidic interior (pH 4.6–5.2) of lysosomes maintained by proton pumps. Their acid-dependent functionality prevents self-degradation in the event of lysosomal leakage or rupture, as the pH of the cell is neutral to slightly alkaline. Moreover, a continuous layer of highly glycosylated membrane proteins, mainly lysosomal-associated membrane protein (LAMP) 1 and 2, shields the luminal side of lysosomal membranes from autodigestion [15]. However, some soluble hydrolases and non-enzyme proteins are transported to lysosomes via the M6PR-independent transport, mediated by alternative receptors. For example, the trafficking of hydrolases including prosaposin and acid sphingomyelinase is mediated by neurotensin receptor 3 [21], while the transport of β-glucocerebrosidase (βGC) depends on lysosomal integral membrane protein type 2 [22].
Lysosomal enzymes degrade intracellular components and extracellular materials that enter the cell via endocytosis. Autophagy is classified into three types based on the mechanisms of cargo delivery: macroautophagy, chaperone-mediated autophagy, and microautophagy [23]. Macroautophagy involves the formation of autophagosomes that fuse with lysosomes, directly engulfing cytoplasmic portions into lysosomes [24], while chaperone-mediated autophagy targets specific proteins recognized by chaperones for direct delivery to lysosomes via LAMP2 receptors. Microautophagy involves the direct engulfment of cytoplasmic material by the lysosomal membrane, occurring through the invagination, protrusion, or septation of the lysosome membrane, allowing for the direct uptake of cargo. Following degradation in autolysosomes, digestion products are transported back to the cytoplasm via lysosomal membranes, facilitated by the vacuolar H+-ATPases (V-ATPase) and amino acid transporters [14].
Endocytic pathways, dynamin-dependent or independent, mediate the uptake of extracellular materials and membrane-bound molecules [25]. Dynamin-dependent endocytosis, including clathrin-coated pits and endophilin-mediated mechanisms, imports extracellular molecules and recycles membrane receptors [26]. Alternatively, dynamin-independent processes like macropinocytosis and phagocytosis specialize in acquiring fluid-phase molecules and ingesting large particles [27]. Once digested within lysosomes, substrates are exported into the cytoplasm through specific membrane transporters, and then utilized either as an energy source through catabolic reactions or as building blocks for anabolic reactions [28].
Lysosomes not only regulate intracellular functions but also are involved in extracellular regulation [28]. After migrating from the perinuclear region to the vicinity of the cell surface, lysosomes fuse with the plasma membrane and release their contents extracellularly [29,30]. In osteoclasts, lysosome exocytosis enables the release and activity of acidic hydrolase cathepsin K at the ruffled border and juxtaposed resorption lacuna, promoting the digestion of the bone matrix and mineral [31]. Lysosomes also play a role in the formation of extracellular vesicles, and the inhibition of the lysosome with different alkaline agents increases extracellular vesicle secretion [32].
The motility of lysosomes is not uniform; some lysosomes remain relatively stationary within the cell, while others exhibit dynamic movement along microtubules [33]. In non-polarized cells, lysosomes are positioned around the center of the microtubule organization or located at the plasma membrane and cell protrusions, while in polarized cells, they are located in the cell body, axon, and dendrites [34]. In neurons, the kinesin mediates the forward transport of lysosome to the end of the axon, and the dynein mediates the reverse transport of lysosome from the distal axon to the cell body. Dendritic microtubules exhibit mixed polarity, where both motor proteins can drive lysosomes in either direction. Lysosomes throughout axons and dendrites exhibit dynamic trafficking and positioning to meet various homeostatic demands [35]. Autophagy depends on lysosome movement [36], and the bidirectional transport of lysosomes mediates the binding of lysosomes to autophagosomes to form autolysosomes, thereby obtaining lysosomal hydrolases and an acidic environment to clear autophagosomes [37]. For example, the activity of TRPML1 has been found to promote the Ca2+-dependent centripetal movement of lysosomes towards the perinuclear region following autophagy induction [38].
3. Lysosomal Ion Channels
Lysosomal ion channels are integral membrane proteins located within the lysosomal membrane, operating alongside transporters to modulate the flow of ions across the lysosomal membrane. They play a crucial role in establishing the concentration gradient for a variety of anions and cations within the lysosomes. The lumen of the lysosome is characterized by high concentrations of H+, Ca2+, and Na+ while exhibiting a low level of K+ in comparison to the cytoplasm (Figure 1). The lysosomal membrane potential is variable [39,40,41]. The concentration gradient of ions inside and outside the lysosomal membrane, as well as the open state of ion channels, determine the direction of ion movement across the cell membrane [42]. Despite their important role in the physiological and pathological processes of cells, the properties of lysosomal ion channels have remained undiscovered due to technical limitations. Recent advancements in electrophysiological techniques, especially in patch-clamp, have shed light on the identity and function of ion channels predominantly localized on lysosomes. These channels, including TRPML1-3, TPC1/2, TMEM175, and chloride channel 7 (ClC7), are activated by pH, voltage, and endogenous ligands. They exhibit varying degrees of permeability toward different ions, such as Ca2+, K+, Na+, H+, and Cl−, and exert a variety of functions in cargo degradation, autophagy regulation, cell signaling, and disease implication (Table 1).

Figure 1.
Function and regulation of lysosomes. Receiving inputs from both endocytic and autophagic pathways, lysosomes play a fundamental role in cellular housekeeping and metabolism, as well as critical and sophisticated roles in the immune and nervous systems. Lysosomal ion channels, by mediating ion gradients and intracellular signaling pathways, participate in various lysosomal functions, including lysosomal membrane trafficking, catabolite export, nutrient sensing, and mTOR signaling. The dynamic interplay of these ion channels not only underscores the complexity of lysosomal regulation but also highlights their indispensability in maintaining cellular health and responding to physiological challenges. (Ion concentrations in cytosol: [K+]Cytosol = 150–180 mM [43], [Na+]Cytosol = 10–110 mM [44,45], [Ca2+]Cytosol = 100 nM [46], [H+] Cytosol = 40–50 nM [47], [Cl−]Cytosol = 25 ± 15 mM [48,49]. Ion concentrations in lysosome: [K+]L = 2–50 mM [41,50], [Na+]L = 80 ± 60 mM [41,50], [Ca2+]L = 500 ± 100 μM [51], [H+]L = 25 μM [52], [Cl−]L= 90 ± 30 mM [53,54]. Lysosomal membrane potential varies from 20 to 114 mV (luminal-side positive) [39,40,41]. The figure was created with BioRender.com.

Table 1.
Major lysosomal ion channels and their functions.
3.1. Ca2+ Channels
Calcium (Ca2+) stands as a pivotal second messenger, orchestrating a multitude of vital cellular processes [55]. Ca2+ homeostasis assumes a critical role in preserving the optimal function of lysosomes, pivotal organelles responsible for cellular waste management and recycling [56]. Channels and transporters delicately manage the influx and efflux of Ca2+, finely tuning essential cellular activities such as autophagy, apoptosis, and gene regulation [57]. The lysosomal Ca2+ concentration ([Ca2+]lumen) is around 500 μM, roughly 5000 times higher than the cytosolic concentration [51], making it the second-largest store of Ca2+ within the mammalian cell. However, the mechanisms of Ca2+ influx and maintenance of high Ca2+ concentration in lysosomes remain unclear. Existing researches suggests the primary source of Ca2+ in lysosomes is the endoplasmic reticulum [58,59]. Several studies indicated that ATP is required for Ca2+ transport into lysosomes in mammalian cells [60,61], and pH, temperature, heavy metals [61,62], sphingosine [63] affect Ca2+ transport as well. Potential lysosomal Ca2+ importers include Ca2+/H+ exchanger (CAX) [64], sarcoendoplasmic reticulum Ca(2+)-ATPase-3 pump (SERCA3) [65], and cation exchanger (solute carrier family 24 member 5) SLC24A5 [66]. Stromal interaction molecule 1 (STIM1) probably acts as a Ca2+ sensor in the acidic store [65]. Recently, transmembrane protein 165 (TMEM165) was identified as a novel Ca2+ importer in lysosomes [67], which significantly advanced understanding of lysosomal calcium entry mechanisms.
The release of Ca2+ from lysosomes has been extensively studied, and a plethora of ion channels are involved, such as transient receptor potential melastatin 2 [68], purinergic ATP-activated cation channel purinergic receptor (P2X4) [69], and calcium voltage-gated channel subunit alpha1 A (CACNA1a) [70]. In this review, we focus on TRPML1-3 and TPC1, 2, which are the most well-characterized.
3.1.1. TRPMLs
The TRPML family consists of three main family members in mammals: TRPML1, TRPML2, and TRPML3, sharing approximately 75% similarity in amino acid sequence [71,72,73]. These ion channels are crucial players in various cellular processes including immune inflammation [74,75,76,77]. The three members of the TRPML family are known to be activated by phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) [71,78], which is a significant component of endo-lysosomal membranes. Additionally, their activation is influenced by H+ concentration, which varies across different endosomal organelles and lysosomes.
TRPML1 primarily localizes the late endolysosomal compartment in mammals [79]. Its activity peaks in the highly acidic environment of lysosomes (pH 4.6–5.2) [80,81], diminishes in weakly acidic conditions (pH 5.5), and is at its lowest under neutral conditions [82]. TRPML1 has been reported to be associated with various cellular processes including particle intake, lysosome exocytosis [83], and is also involved in phagolysosomal biogenesis, facilitating the fusion of phagosomes and lysosomes, ensuring proper cellular disposal [84]. Moreover, TRPML1 participates in autophagy by interacting with mTOR pathway. During starvation, reduced mTOR levels activate TRPML1, which subsequently promotes mTOR activity necessary for autophagic lysosome reformation [85].
TRPML2 exhibits a predominant localization within the long tubular recycling endosome, lysosomes, and plasma membrane [86]. It functions as an inwardly rectifying ion channel that allows calcium ions to permeate and is most active at pH 7.2 [87]. It is primarily regulated by extracytosolic protons, rather than Ca2+ levels [82], or linoleic acid, which is known to activate other transient receptor potential (TRP) [88]. Although it was not directly associated with any disease phenotype, TRPML2 is involved in various cellular processes, including endocytosis, membrane trafficking, and lysosomal function [86]. Recent studies have discovered its new role in host defense against various pathogens, including bacteria, viruses, and fungi. Activation of toll-like receptors (TLRs) such as TLR2, TLR3, TLR4, TLR7, and TLR8 leads to a significant increase in TRPML2 expression [76]. Additionally, TRPML2 activation directly stimulates the secretion of the chemokine CCL2 by bone marrow-derived macrophages, indicating its involvement in chemokine transport and secretion in mouse macrophages [89].
Overexpressed TRPML3 is observed at various cellular locations, including the plasma membrane (PM), ER, autophagosomes, lysosomes, early and late-endosomes [90]. TRPML3 can be activated by PI(3,5)P2 and Phosphatidylinositol-3-phosphate (PI3P) [91,92]. Meanwhile, high luminal pH (pH 6–6.5) and substitution of luminal Na+ with K+ augment TRPML3 activity [93]. It is found within all autophagosomes, including those present in both feeding and stressed cells, and is recruited into newly formed autophagosomes in stressed cells [94], suggesting its important role in autophagy. Unlike TRPML1, TRPML3 appears to inhibit mTOR through a positive feedback mechanism. Xu et al. recently found that under nutrient starvation, reduction in mTOR stimulates TRPML3 through recruitment of TRPML3 from endolysosome onto phagophores where pH and PI3P levels are higher. Ca2+ released via TRPML3 then activates large conductance calcium-activated potassium channels (BK), which in turn facilitates further Ca2+ release through TRPML3, potentially by removing luminal Na+ inhibition. In addition, TRPML3/BK complex facilitates mTOR inhibition and autophagy induction [93]. TRPML3 also plays a role in immunity. With elevated expression of TRPML3 in lung tissue, primarily concentrated within alveolar macrophages (AMΦ). These cells, integral to the immune system, are responsible for clearing harmful particles and microorganisms from the respiratory tract [95].
3.1.2. TPC Channels
TPCs are ligand-gated cation-selective ion channels, located in the membranes of acidic organelles, such as endosomes, lysosomes, and endolysosome [96]. Characterized by their unique structure that includes two pore-forming domains per subunit, TPCs are capable of conducting ions, notably Ca2+ and Na+, in response to changes in membrane potential and intracellular signaling molecules [97].
The TPC family consists of three members, each exhibiting distinct localization and biophysical characteristics. TPC1 and TPC2 are localized in endosomes and lysosomes, while TPC3 is present in plasma membranes [98]. TPC1 and TPC2 act as Ca2+ release channels in response to stimuli such as nicotinic acid adenine dinucleotide phosphate (NAADP) [99]. However, when activated by phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), they release Na+ instead of Ca2+. In addition, Ca2+, pH, ATP, and voltage can regulate the activities of TPC1 and TPC2 [100]. They contribute to processes such as phagocytosis, endocytosis, and lysosomal exocytosis by modulating fusion and fission events within endolysosomal compartments [101]. Dysregulation of TPC activity has been implicated in various physiological and pathological conditions. For example, levels of TPC1 and TPC2 are elevated in left ventricular samples obtained from both ischemic and dilated hearts of patients with heart failure [102], absence of TPC2 in hepatocytes leads to various phenotypes consistent with non-alcoholic fatty liver disease (NAFLD) [103]; knockdown or knockout of either Tpc1 or Tpc2 prevents Ebola virus infection in vitro [104].
In addition, TPCs are widely expressed across various cells in the immune system such as B cells, T cells, dendritic cells, NK cells, and mast cells. Notably, macrophages exhibit a particularly high level of expression of both TPC1 and TPC2, indicating their significant role in the function of these immune cells [105]. As mentioned, TPCs are integral components of this signaling cascade activated by NAADP, which selectively stimulates exocytosis crucial to immune cell function, particularly in cytotoxic T lymphocytes (CTLs) during immune synapse formation [106]. TPCs translocate to immune synapses upon CTL activation, facilitating Ca2+-dependent exocytosis of cytolytic particles for cell killing [107].
Furthermore, TPCs have been implicated in regulating calcium homeostasis within endolysosomal compartments, influencing mast cell reactivity [96]. Mast cells are vital components of the innate immune system and play pivotal roles in defending against pathogens and orchestrating allergic responses. Dysregulation of TPC-mediated calcium signaling can lead to excessive release of histamine and heparin, contributing to allergic reactions [106]. Studies have shown that TPC1 regulates calcium homeostasis, modulating the feedforward cycle involving inositol triphosphate receptors and leading to increased exocytosis and allergic reactions when inhibited [106,107].
3.2. Cl− Channels
As the most abundant physiological anion, chloride (Cl−) is widely distributed in cytoplasm and vesicule/organel [108]. Its transport is involved in numerous physiological processes including regulation of cell excitability, transepithelial material transport, cell volume regulation, and organelle acidification [108,109,110]. The chloride channel (ClC) and ceroid lipofuscinosis neuronal (CLN) family contribute to regulation of Cl− in lysosome.
3.2.1. ClC Family
The ClC family has been extensively studied, mainly acting as Cl− channels or 2Cl−/H+ exchangers. There are nine members present in mammals (ClC1 to ClC7, ClCKa, and ClCKb). Four of these proteins, ClC1, ClC2, ClCKa, and ClCKb, serve as Cl− channels, essential for stabilizing membrane potential and ion homeostasis across the plasma membrane [108]. Other ClC proteins are primarily expressed in intracellular organelles like endosomes and lysosomes, where they are probably important for proper luminal acidification, in synergy with the V-ATPase [111]. ClC3 to ClC5 function as endosomal 2Cl−/H+ exchanger [112], while ClC6 and ClC7 are late endosomal/lysosomal 2Cl−/H+ exchanger [113,114,115,116]. The sorting of endo-lysosomal transmembrane proteins is generally mediated by cytosolic motifs that are recognized by adaptor proteins. For instance, a basic amino acid stretch is crucial for the localization of heterologously expressed ClC-6 to early and/or recycling endosomes [117], and the N terminus of ClC-7 contains binding motifs for both AP and GGA adaptors, which are important for its lysosomal targeting [118]. ClC6 and ClC7 share significant sequence homology and structural similarities [119]. Patients with mutation in ClCn6 (ClC6 encoding gene) exhibit neurodegeneration and West Syndrome [120,121], while mice lacking ClCn6 only develop a mild neuronal lysosomal storage disorder, primarily affecting the initial axon segment, but do not display any discernible phenotype [115]. ClCn7 (ClC7 encoding gene) has been identified as the causal gene in a distinctive phenotype encompassing osteopetrosis [122], renal tubule acidosis [123], and blindness [124].
3.2.2. CLNs
CLN proteins are distributed throughout the endomembrane system and regulate a variety of cellular processes related to ion homeostasis, autophagy, lipid metabolism, and protein trafficking [125]. To date, fourteen CLN proteins (CLN1-14) have been identified and characterized [126,127]. Among these, CLN6 and CLN8 localize to the membrane of the ER, where they regulate lysosomal enzyme recruitment and lysosome biogenesis [128], while CLN1, CLN2, CLN3, CLN5, and CLN10 localize to lysosomes and function as soluble enzymes [129,130,131].
In recent years, CLN7 has been recognized as a novel endolysosomal Cl− channel [132]. Research indicates that overexpression of CLN7 enhances endolysosomal chloride currents and promotes the enlargement of endolysosomes through a Ca2+/calmodulin-dependent mechanism [132]. CLN7 also regulates lysosomal chloride conductance, luminal pH, and lysosomal membrane potential, promoting the release of lysosomal Ca2+ through TRPML1 [132].
3.3. K+ and H+ Channels
TMEM175, also known as the transmembrane protein 175 channel, serves as the principal mediator of K+ and H+ permeability within lysosomes and late endosomes [133,134]. It is a growth-factor-activated and AKT (protein kinase B)-gated lysosomal ion channel [135], which comprises two sets of repeating 6-transmembrane segments organized in an hourglass-like configuration [133]. The absence of TMEM175 in lysosomes results in the loss of K+ conductance, insensitivity to K+ currents, and disruption of lumen pH stability, thereby leading to abnormal fusion events during autophagy [133,136]. Compounds such as arachidonic acid and small synthetic chemicals like DCPIB and ML 67-33 have been identified as activators of TMEM175-mediated H+ and K+ currents [134]. Loss of TMEM175 triggers the rapid buildup of α-synuclein within lysosomes due to accelerated fusion between autophagosomes and lysosomes, ultimately culminating in neuronal cell demise. TMEM175 plays a particularly important role in regulating lysosomal pH homeostasis. When the lysosomal lumen experiences hyperacidification induced by the V-ATPase, TMEM175 is activated and precipitates a rapid surge in H+ efflux [134]. Overexpression of TMEM175 promotes lysosomal alkalization, while its deletion exacerbates lysosomal acidification and diminishes the activity of essential hydrolases, including aspartase, cysteine protease, βGC, cathepsin D, and cathepsin B, resulting in impaired lysosomal autophagy [137,138,139,140].
4. The Role of Lysosomal Ion Channels in the Nervous System
Lysosomes play a pivotal role within the CNS, contributing to various essential functions through degradation and signaling mechanisms. In recent years, lysosomes have garnered significant attention in the study of the late-onset neurodegenerative diseases [141]. The proper operation of lysosomal ion channels is crucial not only for synaptic transmission and plasticity but also holds significance in the pathogenesis and potential treatment of neurodegenerative diseases.
4.1. Physiological Functions of Lysosomal Ion Channels in the Nervous System
TRPML3, identified in sensory neurons, cochlea, and melanocytes, gained prominence due to its association with the Varitint-waddler (Va) mouse phenotype, characterized by hearing loss, circling behaviour, pigmentation defects, and embryonic lethality [142]. In hair cells, TRPML3 is critical for stereocilia bundle formation during development and may function during endocytosis or exocytosis [142]. Mutations in TRPML3 have been identified as the cause of cochlear abnormalities, including disorganization and fusion of stereocilia, distortions in both the apical and distal regions of inner and outer hair cells, as well as loss of pigmented intermediate cells in the stria vascularis, leading to hearing impairment [142]. Notably, conditionally inactivating Trpml3 in mice did not lead to circling behavior, balance impairment, or hearing loss observed in the Va mouse model [143]. In the dorsal root ganglion, TRPML3 shows a very low expression, but exhibits a significant increase in mRNA levels following spared nerve injury. Widespread expression of TRPML3 is observed in nociceptors and mechanoreceptors after spared nerve injury, indicating its potential role in neuropathic pain [144].
In cortical neurons, TRPML1 and TPC2 transcripts are detectable, while the endolysosomal cation channels including TRPML2, TRPML3, and TPC1 were largely undetectable [145]. TRPML1 activation promotes both inward and outward lysosomal trafficking in dendrites through dynein-dependent mechanisms [146]. When induced by disinhibition from LAMTOR1 or pharmacological enhancement, activated TRPML1 enhances lysosomal trafficking, resulting in changes in synaptic plasticity, which favors long-term depression (LTD) induction in the adult hippocampal CA1 region, while impeding long-term potentiation (LTP) consolidation and affecting learning and memory process [146]. TPCs are implicated as central figures in neuronal excitatory signaling driven by NAADP [147]. Elevated [Ca2+] triggers calcium-induced calcium release, promoting critical processes such as LTP and potentially contributing to the pathogenesis of neurodegenerative diseases [148]. Additionally, TPCs may play a pivotal role in glutamate-induced autophagy of glial cells, although there is conflicting evidence regarding their involvement in lysosomal stabilization and the regulation of autophagic flux [149]. Recent studies have shown that TPC2-mediated calcium signaling promotes axon growth in zebrafish’s caudal primary motor neurons [150]. This association was established through the application of the NAADP receptor antagonist trans-Ned 19, along with genetic silencing techniques [150].
TPC2 is implicated in social interaction. In hypothalamic neurons, TPC provides critical Ca2+ signals to increase the releasable large dense-cored vesicles for exocytosis, which stores oxytocin, a prominent regulator of many aspects of mammalian social behaviors [151]. Knockout or inhibition of TPC in mice leads to a significant reduction in plasma oxytocin levels, impaired oxytocin secretion, and social defects, highlighting the importance of neuropeptide vesicle priming for activity-dependent release [151]. Furthermore, activation of type 1 metabotropic glutamate receptors sustains somatodendritic oxytocin release by recruiting TPCs [151].
Overall, lysosomal ion channels play diverse and essential roles in the nervous system, impacting various physiological processes and contributing to the pathogenesis of neurodegenerative diseases.
4.2. Role of Lysosomal Ion Channel in Nervous Disease
4.2.1. Lysosomal Storage Disorders (LSDs)
The LSDs are inherited metabolic disorders characterized by abnormal accumulation of macromolecular substrates due to lysosomal dysfunction [152]. Neurological symptoms or signs are prevalent in most LSDs, including developmental delays, seizures, acroparesthesia, motor weakness, premature mortality, and extrapyramidal manifestations [153]. Emerging evidence suggests that ion channels in the endolysosomal system play a crucial role in the pathology of neurodegenerative LSDs [154].
LSDs can be categorized based on the type of accumulated storage material, including sphingolipidoses, mucopolysaccharidoses, mucolipidoses (ML), and neuronal ceroid lipofuscinoses (NCL) [155]. Within the ML subgroup, there are four distinct types: MLI, MLII, MLIII, and MLIV. MLIV is a debilitating disease marked by infantile-onset visual impairment, motor dysfunction, and neurodegeneration, constituting the most direct link between defective lysosomal Ca2+ release and neurodegeneration, attributed to dysfunction of the lysosomal cation channel TRPML1 [156].
TPC2, sharing several features with TRPML1, is also implicated in LSDs. Both channels are permeable for Ca2+ and Na+, reside in endolysosomal membranes, are activated by PI(3,5)P2, cause trafficking defects when lost, interact with mTOR/Transcription factor EB (TFEB)/autophagy pathways, and promote lysosomal exocytosis [145]. TPC2 is widely expressed in the CNS, with both neurons and glia expressing TPC2 in various brain regions, including the corpus callosum, hippocampus, and cerebellum [145]. Intravenous administration of a TPC2 agonist reduces the accumulation of storage materials in the CNS and restores motor performance in mouse model of the MLIV [145,157].
NCLs also known as batten disease, are characterized by seizures and visual, cognitive decline, and motor deficits, culminating in premature death [158]. The important roles of ClC6 and ClC7 in NCL have been revealed, with their influence on Cl−accumulation which consequently affects protein degradation [41,159,160,161,162,163]. Loss of Clc6 lead to the mild neuropathologic features, such as the accumulation of autofluorescent [164], while NCL phenotypes caused by ClC7 are more typical including lysosomal storage, retinal and optic nerve degeneration, and selective loss of hippocampal and cerebellar neurons [159,164]. Addtionally, CLCN7 variant is associated with developmental delay and hypopigmentation due to lysosomal swelling, abnormal storage and hyperacidity [165,166]. The participation of V-ATPase in the lysosomal acidification process is also noteworthy in NCL. Dysfunction of V-ATPase is observed in Cln1−/− mice, an infantile NCL model [167]. Furthermore, TFEB-V-ATPase axis is involved in lysosomal biogenesis [168] and regulation of microglia and immune response in tauopathy and Alzheimer’s disease (AD) [169]. Besides, each CLN gene is associated with a specific subtype of the disease, varying in severity and age of onset [127,158]. For instance, homozygous frameshift mutation in the CLN7 gene of macaque recapitulates key features of human NCLs, including behavioral and neuropathological deficits such as visual impairment, tremor, incoordination, ataxia, and impaired balance [170]. In Cln7-deficient mouse embryonic fibroblasts (MEFs), CLN5 protein levels are significantly reduced, suggesting a potential role of CLN5 in the pathogenesis of CLN7 disease [171]. TFEB activation by tamoxifen reduces lysosomal accumulation of storage material in CLN7 type NCL patient-derived induced pluripotent stem cells (iPSC), indicating a therapeutic potential for TFEB modulation [172].
4.2.2. Niemann Pick Disease (NPD)
NPD is a rare congenital recessive disorder characterized by lipid accumulation in various organs [173], leading to a range of clinical manifestations. Patients may exhibit lipid storage and foam cell infiltration in tissues, along with overlapping features including hepatosplenomegaly, pulmonary insufficiency, and/or CNS involvement. NPD has four subgroups (A, B, C1, C2) distinguished by age of onset, clinical presentation, and sphingomyelin storage patterns [174].
Types A and B result from mutations in the SMPD1 gene [175]. Type A NPD patients typically manifest within the first year of life with hepatosplenomegaly and failure to thrive. It follows a rapidly progressive neurodegenerative course, marked by profound hypotonia and developmental delays, often resulting in early mortality [176]. In contrast, NPC is characterized by mutations in the NPC1 gene (95% of cases), with a minority attributable to NPC2 gene (4%), and potentially other genes unidentified genes (1%) [177]. NPC patients do not exhibit severe sphingomyelinase deficiency, but present with sub-acute nervous system involvement and moderate visceral storage pathology [178]. While NPC1 and NPC2 proteins are essential for proper transport and metabolism of cholesterol and other lipids within cells [179], accumulation of cholesterol is observed in about 80% of the NPC cases [178].
Neurological symptoms of NPC commonly include cerebellar ataxia, dysarthria, dysphagia, progressive dementia, and occasionally seizures [178]. Recent research has shown promising approaches for the treatment, including pharmacological and genetic activation of TRPML1 to ameliorate lipid trafficking defects and cholesterol accumulation associated with NPC1 [180]. Additionally, activation of TPC2 using the small molecule agonist has demonstrated efficacy in reducing cholesterol accumulation in patient fibroblasts while promoting lysosomal exocytosis and autophagy, processes often impaired in NPC1 [145].
4.2.3. Neurodegenerative Disease
The dysfunction of these ion channels can lead to impaired lysosomal function, which is increasingly recognized as a significant contributor to the pathology of neurodegenerative diseases. Modulating the activity of these channels holds promise for restoring proper lysosomal function, potentially slowing or even halting the progression of these debilitating diseases.
- (a)
- Alzheimer’s disease (AD)
AD is the predominant form of dementia, accounting for 60–70% of all diagnosed cases of cognitive decline [181]. AD manifests with symptoms including memory loss, repetitive behavior, misplacing items, and difficulties in daily activities. Genetic mutations in genes such as APP, PSEN1, PSEN2, and APOE can contribute to both early and late-onset forms of AD [182,183]. Key hallmarks of AD pathology include the accumulation of amyloid-beta plaques and hyperphosphorylated tau protein in neurofibrillary tangles.
The relationship between lysosomes and AD is complex and involves multiple mechanisms. Various AD risk genes, including BIN1, CD2AP, PICALM, PLD3, SORL1, and TREM2, are linked to the endosomal-autophagic-lysosomal (EAL) system. Early studies have suggested that enhancing lysosomal function could potentially mitigate key AD hallmarks, such as tau accumulation [184]. Recent research investigates the role of TRPML1 in EAL dysfunction associated with AD. Abnormal TRPML1-mediated Ca2+ release and endolysosomal abnormalities have been observed in AD brains and neurons expressing APOE ε4-, a major AD risk factor. Furthermore, inhibiting phosphoinositide kinase FYVE (PIKfyve) induces AD-like EAL defects in neurons, which can be rescued by TRPML1 agonist ML-SA1 [185]. Additionally, activation of TRPML1 has been shown to facilitate the expulsion of accumulated tau through lysosomal exocytosis [186] and eliminate amyloid-β from lysosomal compartments in neurons [187]. These findings highlight TRPML1 as a promising therapeutic target for AD.
- (b)
- Parkinson’s disease (PD)
PD is a prevalent neurodegenerative disorder characterized by symptoms such as tremors, bradykinesia (slowed movement), muscle stiffness, impaired balance, and cognitive impairment [188]. It can be caused by genetic mutations in various genes, including SCNA, LRRK2, PINK1, and PLA2G6 [189], or by exposure to certain chemicals such as phenothiazines [190], polychlorinated biphenyls (PCBs) [190], and organochlorines [191]. Mitochondrial dysfunction, oxidative stress, and the accumulation of α-synuclein protein contribute to the formation of Lewy bodies and progressive degeneration of the dopaminergic system in the brain [192]. The lysosome, responsible for protein degradation and recycling, plays a crucial role in PD pathology [193].
Studies using fibroblasts from PD patients have revealed an expansion of the lysosomal compartment in these cells, a phenomenon mitigated by treatment with TPC/NAADP antagonists Ned19 or genetic knockdown of Tpc2 [194]. Moreover, overexpression of dominant-negative TPC2 or NAADP antagonism hindered autophagic induction mediated by the PD-associated LRRK2 protein [195]. However, further investigations are needed to elucidate the role of TPC2 in neural cells, particularly in iPSC-derived human neurons, and its impact on PD-associated α-synuclein aggregation and Lewy body formation [196]. Additionally, exploring the effects of different mechanisms of TPC2 activation, such as by PI(3,5)P2 or NAADP, on lysosomal function, autophagy, and cell viability in PD and lysosomal storage disorders is essential. While TPC2 antagonists could restore lysosomal Ca2+ and pH and rescue autophagy [197], prolonged inhibition of TPC2 may result in trafficking defects and cholesterol overload, posing potential risks for neurodegenerative disease development [103]. Therefore, understanding the complex role of TPC2 in lysosomal function and neurodegeneration is critical for developing targeted therapeutic interventions for PD and related disorders.
Earlier genome-wide association studies have uncovered the potential significance of genetic variations in TMEM175 concerning PD [198]. Recent research has demonstrated that elevated expression or activity of TMEM175 may suppress autophagy and promote apoptosis, potentially leading to neuronal demise in PD. Conversely, knockout of Tmem175 has shown significant neuroprotection in a mouse model of PD induced by MPTP without causing notable adverse effects [199]. However, contrasting findings indicate that deficiency in TMEM175 results in the loss of dopaminergic neurons and impairs motor function in mice. Additionally, a TMEM175 loss-of-function variant has been nominally associated with accelerated rates of cognitive and motor decline in humans with PD [135].
- (c)
- Amyotrophic lateral sclerosis (ALS)
ALS is a rare neurodegenerative disease of the motor neurons, characterized by progressive voluntary muscle weakness [200]. Onset and development of ALS are associated with lysosome/autophagy pathway. TRPML1 agonist ML-SA1 induces lysosomal Ca2+ in motor neuronal cells and protects motor neurons from the neurotoxicity of cyanobacterial toxin beta-methylamino-L-alanine by promoting autophagic clearance and counteracting ER stress [201], which accumulates in CNS of the ALS/Parkinson-dementia complex patients [202]. Besides, genetic studies identify mutations in the factor-induced gene 4 (FIG4) as a cause of familial ALS [203]. FIG4 regulates cellular levels of PI(3,5)P2 [204], with its deficiency impairing biosynthesis of PI(3,5)P2, the endogenous ligand of endolysosomal TRPML, and two-pore channels. It is worth to note that mutations in the FIG4 gene present a comparatively rare etiological factor in ALS pathology, particularly compared to other contributors related to lysosome and autophagy function, such as gene C9orf72 and FUS [205,206].
4.2.4. Stroke
Stroke, a prevalent cerebrovascular condition worldwide, is characterized by post-ischemic reperfusion, which stands out as a significant contributor to delayed secondary brain injury [207]. In the oxygen-glucose deprivation/reoxygenation (OGD/R) model, a reduction in TMEM175 levels was observed in neurons, while upregulation of TMEM175 demonstrated a dual effect by mitigating mitochondrial inactivation and protecting neurons from ischemic injury. Moreover, TMEM175 restoration facilitated lysosomal internal environment homeostasis and reinstated hydrolase activity. Consequently, heightened cathepsin D activity ensued, rendering neurons resistant to OGD-induced damage [139]. While the ramifications of TMEM175 deficiency in ischemia-reperfusion injury remain unknown, it is plausible to speculate its crucial involvement in this pathological process.
During ischemic reperfusion, the autophagic pathways in various brain cells are activated [208], leading to neuronal necrosis or apoptosis. Considering that ischemia results in hypoxia and excessive glutamate release, excessive activation of autophagic flux occurs [209]. In a transient focal ischemia model, pharmacological inhibition of the TPC2 can rescue primary cortical neurons from hypoxia-induced death, interfere with autophagy, impede excessive autophagic flux, and reduce the infarct area in rats with focal ischemia [210]. Post-ischemic hypoxic neuronal protein homeostasis dysfunction, endoplasmic reticulum stress, and glutamate receptor-mediated excitotoxicity may be improved following TPC2 inhibition. Moreover, the TPC2 lysosomal channel is likely a putative target in this cascade reaction [210].
5. Conclusions and Future Perspectives
Lysosomes regulate a broad spectrum of vital cellular functions, including cellular clearance, autophagy, recycling and gene expression [17]. Within lysosomes, ion channels fulfill distinct functions (Figure 1). For instance, Ca2+ channels, play a crucial role in regulating processes such as autophagy, membrane fusion and division. Meanwhile, Na+ and K+ channels primarily modulate the membrane potential of lysosomes, thereby influencing their activity and function. Additionally, H+ channels are primarily involved in regulating the pH value of lysosomes, ensuring optimal conditions for lysosomal enzyme activity and cellular degradation processes. Furthermore, interplay between lysosomal ion channels and transporters has been illuminated based on mathematical models: ClC7 function as 2Cl−/H+ exchanger, interestingly, its disruption impinge on luminal concentrations of Ca2+, Na+, K+ [211], affecting acidification. A counterion flux, involving anions (like Cl− entering the lumen) and/or cations (like K+ and Na+ leaving the lumen), is necessary to neutralize the membrane potential generated by the V-ATPase [212].
Lysosomal ion channels play multifaceted roles in the CNS, as well as in the immune systems, exerting significant influence on various physiological and pathological processes. Through their regulation of ion homeostasis, pH balance, and vesicular trafficking [101,213], these channels contribute to fundamental cellular functions such as autophagy [214] and immune response modulation [105]. Dysregulation of these channels has been implicated in a myriad of neurological and immunological disorders, emphasizing their potential as therapeutic targets for intervention.
Certain diseases directly result from mutations or variations in genes encoding lysosomal ion channels, correcting the function of these channels is a logical and promising approach to treatment. For example, mutations in Trpml1 can result in MLIV [191], a severe lysosomal storage disorder characterized by developmental delays, intellectual disability, and impaired vision and motor function. Although existing studies have utilized compounds such as small molecules, peptides, micro-RNAs, and antibodies to target plasma membrane ion channels, organelle channels, particularly lysosomal ion channels, remain largely unexplored as targets for drug development [215]. However, promising research has shown that synthetic TRPML1 agonist ML-SA1 can repair muscle cell damage, alleviate abnormal lysosomal storage [216], and rescue AD-like EAL pathogenesis [185]. An enzyme replacement therapy represents the first treatment to delay the progression of CLN2 NCL, it involves the administration of the enzyme, a recombinant lysosomal enzyme named cerliponase alfa, directly into the lateral cerebral ventricles to target the central nervous system [217]. Concurrently with this significant milestone, numerous research groups are employing diverse therapeutic approaches to expedite the creation of innovative treatments for different types of NCL at an unprecedented speed.
A deeper understanding of the physiological roles of lysosomal ion channels could unveil novel therapeutic strategies for conditions characterized by dysregulated immune responses and neuroinflammation [218]. Considering the distinct physiological functions and substantial potential for drug development associated with lysosomal ion channels, future research in this realm is poised to advance, paving the way for breakthroughs not only in the field of ion channels but also in broader areas of cellular physiology, neurobiology, and therapeutic interventions.
Author Contributions
J.C., N.H. and J.S. wrote the manuscript and drew the figure. H.L. and Y.G. designed and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China 32100806 and 82271256.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The figure was created with BioRender.com.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Abbreviations
AD, Alzheimer’s Disease; AMΦ, Alveolar Macrophages; BMDM, Bone Marrow-Derived Macrophages; ClC, Chloride Channel; CLN, Ceroid Lipofuscinosis Neuronal; CNS, Central Nervous System; CMA, Chaperone-Mediated Autophagy; CTLs, Cytotoxic T Lymphocytes; EAL, Eal Domain; EE, Early endosome; ER, Endoplasmic Reticulum; FIG4, Factor-induced Gene 4; iPSC, induced Pluripotent Stem Cells; LacCer, Lactosylceramide; LAMP, Lysosomal-associated membrane protein; LE, Late endosome; LY, Lysosome; LSDs, lysosomal storage disorders; LTD, Long-Term Depression; LTP, Long-Term Potentiation; M6P, Mannose 6-Phosphate; M6PR, Mannose-6-phosphate receptor; MEFs, Mouse Embryonic Fibroblasts; ML, Mucolipidosis; NAADP, Nicotinic Acid Adenine Dinucleotide Phosphate; NAFLD, Non-Alcoholic Fatty Liver Disease; NCL, Neuronal Ceroid Lipofuscinoses; NPA, Niemann-Pick disease type A; NPC, Niemann-Pick disease type C; OGD/R, Oxygen-Glucose Deprivation/Reperfusion; PD, Parkinson’s Disease; PI(3,5)P2, Phosphatidylinositol 3,5-bisphosphate; PI(4,5)P2, Phosphatidylinositol 4,5-bisphosphate; PI3P, Phosphatidylinositol-3-phosphate; PM, Plasma Membrane; PCBs, Polychlorinated Biphenyls; PIKfyve, Phosphoinositide Kinase, FYVE-type Zinc Finger Containing; RE, Recycling endosome; SNI, Spared Nerve Injury; TFEB, Transcription Factor EB; TPC, Two-Pore Channel; TMEM175, Transmembrane Protein 175; TRPML, Transient Receptor Potential Mucolipin; Va, Varitint-waddler; V-ATPase, vacuolar H+-ATPases; βGC, β-glucocerebrosidase.
References
- Cui, L.; Li, H.; Xi, Y.; Hu, Q.; Liu, H.; Fan, J.; Xiang, Y.; Zhang, X.; Shui, W.; Lai, Y. Vesicle trafficking and vesicle fusion: Mechanisms, biological functions, and their implications for potential disease therapy. Mol. Biomed. 2022, 3, 29. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef]
- Cai, W.; Li, P.; Gu, M.; Xu, H. Lysosomal Ion Channels and Lysosome-Organelle Interactions. Handb. Exp. Pharmacol. 2023, 278, 93–108. [Google Scholar] [CrossRef]
- Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Bagshaw, R.D.; Mahuran, D.J.; Callahan, J.W. A proteomic analysis of lysosomal integral membrane proteins reveals the diverse composition of the organelle. Mol. Cell. Proteom. 2005, 4, 133–143. [Google Scholar] [CrossRef]
- Riederer, E.; Cang, C.; Ren, D. Lysosomal Ion Channels: What Are They Good For and Are They Druggable Targets? Annu. Rev. Pharmacol. Toxicol. 2023, 63, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Zhu, M.X. Regulation of lysosomal ion homeostasis by channels and transporters. Sci. China. Life Sci. 2016, 59, 777–791. [Google Scholar] [CrossRef]
- Patel, S.; Kilpatrick, B.S. Two-pore channels and disease. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865 Pt B, 1678–1686. [Google Scholar] [CrossRef]
- Wu, L.; Lin, Y.; Song, J.; Li, L.; Rao, X.; Wan, W.; Wei, G.; Hua, F.; Ying, J. TMEM175: A lysosomal ion channel associated with neurological diseases. Neurobiol. Dis. 2023, 185, 106244. [Google Scholar] [CrossRef]
- Kendall, R.L.; Holian, A. The role of lysosomal ion channels in lysosome dysfunction. Inhal. Toxicol. 2021, 33, 41–54. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef]
- Schwake, M.; Schröder, B.; Saftig, P. Lysosomal membrane proteins and their central role in physiology. Traffic 2013, 14, 739–748. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, N.; Cai, W.; Xu, H. Lysosomal solute and water transport. J. Cell Biol. 2022, 221, e202109133. [Google Scholar] [CrossRef]
- Gros, F.; Muller, S. The role of lysosomes in metabolic and autoimmune diseases. Nat. Rev. Nephrol. 2023, 19, 366–383. [Google Scholar] [CrossRef]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.; Griffiths, G.M. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 605–614. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Prata, M.J.; Alves, S. Mannose-6-phosphate pathway: A review on its role in lysosomal function and dysfunction. Mol. Genet. Metab. 2012, 105, 542–550. [Google Scholar] [CrossRef]
- Ni, X.; Morales, C.R. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic 2006, 7, 889–902. [Google Scholar] [CrossRef]
- Zunke, F.; Andresen, L.; Wesseler, S.; Groth, J.; Arnold, P.; Rothaug, M.; Mazzulli, J.R.; Krainc, D.; Blanz, J.; Saftig, P.; et al. Characterization of the complex formed by β-glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc. Natl. Acad. Sci. USA 2016, 113, 3791–3796. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- Nowosad, A.; Besson, A. Lysosomes at the Crossroads of Cell Metabolism, Cell Cycle, and Stemness. Int. J. Mol. Sci. 2022, 23, 2290. [Google Scholar] [CrossRef]
- Cheng, X.; Chen, K.; Dong, B.; Yang, M.; Filbrun, S.L.; Myoung, Y.; Huang, T.X.; Gu, Y.; Wang, G.; Fang, N. Dynamin-dependent vesicle twist at the final stage of clathrin-mediated endocytosis. Nat. Cell Biol. 2021, 23, 859–869. [Google Scholar] [CrossRef]
- Rennick, J.J.; Johnston, A.P.R.; Parton, R.G. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef]
- Tancini, B.; Buratta, S.; Delo, F.; Sagini, K.; Chiaradia, E.; Pellegrino, R.M.; Emiliani, C.; Urbanelli, L. Lysosomal Exocytosis: The Extracellular Role of an Intracellular Organelle. Membranes 2020, 10, 406. [Google Scholar] [CrossRef]
- Andrews, N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000, 10, 316–321. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef]
- Lacombe, J.; Karsenty, G.; Ferron, M. Regulation of lysosome biogenesis and functions in osteoclasts. Cell Cycle 2013, 12, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Matteoni, R.; Kreis, T.E. Translocation and clustering of endosomes and lysosomes depends on microtubules. J. Cell Biol. 1987, 105, 1253–1265. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Roney, J.C.; Cheng, X.T.; Sheng, Z.H. Neuronal endolysosomal transport and lysosomal functionality in maintaining axonostasis. J. Cell Biol. 2022, 221, e202111077. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Saminathan, A.; Devany, J.; Veetil, A.T.; Suresh, B.; Pillai, K.S.; Schwake, M.; Krishnan, Y. A DNA-based voltmeter for organelles. Nat. Nanotechnol. 2021, 16, 96–103. [Google Scholar] [CrossRef]
- Koivusalo, M.; Steinberg, B.E.; Mason, D.; Grinstein, S. In situ measurement of the electrical potential across the lysosomal membrane using FRET. Traffic 2011, 12, 972–982. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Huynh, K.K.; Brodovitch, A.; Jabs, S.; Stauber, T.; Jentsch, T.J.; Grinstein, S. A cation counterflux supports lysosomal acidification. J. Cell Biol. 2010, 189, 1171–1186. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gu, M.; Xu, H. Lysosomal Ion Channels as Decoders of Cellular Signals. Trends Biochem. Sci. 2019, 44, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D. Cation transport in mitochondria--the potassium cycle. Biochim. Biophys. Acta (BBA)-Bioenerg. 1996, 1275, 123–126. [Google Scholar] [CrossRef] [PubMed]
- O‘Doherty, J.; Stark, R.J. Stimulation of pancreatic acinar secretion: Increases in cytosolic calcium and sodium. Am. J. Physiol. 1982, 242, G513–G521. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Saito, Y.; Nishiyama, A.; Takasaka, T. Na+-Ca2+ exchange in the isolated cochlear outer hair cells of the guinea-pig studied by fluorescence image microscopy. Pflug. Arch. 1992, 420, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Mogami, H.; Tepikin, A.V.; Petersen, O.H. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO J. 1998, 17, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Untiet, V.; Kovermann, P.; Gerkau, N.J.; Gensch, T.; Rose, C.R.; Fahlke, C. Glutamate transporter-associated anion channels adjust intracellular chloride concentrations during glial maturation. Glia 2017, 65, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Untiet, V.; Moeller, L.M.; Ibarra-Soria, X.; Sánchez-Andrade, G.; Stricker, M.; Neuhaus, E.M.; Logan, D.W.; Gensch, T.; Spehr, M. Elevated Cytosolic Cl- Concentrations in Dendritic Knobs of Mouse Vomeronasal Sensory Neurons. Chem. Senses 2016, 41, 669–676. [Google Scholar] [CrossRef]
- Bouhamdani, N.; Comeau, D.; Turcotte, S. A Compendium of Information on the Lysosome. Front. Cell Dev. Biol. 2021, 9, 798262. [Google Scholar] [CrossRef]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115 Pt 3, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Ostrowski, P.; Jaumouillé, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Leung, K.; Krishnan, Y. High lumenal chloride in the lysosome is critical for lysosome function. Elife 2017, 6, e28862. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.; Chakraborty, K.; Saminathan, A.; Krishnan, Y. A DNA nanomachine chemically resolves lysosomes in live cells. Nat. Nanotechnol. 2019, 14, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Waller-Evans, H. Lysosomal Ca2+ Homeostasis and Signaling in Health and Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035311. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, P.; Nascimento Da Conceicao, V.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife 2016, 5, e15887. [Google Scholar] [CrossRef] [PubMed]
- López Sanjurjo, C.I.; Tovey, S.C.; Taylor, C.W. Rapid recycling of Ca2+ between IP3-sensitive stores and lysosomes. PLoS ONE 2014, 9, e111275. [Google Scholar] [CrossRef]
- Adachi, T.; Arai, K.; Ohkuma, S. A comparative study of (Ca2+-Mg2+)-ATPase on the lysosomal membrane and ecto-ATPase on the plasma membrane from rat liver. Biol. Pharm. Bull. 1996, 19, 1291–1297. [Google Scholar] [CrossRef]
- Klemper, M.S. An adenosine triphosphate-dependent calcium uptake pump in human neutrophil lysosomes. J. Clin. Investig. 1985, 76, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Lemons, R.M.; Thoene, J.G. Mediated calcium transport by isolated human fibroblast lysosomes. J. Biol. Chem. 1991, 266, 14378–14382. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, M.; Pittman, J.K.; Mayor, R.; Patel, S. Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J. Cell Biol. 2016, 212, 803–813. [Google Scholar] [CrossRef] [PubMed]
- López, J.J.; Jardín, I.; Bobe, R.; Pariente, J.A.; Enouf, J.; Salido, G.M.; Rosado, J.A. STIM1 regulates acidic Ca2+ store refilling by interaction with SERCA3 in human platelets. Biochem. Pharmacol. 2008, 75, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Lamason, R.L.; Mohideen, M.A.; Mest, J.R.; Wong, A.C.; Norton, H.L.; Aros, M.C.; Jurynec, M.J.; Mao, X.; Humphreville, V.R.; Humbert, J.E.; et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 2005, 310, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Zajac, M.; Mukherjee, S.; Anees, P.; Oettinger, D.; Henn, K.; Srikumar, J.; Zou, J.; Saminathan, A.; Krishnan, Y. A mechanism of lysosomal calcium entry. Sci. Adv. 2024, 10, eadk2317. [Google Scholar] [CrossRef] [PubMed]
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sánchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zou, Y.; Zhong, X.Z.; Cao, Q.; Zhao, K.; Zhu, M.X.; Murrell-Lagnado, R.; Dong, X.P. P2X4 forms functional ATP-activated cation channels on lysosomal membranes regulated by luminal pH. J. Biol. Chem. 2014, 289, 17658–17667. [Google Scholar] [CrossRef]
- Zhu, F.; Miao, Y.; Cheng, M.; Ye, X.; Chen, A.; Zheng, G.; Tian, X. The CACNA1A Mutant Disrupts Lysosome Calcium Homeostasis in Cerebellar Neurons and the Resulting Endo-Lysosomal Fusion Defect Can be Improved by Calcium Modulation. Neurochem. Res. 2022, 47, 249–263. [Google Scholar] [CrossRef]
- Jaślan, D.; Böck, J.; Krogsaeter, E.; Grimm, C. Evolutionary Aspects of TRPMLs and TPCs. Int. J. Mol. Sci. 2020, 21, 4181. [Google Scholar] [CrossRef] [PubMed]
- Fine, M.; Li, X. A Structural Overview of TRPML1 and the TRPML Family. In Endolysosomal Voltage-Dependent Cation Channels; Handbook of Experimental Parhmacology Series; Springer: Cham, Switzerland, 2023; Volume 278, pp. 181–198. [Google Scholar] [CrossRef]
- Puertollano, R.; Kiselyov, K. TRPMLs: In sickness and in health. Am. J. Physiol. Ren. Physiol. 2009, 296, F1245–F1254. [Google Scholar] [CrossRef] [PubMed]
- Spix, B.; Chao, Y.K.; Abrahamian, C.; Chen, C.C.; Grimm, C. TRPML Cation Channels in Inflammation and Immunity. Front. Immunol. 2020, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Xie, L.; Li, D.; Hong, X.; Qin, C. Gene expression of TRPMLs and its regulation by pathogen stimulation. Gene 2023, 864, 147291. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Morelli, M.B.; Amantini, C.; Nabissi, M.; Santoni, M.; Santoni, A. Involvement of the TRPML Mucolipin Channels in Viral Infections and Anti-viral Innate Immune Responses. Front. Immunol. 2020, 11, 739. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, A.F.; Parrington, J. Deciphering the Role of Endolysosomal Ca2+ Channels in Immunity. Front. Immunol. 2021, 12, 656965. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Jörs, S.; Guo, Z.; Obukhov, A.G.; Heller, S. Constitutive activity of TRPML2 and TRPML3 channels versus activation by low extracellular sodium and small molecules. J. Biol. Chem. 2012, 287, 22701–22708. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, X.; Gao, Q.; Xu, H. TRPML1: An ion channel in the lysosome. Handb. Exp. Pharmacol. 2014, 222, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, P.L. Lysosomal TRPML1 Channel: Implications in Cardiovascular and Kidney Diseases. Adv. Exp. Med. Biol. 2021, 1349, 275–301. [Google Scholar] [CrossRef]
- Swetha, M.G.; Sriram, V.; Krishnan, K.S.; Oorschot, V.M.; ten Brink, C.; Klumperman, J.; Mayor, S. Lysosomal membrane protein composition, acidic pH and sterol content are regulated via a light-dependent pathway in metazoan cells. Traffic 2011, 12, 1037–1055. [Google Scholar] [CrossRef]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Samie, M.; Wang, X.; Zhang, X.; Goschka, A.; Li, X.; Cheng, X.; Gregg, E.; Azar, M.; Zhuo, Y.; Garrity, A.G.; et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell 2013, 26, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Dayam, R.M.; Saric, A.; Shilliday, R.E.; Botelho, R.J. The Phosphoinositide-Gated Lysosomal Ca2+ Channel, TRPML1, Is Required for Phagosome Maturation. Traffic 2015, 16, 1010–1026. [Google Scholar] [CrossRef] [PubMed]
- Onyenwoke, R.U.; Sexton, J.Z.; Yan, F.; Díaz, M.C.; Forsberg, L.J.; Major, M.B.; Brenman, J.E. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem. J. 2015, 470, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Cuajungco, M.P.; Silva, J.; Habibi, A.; Valadez, J.A. The mucolipin-2 (TRPML2) ion channel: A tissue-specific protein crucial to normal cell function. Pflug. Arch. 2016, 468, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Plesch, E.; Chen, C.C.; Butz, E.; Scotto Rosato, A.; Krogsaeter, E.K.; Yinan, H.; Bartel, K.; Keller, M.; Robaa, D.; Teupser, D.; et al. Selective agonist of TRPML2 reveals direct role in chemokine release from innate immune cells. Elife 2018, 7, e39720. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.M.; Ruparel, S. Role of Oxidized Lipids and TRP Channels in Orofacial Pain and Inflammation. J. Dent. Res. 2016, 95, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Hua, Y.; Vergarajauregui, S.; Diab, H.I.; Puertollano, R. Novel Role of TRPML2 in the Regulation of the Innate Immune Response. J. Immunol. 2015, 195, 4922–4932. [Google Scholar] [CrossRef]
- Kim, H.J.; Soyombo, A.A.; Tjon-Kon-Sang, S.; So, I.; Muallem, S. The Ca2+ channel TRPML3 regulates membrane trafficking and autophagy. Traffic 2009, 10, 1157–1167. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, M.K.; Hong, S.; Choi, A.; Choi, J.H.; Muallem, S.; So, I.; Yang, D.; Kim, H.J. The intracellular Ca2+ channel TRPML3 is a PI3P effector that regulates autophagosome biogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2200085119. [Google Scholar] [CrossRef]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhong, X.Z.; Huang, P.; Jaślan, D.; Wang, P.; Sun, X.; Weiden, E.M.; El Hiani, Y.; Grimm, C.; Dong, X.P. TRPML3/BK complex promotes autophagy and bacterial clearance by providing a positive feedback regulation of mTOR via PI3P. Proc. Natl. Acad. Sci. USA 2023, 120, e2215777120. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Kim, D.H.; Park, K.S.; Kim, M.K.; Park, Y.M.; Muallem, S.; So, I.; Kim, H.J. Palmitoylation controls trafficking of the intracellular Ca2+ channel MCOLN3/TRPML3 to regulate autophagy. Autophagy 2019, 15, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Spix, B.; Butz, E.S.; Chen, C.C.; Rosato, A.S.; Tang, R.; Jeridi, A.; Kudrina, V.; Plesch, E.; Wartenberg, P.; Arlt, E.; et al. Lung emphysema and impaired macrophage elastase clearance in mucolipin 3 deficient mice. Nat. Commun. 2022, 13, 318. [Google Scholar] [CrossRef] [PubMed]
- Steiner, P.; Arlt, E.; Boekhoff, I.; Gudermann, T.; Zierler, S. Two-Pore Channels Regulate Inter-Organellar Ca2+ Homeostasis in Immune Cells. Cells 2022, 11, 1465. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zeng, W.; Jiang, Y. Tuning the ion selectivity of two-pore channels. Proc. Natl. Acad. Sci. USA 2017, 114, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Ramos, I.; Reich, A.; Wessel, G.M. Two-pore channels function in calcium regulation in sea star oocytes and embryos. Development 2014, 141, 4598–4609. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Casali, C.; Negri, S.; Iengo, L.; Biggiogera, M.; Maione, A.S.; Moccia, F. Nicotinic Acid Adenine Dinucleotide Phosphate Induces Intracellular Ca2+ Signalling and Stimulates Proliferation in Human Cardiac Mesenchymal Stromal Cells. Front. Cell Dev. Biol. 2022, 10, 874043. [Google Scholar] [CrossRef]
- Feijóo-Bandín, S.; García-Vence, M.; García-Rúa, V.; Roselló-Lletí, E.; Portolés, M.; Rivera, M.; González-Juanatey, J.R.; Lago, F. Two-pore channels (TPCs): Novel voltage-gated ion channels with pleiotropic functions. Channels 2017, 11, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Rosato, A.S.; Tang, R.; Grimm, C. Two-pore and TRPML cation channels: Regulators of phagocytosis, autophagy and lysosomal exocytosis. Pharmacol. Ther. 2021, 220, 107713. [Google Scholar] [CrossRef]
- García-Rúa, V.; Feijóo-Bandín, S.; Rodríguez-Penas, D.; Mosquera-Leal, A.; Abu-Assi, E.; Beiras, A.; María Seoane, L.; Lear, P.; Parrington, J.; Portolés, M.; et al. Endolysosomal two-pore channels regulate autophagy in cardiomyocytes. J. Physiol. 2016, 594, 3061–3077. [Google Scholar] [CrossRef]
- Grimm, C.; Holdt, L.M.; Chen, C.C.; Hassan, S.; Müller, C.; Jörs, S.; Cuny, H.; Kissing, S.; Schröder, B.; Butz, E.; et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat. Commun. 2014, 5, 4699. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Butz, E.; Chen, C.C.; Wahl-Schott, C.; Biel, M. From mucolipidosis type IV to Ebola: TRPML and two-pore channels at the crossroads of endo-lysosomal trafficking and disease. Cell Calcium 2017, 67, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.C.; Morgan, A.J.; Galione, A. Acidic Ca2+ stores and immune-cell function. Cell Calcium 2022, 101, 102516. [Google Scholar] [CrossRef]
- Arlt, E.; Fraticelli, M.; Tsvilovskyy, V.; Nadolni, W.; Breit, A.; O‘Neill, T.J.; Resenberger, S.; Wennemuth, G.; Wahl-Schott, C.; Biel, M.; et al. TPC1 deficiency or blockade augments systemic anaphylaxis and mast cell activity. Proc. Natl. Acad. Sci. USA 2020, 117, 18068–18078. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.C.; Galione, A. Cytolytic granules supply Ca2+ for their own exocytosis via NAADP and resident two-pore channels. Commun. Integr. Biol. 2013, 6, e24175. [Google Scholar] [CrossRef]
- Stauber, T.; Jentsch, T.J. Chloride in vesicular trafficking and function. Annu. Rev. Physiol. 2013, 75, 453–477. [Google Scholar] [CrossRef] [PubMed]
- Pressey, J.C.; de Saint-Rome, M.; Raveendran, V.A.; Woodin, M.A. Chloride transporters controlling neuronal excitability. Physiol. Rev. 2023, 103, 1095–1135. [Google Scholar] [CrossRef]
- Valdivieso, Á.G.; Santa-Coloma, T.A. The chloride anion as a signalling effector. Biol. Rev. Camb. Philos. Soc. 2019, 94, 1839–1856. [Google Scholar] [CrossRef]
- Zifarelli, G.; Pusch, M. CLC chloride channels and transporters: A biophysical and physiological perspective. Rev. Physiol. Biochem. Pharmacol. 2007, 158, 23–76. [Google Scholar] [CrossRef]
- Bose, S.; He, H.; Stauber, T. Neurodegeneration Upon Dysfunction of Endosomal/Lysosomal CLC Chloride Transporters. Front. Cell Dev. Biol. 2021, 9, 639231. [Google Scholar] [CrossRef] [PubMed]
- Kornak, U.; Kasper, D.; Bösl, M.R.; Kaiser, E.; Schweizer, M.; Schulz, A.; Friedrich, W.; Delling, G.; Jentsch, T.J. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001, 104, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Leisle, L.; Ludwig, C.F.; Wagner, F.A.; Jentsch, T.J.; Stauber, T. ClC-7 is a slowly voltage-gated 2Cl−/1H+-exchanger and requires Ostm1 for transport activity. EMBO J. 2011, 30, 2140–2152. [Google Scholar] [CrossRef] [PubMed]
- Poët, M.; Kornak, U.; Schweizer, M.; Zdebik, A.A.; Scheel, O.; Hoelter, S.; Wurst, W.; Schmitt, A.; Fuhrmann, J.C.; Planells-Cases, R.; et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. USA 2006, 103, 13854–13859. [Google Scholar] [CrossRef] [PubMed]
- Neagoe, I.; Stauber, T.; Fidzinski, P.; Bergsdorf, E.Y.; Jentsch, T.J. The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J. Biol. Chem. 2010, 285, 21689–21697. [Google Scholar] [CrossRef] [PubMed]
- Ignoul, S.; Simaels, J.; Hermans, D.; Annaert, W.; Eggermont, J. Human ClC-6 is a late endosomal glycoprotein that associates with detergent-resistant lipid domains. PLoS ONE 2007, 2, e474. [Google Scholar] [CrossRef] [PubMed]
- Stauber, T.; Jentsch, T.J. Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J. Biol. Chem. 2010, 285, 34537–34548. [Google Scholar] [CrossRef] [PubMed]
- Coppola, M.A.; Gavazzo, P.; Zanardi, I.; Tettey-Matey, A.; Liantonio, A.; Fong, P.; Pusch, M. Distinct ClC-6 and ClC-7 Cl− sensitivities provide insight into ClC-7’s role in lysosomal Cl− homeostasis. J. Physiol. 2023, 601, 5635–5653. [Google Scholar] [CrossRef]
- He, H.; Cao, X.; Yin, F.; Wu, T.; Stauber, T.; Peng, J. West Syndrome Caused By a Chloride/Proton Exchange-Uncoupling CLCN6 Mutation Related to Autophagic-Lysosomal Dysfunction. Mol. Neurobiol. 2021, 58, 2990–2999. [Google Scholar] [CrossRef]
- Polovitskaya, M.M.; Barbini, C.; Martinelli, D.; Harms, F.L.; Cole, F.S.; Calligari, P.; Bocchinfuso, G.; Stella, L.; Ciolfi, A.; Niceta, M.; et al. A Recurrent Gain-of-Function Mutation in CLCN6, Encoding the ClC-6 Cl−/H+-Exchanger, Causes Early-Onset Neurodegeneration. Am. J. Hum. Genet. 2020, 107, 1062–1077. [Google Scholar] [CrossRef]
- Stauber, T.; Wartosch, L.; Vishnolia, S.; Schulz, A.; Kornak, U. CLCN7, a gene shared by autosomal recessive and autosomal dominant osteopetrosis. Bone 2023, 168, 116639. [Google Scholar] [CrossRef] [PubMed]
- Piret, S.E.; Gorvin, C.M.; Trinh, A.; Taylor, J.; Lise, S.; Taylor, J.C.; Ebeling, P.R.; Thakker, R.V. Autosomal dominant osteopetrosis associated with renal tubular acidosis is due to a CLCN7 mutation. Am. J. Med. Genet. Part. A 2016, 170, 2988–2992. [Google Scholar] [CrossRef] [PubMed]
- Steward, C.G. Neurological aspects of osteopetrosis. Neuropathol. Appl. Neurobiol. 2003, 29, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Remtulla, A.A.N.; Huber, R.J. The conserved cellular roles of CLN proteins: Novel insights from Dictyostelium discoideum. Eur. J. Cell Biol. 2023, 102, 151305. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J. Recent insights into the networking of CLN genes and proteins in mammalian cells. J. Neurochem. 2023, 165, 643–659. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.; Stenzel, W.; Goebel, H.H. Human NCL Neuropathology. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852 Pt B, 2262–2266. [Google Scholar] [CrossRef]
- Bajaj, L.; Sharma, J.; di Ronza, A.; Zhang, P.; Eblimit, A.; Pal, R.; Roman, D.; Collette, J.R.; Booth, C.; Chang, K.T.; et al. A CLN6-CLN8 complex recruits lysosomal enzymes at the ER for Golgi transfer. J. Clin. Investig. 2020, 130, 4118–4132. [Google Scholar] [CrossRef] [PubMed]
- Luebben, A.V.; Bender, D.; Becker, S.; Crowther, L.M.; Erven, I.; Hofmann, K.; Söding, J.; Klemp, H.; Bellotti, C.; Stäuble, A.; et al. Cln5 represents a new type of cysteine-based S-depalmitoylase linked to neurodegeneration. Sci. Adv. 2022, 8, eabj8633. [Google Scholar] [CrossRef]
- Yasa, S.; Sauvageau, E.; Modica, G.; Lefrancois, S. CLN5 and CLN3 function as a complex to regulate endolysosome function. Biochem. J. 2021, 478, 2339–2357. [Google Scholar] [CrossRef]
- Cárcel-Trullols, J.; Kovács, A.D.; Pearce, D.A. Cell biology of the NCL proteins: What they do and don’t do. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852 Pt B, 2242–2255. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, W.; Lin, B.; Yao, Y.; Li, C.; Hu, W.; Wu, H.; Huang, J.; Zhang, M.; Xue, T.; et al. CLN7 is an organellar chloride channel regulating lysosomal function. Sci. Adv. 2021, 7, eabj9608. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Aranda, K.; Seo, Y.J.; Gasnier, B.; Ren, D. TMEM175 Is an Organelle K+ Channel Regulating Lysosomal Function. Cell 2015, 162, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, P.; Wang, C.; Feng, X.; Geng, Q.; Chen, W.; Marthi, M.; Zhang, W.; Gao, C.; Reid, W.; et al. Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell 2022, 185, 2292–2308.e20. [Google Scholar] [CrossRef]
- Wie, J.; Liu, Z.; Song, H.; Tropea, T.F.; Yang, L.; Wang, H.; Liang, Y.; Cang, C.; Aranda, K.; Lohmann, J.; et al. A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Nature 2021, 591, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zeng, W.; Han, Y.; Lee, W.R.; Liou, J.; Jiang, Y. Lysosomal LAMP proteins regulate lysosomal pH by direct inhibition of the TMEM175 channel. Mol. Cell 2023, 83, 2524–2539.e7. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.Q.; Han, X.L.; Kang, X.L.; Wang, D.; Chen, C.H.; Wang, J.X.; Zhao, X.F. Autophagy triggers CTSD (cathepsin D) maturation and localization inside cells to promote apoptosis. Autophagy 2021, 17, 1170–1192. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.D. Regulation of lysosomal dynamics and autophagy by CTSB/cathepsin B. Autophagy 2016, 12, 2504–2505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lu, H.; Xie, X.; Shen, H.; Li, X.; Zhang, Y.; Wu, J.; Ni, J.; Li, H.; Chen, G. TMEM175 mediates Lysosomal function and participates in neuronal injury induced by cerebral ischemia-reperfusion. Mol. Brain 2020, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef]
- Sharma, J.; di Ronza, A.; Lotfi, P.; Sardiello, M. Lysosomes and Brain Health. Annu. Rev. Neurosci. 2018, 41, 255–276. [Google Scholar] [CrossRef]
- Atiba-Davies, M.; Noben-Trauth, K. TRPML3 and hearing loss in the varitint-waddler mouse. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2007, 1772, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Jörs, S.; Grimm, C.; Becker, L.; Heller, S. Genetic inactivation of Trpml3 does not lead to hearing and vestibular impairment in mice. PLoS ONE 2010, 5, e14317. [Google Scholar] [CrossRef] [PubMed]
- Staaf, S.; Oerther, S.; Lucas, G.; Mattsson, J.P.; Ernfors, P. Differential regulation of TRP channels in a rat model of neuropathic pain. Pain 2009, 144, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Scotto Rosato, A.; Krogsaeter, E.K.; Jaślan, D.; Abrahamian, C.; Montefusco, S.; Soldati, C.; Spix, B.; Pizzo, M.T.; Grieco, G.; Böck, J.; et al. TPC2 rescues lysosomal storage in mucolipidosis type IV, Niemann-Pick type C1, and Batten disease. EMBO Mol. Med. 2022, 14, e15377. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Hao, X.; Lin, W.; Su, W.; Chiang, E.; Baudry, M.; Bi, X. LAMTOR1 inhibition of TRPML1-dependent lysosomal calcium release regulates dendritic lysosome trafficking and hippocampal neuronal function. EMBO J. 2022, 41, e108119. [Google Scholar] [CrossRef]
- Ruas, M.; Davis, L.C.; Chen, C.C.; Morgan, A.J.; Chuang, K.T.; Walseth, T.F.; Grimm, C.; Garnham, C.; Powell, T.; Platt, N.; et al. Expression of Ca2+-permeable two-pore channels rescues NAADP signalling in TPC-deficient cells. EMBO J. 2015, 34, 1743–1758. [Google Scholar] [CrossRef] [PubMed]
- Endo, M. Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 2009, 89, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.J.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Webb, S.E.; Chan, C.M.; Miller, A.L. TPC2-mediated Ca2+ signaling is required for axon extension in caudal primary motor neurons in zebrafish embryos. J. Cell Sci. 2020, 133, jcs244780. [Google Scholar] [CrossRef]
- Martucci, L.L.; Launay, J.M.; Kawakami, N.; Sicard, C.; Desvignes, N.; Dakouane-Giudicelli, M.; Spix, B.; Têtu, M.; Gilmaire, F.O.; Paulcan, S.; et al. Endolysosomal TPCs regulate social behavior by controlling oxytocin secretion. Proc. Natl. Acad. Sci. USA 2023, 120, e2213682120. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Edelmann, M.J.; Maegawa, G.H.B. CNS-Targeting Therapies for Lysosomal Storage Diseases: Current Advances and Challenges. Front. Mol. Biosci. 2020, 7, 559804. [Google Scholar] [CrossRef] [PubMed]
- Prat Castro, S.; Kudrina, V.; Jaślan, D.; Böck, J.; Scotto Rosato, A.; Grimm, C. Neurodegenerative Lysosomal Storage Disorders: TPC2 Comes to the Rescue! Cells 2022, 11, 2807. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Feng, X.; Yang, J. Lysosomal Calcium in Neurodegeneration. Messenger 2016, 5, 56–66. [Google Scholar] [CrossRef]
- Gerndt, S.; Chen, C.C.; Chao, Y.K.; Yuan, Y.; Burgstaller, S.; Scotto Rosato, A.; Krogsaeter, E.; Urban, N.; Jacob, K.; Nguyen, O.N.P.; et al. Agonist-mediated switching of ion selectivity in TPC2 differentially promotes lysosomal function. Elife 2020, 9, e54712. [Google Scholar] [CrossRef]
- Johnson, T.B.; Cain, J.T.; White, K.A.; Ramirez-Montealegre, D.; Pearce, D.A.; Weimer, J.M. Therapeutic landscape for Batten disease: Current treatments and future prospects. Nat. Rev. Neurol. 2019, 15, 161–178. [Google Scholar] [CrossRef]
- Kasper, D.; Planells-Cases, R.; Fuhrmann, J.C.; Scheel, O.; Zeitz, O.; Ruether, K.; Schmitt, A.; Poët, M.; Steinfeld, R.; Schweizer, M.; et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005, 24, 1079–1091. [Google Scholar] [CrossRef]
- Weinert, S.; Jabs, S.; Supanchart, C.; Schweizer, M.; Gimber, N.; Richter, M.; Rademann, J.; Stauber, T.; Kornak, U.; Jentsch, T.J. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl- accumulation. Science 2010, 328, 1401–1403. [Google Scholar] [CrossRef]
- Leray, X.; Hilton, J.K.; Nwangwu, K.; Becerril, A.; Mikusevic, V.; Fitzgerald, G.; Amin, A.; Weston, M.R.; Mindell, J.A. Tonic inhibition of the chloride/proton antiporter ClC-7 by PI(3,5)P2 is crucial for lysosomal pH maintenance. Elife 2022, 11, e74136. [Google Scholar] [CrossRef]
- Wartosch, L.; Fuhrmann, J.C.; Schweizer, M.; Stauber, T.; Jentsch, T.J. Lysosomal degradation of endocytosed proteins depends on the chloride transport protein ClC-7. FASEB J. 2009, 23, 4056–4068. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Z.; Zeziulia, M.; Kwon, W.; Jentsch, T.J.; Grinstein, S.; Freeman, S.A. ClC-7 drives intraphagosomal chloride accumulation to support hydrolase activity and phagosome resolution. J. Cell Biol. 2023, 222, e202208155. [Google Scholar] [CrossRef] [PubMed]
- Pressey, S.N.; O‘Donnell, K.J.; Stauber, T.; Fuhrmann, J.C.; Tyynelä, J.; Jentsch, T.J.; Cooper, J.D. Distinct neuropathologic phenotypes after disrupting the chloride transport proteins ClC-6 or ClC-7/Ostm1. J. Neuropathol. Exp. Neurol. 2010, 69, 1228–1246. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, E.R.; Weston, M.R.; Hackbarth, M.; Becerril, A.; Larson, A.; Zein, W.M.; Baker, P.R., 2nd; Burke, J.D.; Dorward, H.; Davids, M.; et al. Lysosomal Storage and Albinism Due to Effects of a De Novo CLCN7 Variant on Lysosomal Acidification. Am. J. Hum. Genet. 2019, 104, 1127–1138. [Google Scholar] [CrossRef]
- Bose, S.; de Heus, C.; Kennedy, M.E.; Wang, F.; Jentsch, T.J.; Klumperman, J.; Stauber, T. Impaired Autophagic Clearance with a Gain-of-Function Variant of the Lysosomal Cl−/H+ Exchanger ClC-7. Biomolecules 2023, 13, 1799. [Google Scholar] [CrossRef]
- Bagh, M.B.; Peng, S.; Chandra, G.; Zhang, Z.; Singh, S.P.; Pattabiraman, N.; Liu, A.; Mukherjee, A.B. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat. Commun. 2017, 8, 14612. [Google Scholar] [CrossRef]
- Tognon, E.; Kobia, F.; Busi, I.; Fumagalli, A.; De Masi, F.; Vaccari, T. Control of lysosomal biogenesis and Notch-dependent tissue patterning by components of the TFEB-V-ATPase axis in Drosophila melanogaster. Autophagy 2016, 12, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Martini-Stoica, H.; Qi, C.; Lu, T.C.; Wang, S.; Xiong, W.; Qi, Y.; Xu, Y.; Sardiello, M.; Li, H.; et al. TFEB-vacuolar ATPase signaling regulates lysosomal function and microglial activation in tauopathy. Nat. Neurosci. 2024, 27, 48–62. [Google Scholar] [CrossRef]
- McBride, J.L.; Neuringer, M.; Ferguson, B.; Kohama, S.G.; Tagge, I.J.; Zweig, R.C.; Renner, L.M.; McGill, T.J.; Stoddard, J.; Peterson, S.; et al. Discovery of a CLN7 model of Batten disease in non-human primates. Neurobiol. Dis. 2018, 119, 65–78. [Google Scholar] [CrossRef]
- Danyukova, T.; Ariunbat, K.; Thelen, M.; Brocke-Ahmadinejad, N.; Mole, S.E.; Storch, S. Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation. Hum. Mol. Genet. 2018, 27, 1711–1722. [Google Scholar] [CrossRef]
- Soldati, C.; Lopez-Fabuel, I.; Wanderlingh, L.G.; Garcia-Macia, M.; Monfregola, J.; Esposito, A.; Napolitano, G.; Guevara-Ferrer, M.; Scotto Rosato, A.; Krogsaeter, E.K.; et al. Repurposing of tamoxifen ameliorates CLN3 and CLN7 disease phenotype. EMBO Mol. Med. 2021, 13, e13742. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Lau, H.A. Niemann–Pick Disease. In Encyclopedia of the Neurological Sciences, 2nd ed.; Aminoff, M.J., Daroff, R.B., Eds.; Academic Press: Oxford, UK, 2014; pp. 590–591. [Google Scholar]
- Kundu, G.K.; Anwar, S.S.; Liza, N.A.S.; Alam, S.T.; Ahmed, S.; Sarker, M.A.; Nishi, M.N.; Afrin, S.; Quddus, M.A. Niemann Pick disease: A rare lysosomal storage disease. Bangabandhu Sheikh Mujib Med. Univ. J. 2023, 15, 141–144. [Google Scholar] [CrossRef]
- Kheder, A.; Scott, C.; Olpin, S.; Hadjivassiliou, M. Niemann-Pick type C: A potentially treatable disorder? Pract. Neurol. 2013, 13, 382–385. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.M.; Aron, A.; Brodie, S.E.; Desnick, R.J.; Wasserstein, M.P. Natural history of Type A Niemann-Pick disease: Possible endpoints for therapeutic trials. Neurology 2006, 66, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Zielinski, S.E.; Crowley, J.R.; Dudley-Rucker, N.; Schaffer, J.E.; Ory, D.S. NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J. Biol. Chem. 2003, 278, 25517–25525. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 178–191. [Google Scholar] [CrossRef]
- Almkvist, O.; Johansson, C.; Laffita-Mesa, J.; Thordardottir, S.; Graff, C. APOE ε4 influences cognitive decline positively in APP and negatively in PSEN1 mutation carriers with autosomal-dominant Alzheimer’s disease. Eur. J. Neurol. 2022, 29, 3580–3589. [Google Scholar] [CrossRef] [PubMed]
- Bendiske, J.; Bahr, B.A. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis--an approach for slowing Alzheimer disease? J. Neuropathol. Exp. Neurol. 2003, 62, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Somogyi, A.; Kirkham, E.D.; Lloyd-Evans, E.; Winston, J.; Allen, N.D.; Mackrill, J.J.; Anderson, K.E.; Hawkins, P.T.; Gardiner, S.E.; Waller-Evans, H.; et al. The synthetic TRPML1 agonist ML-SA1 rescues Alzheimer-related alterations of the endosomal-autophagic-lysosomal system. J. Cell Sci. 2023, 136, jcs259875. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Du, S.; Marsh, J.A.; Horie, K.; Sato, C.; Ballabio, A.; Karch, C.M.; Holtzman, D.M.; Zheng, H. TFEB regulates lysosomal exocytosis of tau and its loss of function exacerbates tau pathology and spreading. Mol. Psychiatry 2021, 26, 5925–5939. [Google Scholar] [CrossRef]
- Bae, M.; Patel, N.; Xu, H.; Lee, M.; Tominaga-Yamanaka, K.; Nath, A.; Geiger, J.; Gorospe, M.; Mattson, M.P.; Haughey, N.J. Activation of TRPML1 clears intraneuronal Aβ in preclinical models of HIV infection. J. Neurosci. 2014, 34, 11485–11503. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Vetchinova, A.S.; Kapkaeva, M.R.; Ivanov, M.V.; Kutukova, K.A.; Mudzhiri, N.M.; Frumkina, L.E.; Brydun, A.V.; Sukhorukov, V.S.; Illarioshkin, S.N. Mitochondrial Dysfunction in Dopaminergic Neurons Derived from Patients with LRRK2- and SNCA-Associated Genetic Forms of Parkinson’s Disease. Curr. Issues Mol. Biol. 2023, 45, 8395–8411. [Google Scholar] [CrossRef]
- Tapias, V.; McCoy, J.L.; Greenamyre, J.T. Phenothiazine normalizes the NADH/NAD+ ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson’s disease. Redox Biol. 2019, 24, 101164. [Google Scholar] [CrossRef] [PubMed]
- Nandipati, S.; Litvan, I. Environmental Exposures and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2016, 13, 881. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Blumenreich, S.; Barav, O.B.; Jenkins, B.J.; Futerman, A.H. Lysosomal Storage Disorders Shed Light on Lysosomal Dysfunction in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 4966. [Google Scholar] [CrossRef] [PubMed]
- Hockey, L.N.; Kilpatrick, B.S.; Eden, E.R.; Lin-Moshier, Y.; Brailoiu, G.C.; Brailoiu, E.; Futter, C.E.; Schapira, A.H.; Marchant, J.S.; Patel, S. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 2015, 128, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Ríos, P.; Gómez-Suaga, P.; Fernández, B.; Madero-Pérez, J.; Schwab, A.J.; Ebert, A.D.; Hilfiker, S. Alterations in late endocytic trafficking related to the pathobiology of LRRK2-linked Parkinson’s disease. Biochem. Soc. Trans. 2015, 43, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Montano, M.; Corley, M.J.; Helmy, E.; Kobayashi, H.; Kinisu, M.; Suryawanshi, R.; Luo, X.; Royer, L.A.; Roan, N.R.; et al. Neuropilin-1 Mediates SARS-CoV-2 Infection of Astrocytes in Brain Organoids, Inducing Inflammation Leading to Dysfunction and Death of Neurons. mBio 2022, 13, e0230822. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.C.; Wu, A.J.; Huang, A.S.; Dong, R.; Malampati, S.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Zhu, Z.; Su, C.; et al. Lysosomal TPCN (two pore segment channel) inhibition ameliorates beta-amyloid pathology and mitigates memory impairment in Alzheimer disease. Autophagy 2022, 18, 624–642. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Lin, B.; Zeng, W.; Fan, C.; Wu, H.; Ge, Y.; Li, Q.; Li, C.; Wei, Y.; Xin, J.; et al. Lysosomal K+ channel TMEM175 promotes apoptosis and aggravates symptoms of Parkinson’s disease. EMBO Rep. 2022, 23, e53234. [Google Scholar] [CrossRef] [PubMed]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.A.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, V.; Petrozziello, T.; Sisalli, M.J.; Boscia, F.; Canzoniero, L.M.T.; Secondo, A. The activation of Mucolipin TRP channel 1 (TRPML1) protects motor neurons from L-BMAA neurotoxicity by promoting autophagic clearance. Sci. Rep. 2019, 9, 10743. [Google Scholar] [CrossRef]
- Murch, S.J.; Cox, P.A.; Banack, S.A.; Steele, J.C.; Sacks, O.W. Occurrence of beta-methylamino-l-alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol. Scand. 2004, 110, 267–269. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Mironova, Y.A.; Lin, J.P.; Kalinski, A.L.; Huffman, L.D.; Lenk, G.M.; Havton, L.A.; Meisler, M.H.; Giger, R.J. Protective role of the lipid phosphatase Fig4 in the adult nervous system. Hum. Mol. Genet. 2018, 27, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet. Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Beckers, J.; Tharkeshwar, A.K.; Van Damme, P. C9orf72 ALS-FTD: Recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 2021, 17, 3306–3322. [Google Scholar] [CrossRef] [PubMed]
- Boursin, P.; Paternotte, S.; Dercy, B.; Sabben, C.; Maïer, B. Semantics, epidemiology and semiology of stroke. Soins Rev. Ref. Infirm. 2018, 63, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, Y.; Liu, C. Autophagy and Ischemic Stroke. Adv. Exp. Med. Biol. 2020, 1207, 111–134. [Google Scholar] [CrossRef] [PubMed]
- Krzyżanowska, W.; Pomierny, B.; Filip, M.; Pera, J. Glutamate transporters in brain ischemia: To modulate or not? Acta Pharmacol. Sin. 2014, 35, 444–462. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, V.; Vinciguerra, A.; Sisalli, M.J.; Pignataro, G.; Secondo, A. Pharmacological inhibition of lysosomal two-pore channel 2 (TPC2) confers neuroprotection in stroke via autophagy regulation. Neurobiol. Dis. 2023, 178, 106020. [Google Scholar] [CrossRef] [PubMed]
- Astaburuaga, R.; Quintanar Haro, O.D.; Stauber, T.; Relógio, A. A Mathematical Model of Lysosomal Ion Homeostasis Points to Differential Effects of Cl− Transport in Ca2+ Dynamics. Cells 2019, 8, 1263. [Google Scholar] [CrossRef]
- Ishida, Y.; Nayak, S.; Mindell, J.A.; Grabe, M. A model of lysosomal pH regulation. J. Gen. Physiol. 2013, 141, 705–720. [Google Scholar] [CrossRef]
- Chen, C.C.; Krogsaeter, E.; Grimm, C. Two-pore and TRP cation channels in endolysosomal osmo-/mechanosensation and volume regulation. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118921. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.K.; Agarwal, S.; Kuo, C.H.; Kung, Y.L.; Day, C.H.; Lin, P.Y.; Lin, S.Z.; Hsieh, D.J.; Huang, C.Y.; Chiang, C.Y. Artemisia Leaf Extract protects against neuron toxicity by TRPML1 activation and promoting autophagy/mitophagy clearance in both in vitro and in vivo models of MPP+/MPTP-induced Parkinson’s disease. Phytomedicine 2022, 104, 154250. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; Brown, A.D.; Cox, P.J.; Omoto, K.; Owen, R.M.; Pryde, D.C.; Sidders, B.; Skerratt, S.E.; Stevens, E.B.; Storer, R.I.; et al. Ion channels as therapeutic targets: A drug discovery perspective. J. Med. Chem. 2013, 56, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, X.; Yang, Y.; Li, D.; Tang, K.; Zhao, Z.; He, W.; Wang, C.; Sahoo, N.; Converso-Baran, K.; et al. Small-molecule activation of lysosomal TRP channels ameliorates Duchenne muscular dystrophy in mouse models. Sci. Adv. 2020, 6, eaaz2736. [Google Scholar] [CrossRef] [PubMed]
- Schwering, C.; Kammler, G.; Wibbeler, E.; Christner, M.; Knobloch, J.K.; Nickel, M.; Denecke, J.; Baehr, M.; Schulz, A. Development of the “Hamburg Best Practice Guidelines for ICV-Enzyme Replacement therapy (ERT) in CLN2 Disease” Based on 6 Years Treatment Experience in 48 Patients. J. Child Neurol. 2021, 36, 635–641. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, W.; Shin, N.K.; Bae, Y.M.; Wie, J. Unveiling the impact of lysosomal ion channels: Balancing ion signaling and disease pathogenesis. Korean J. Physiol. Pharmacol. 2023, 27, 311–323. [Google Scholar] [CrossRef]
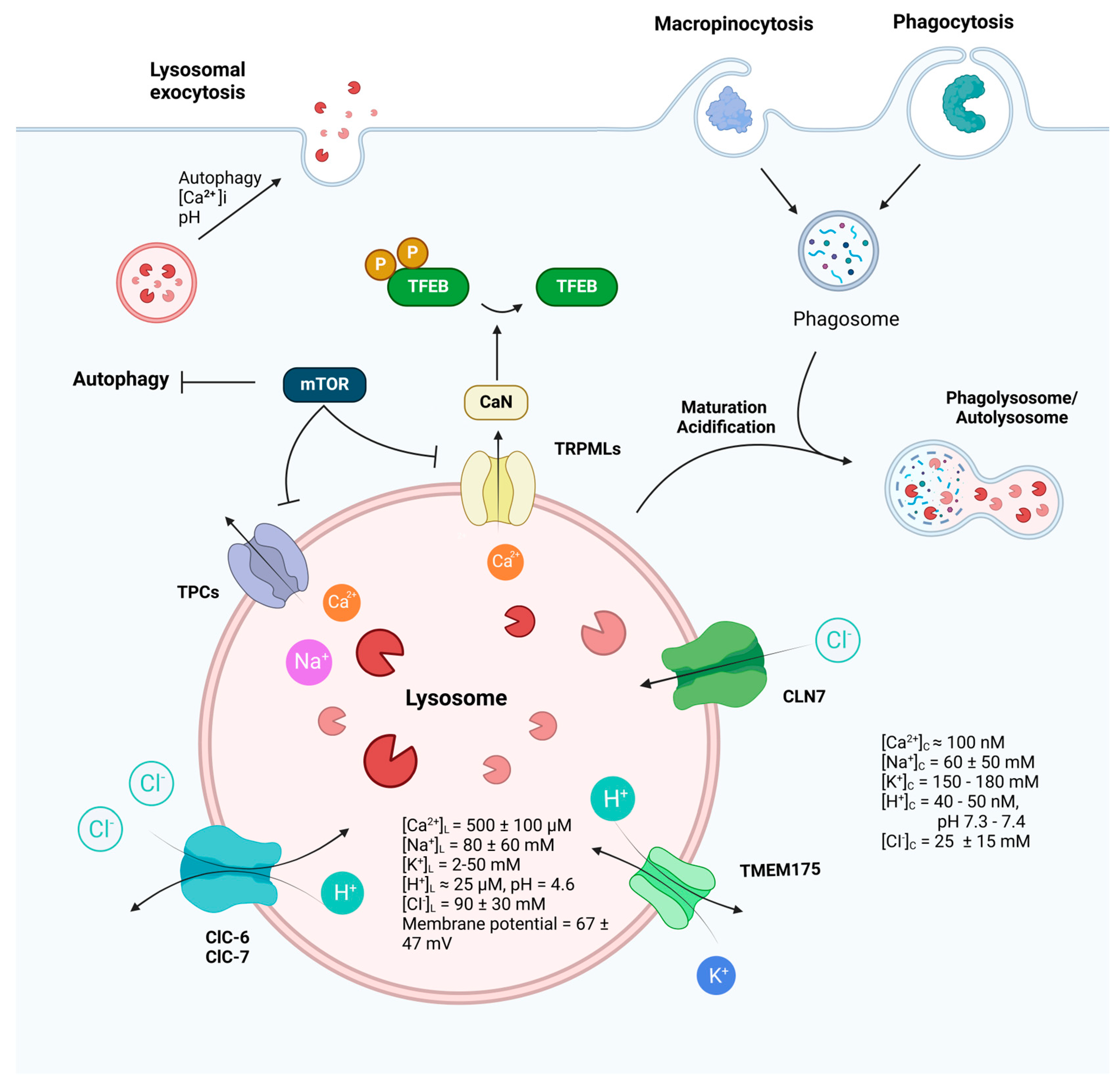
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).