
Expression of Stress-Induced Genes in Bronchoalveolar Lavage Cells and Lung Fibroblasts from Healthy and COPD Subjects

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
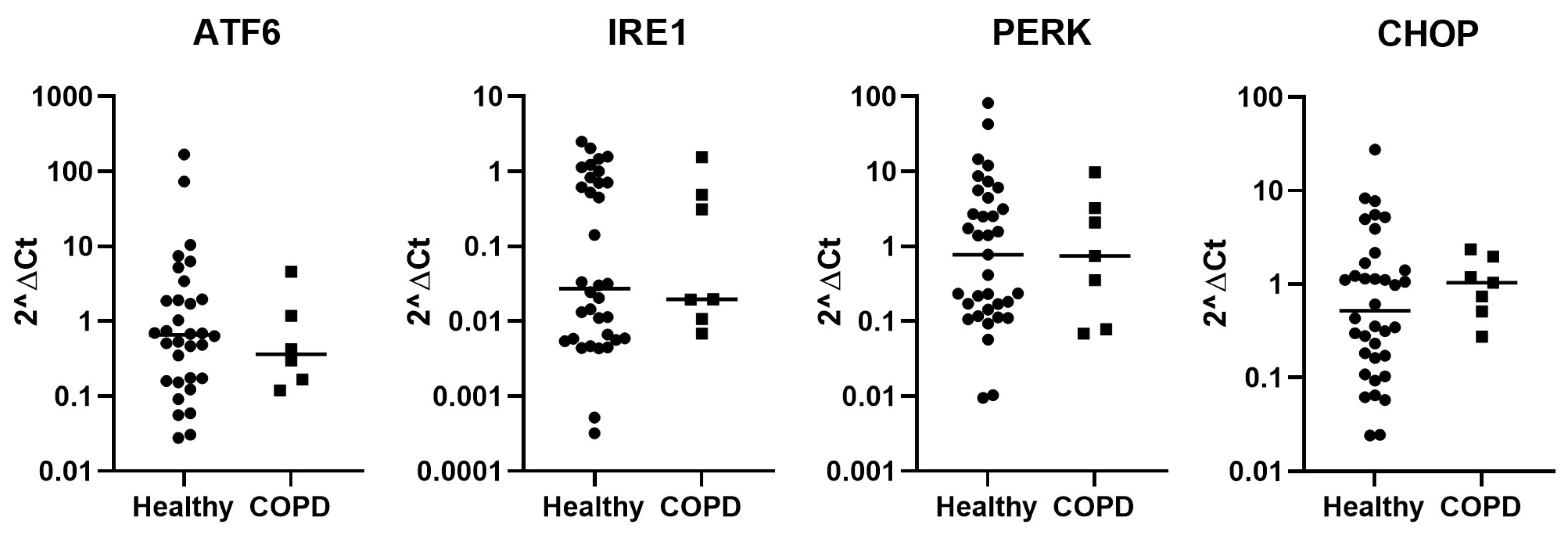
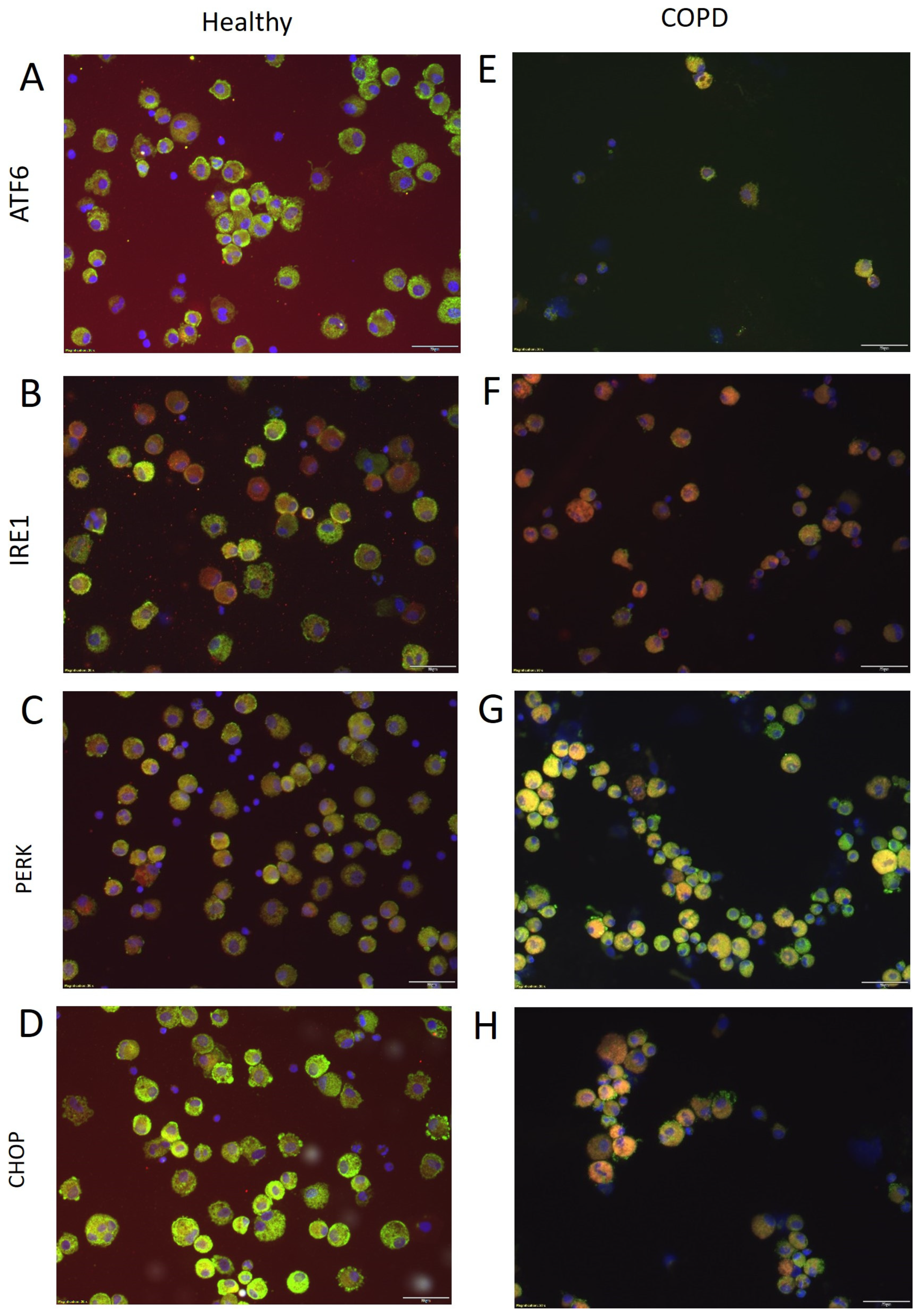
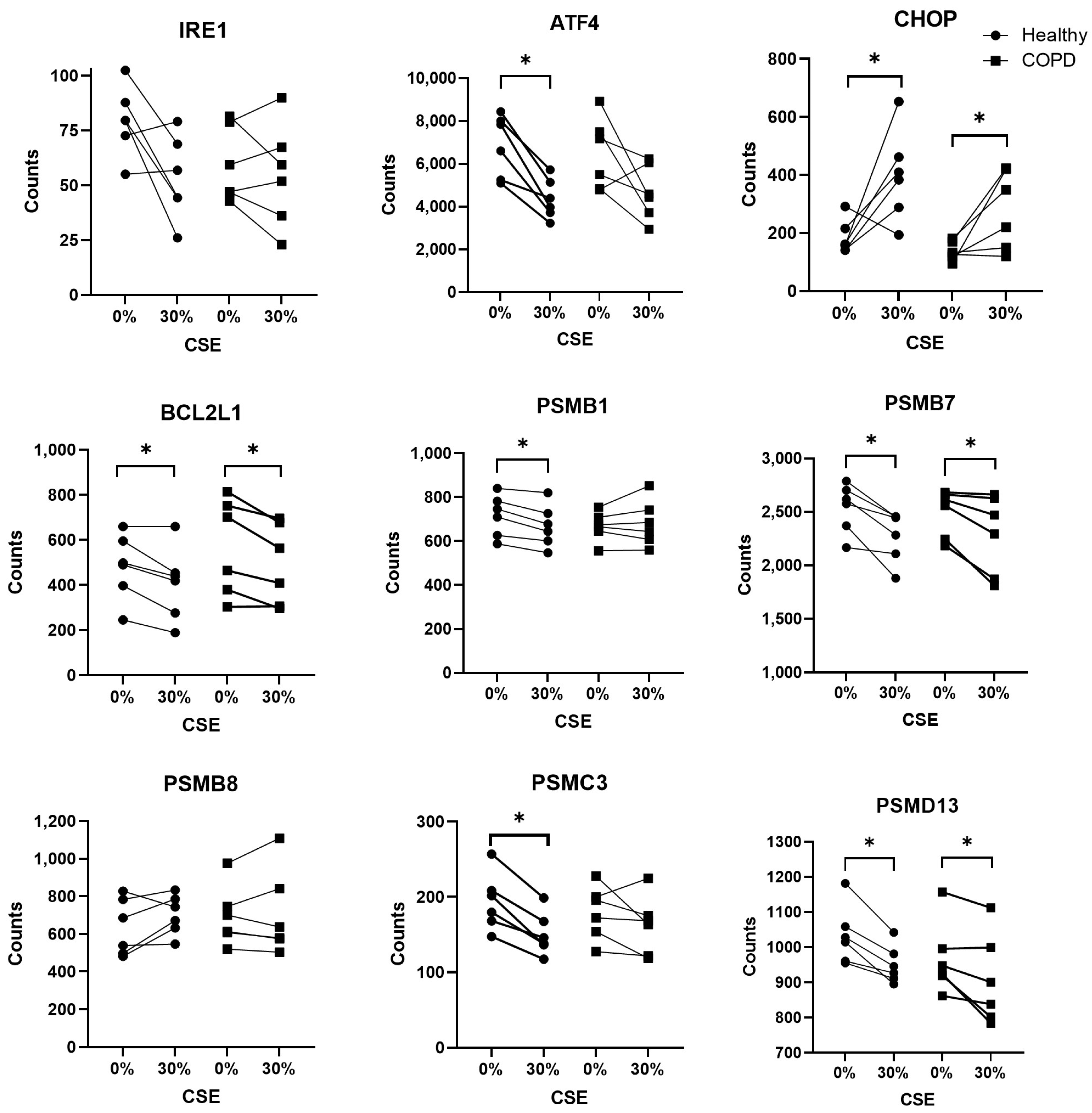
2.1. Expression of Stress-Related Genes in BAL Cells
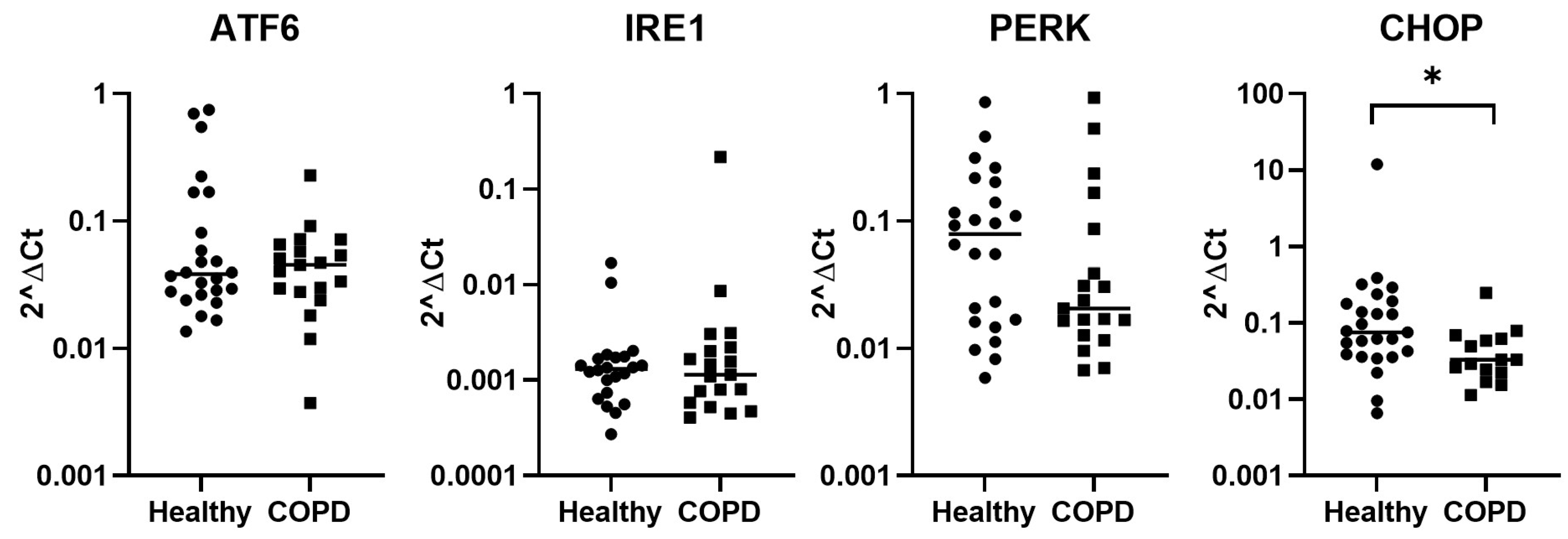
2.2. Expression of Stress-Related Genes in Lung Fibroblasts
2.3. The Effect of CSE on Cell Proliferation
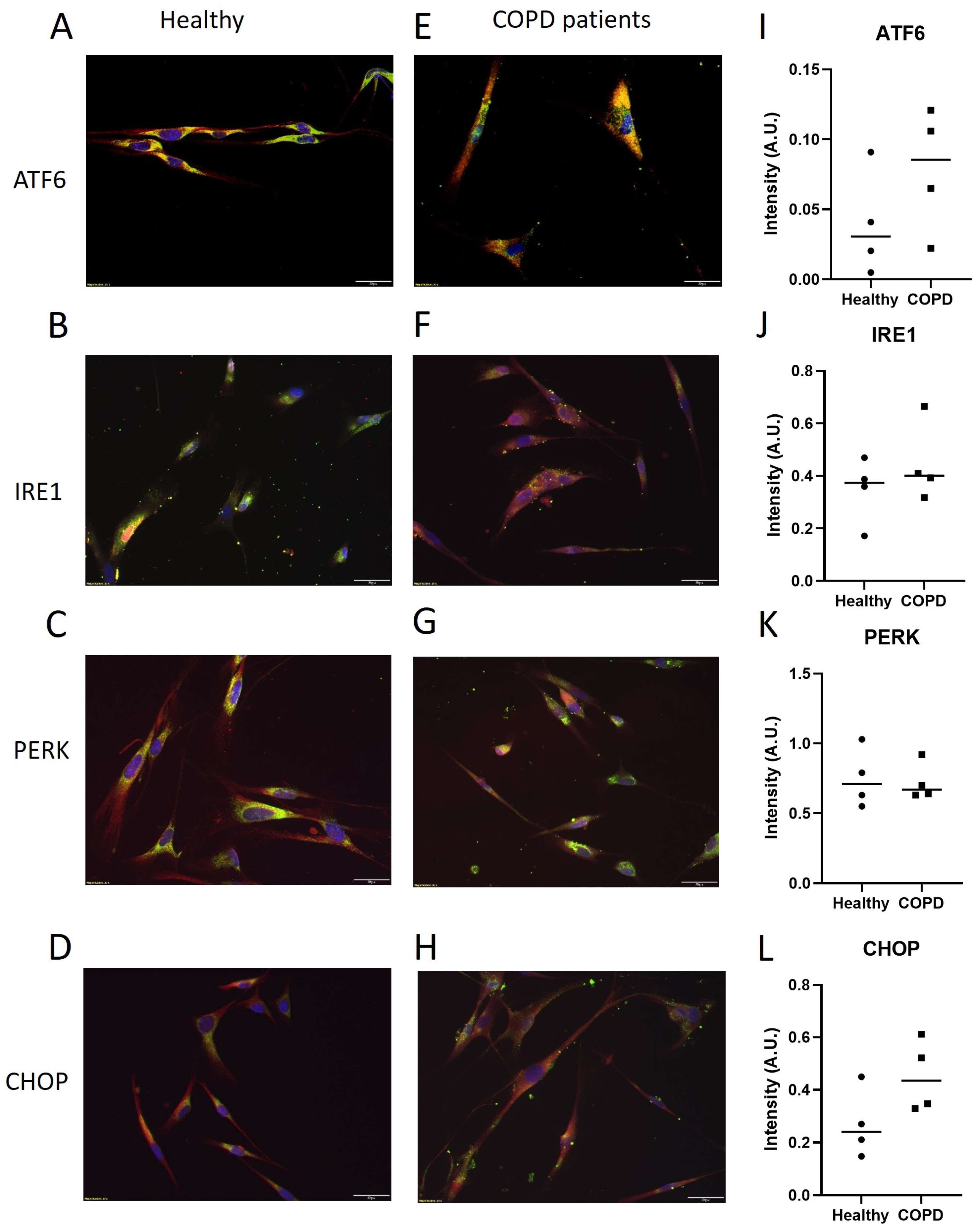
2.4. Protein Expression of ER Stress Mediators after CSE Exposure
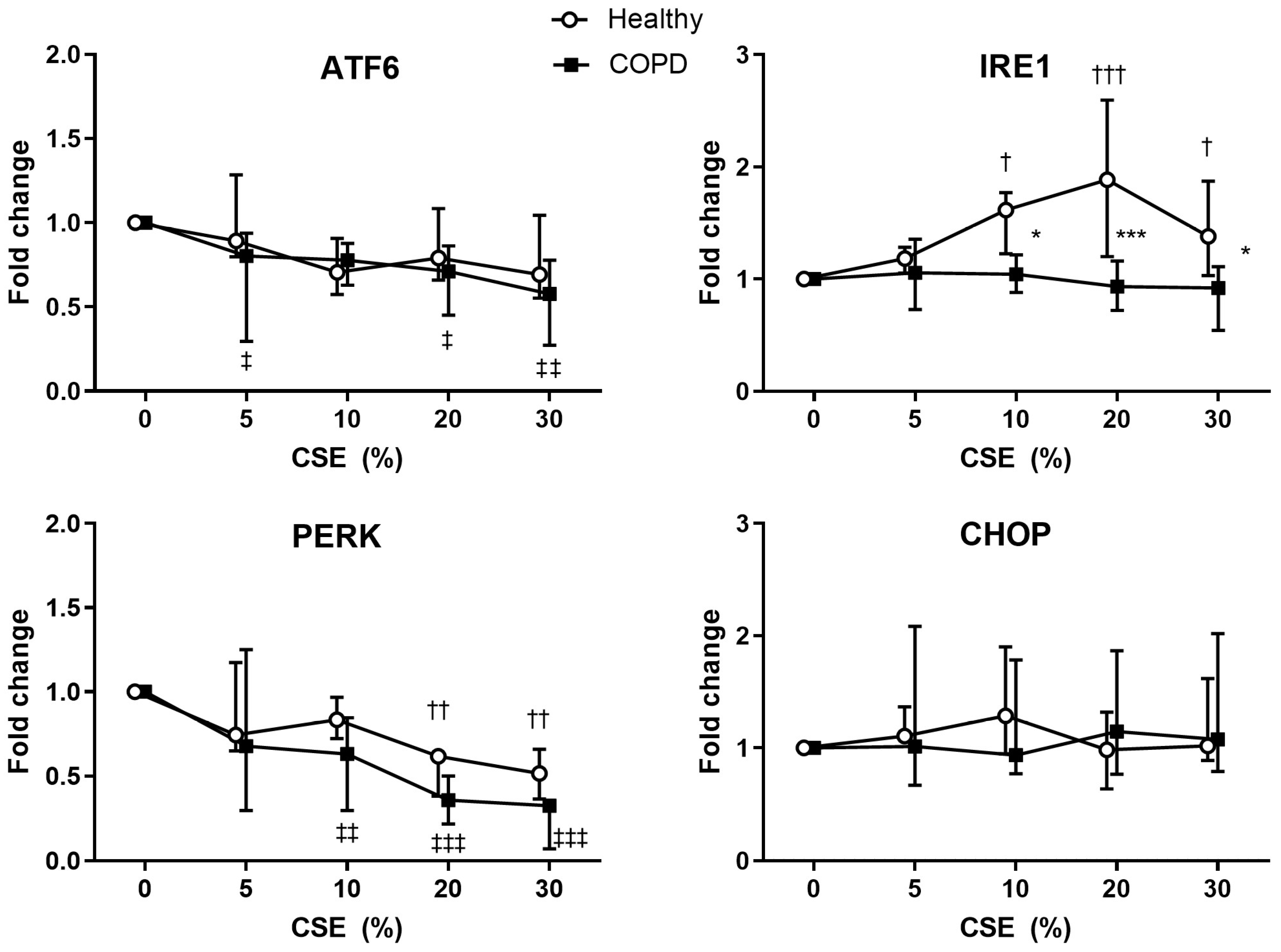
2.5. Expression of ER Stress-Related Genes in CSE Stimulated Lung Fibroblasts
3. Discussion
4. Material and Methods
4.1. Subject Characteristics
4.2. Bronchoalveolar Lavage
4.3. Cell Culture of Fibroblasts
4.4. Cigarette Smoke Extract Preparation
4.5. Cigarette Smoke Extract Exposure
4.6. RNA Purification and qPCR
4.7. Immunofluorescent Staining
4.8. Western Blot
4.9. Analyses of Cell Proliferation Using HoloMonitor
4.10. NanoString Gene Expression Analysis
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Li, T.; Pan, M.; Wang, W.; Huang, W.; Yuan, Y.; Xie, Z.; Chen, Y.; Peng, J.; Li, X.; et al. SIRT1 prevents cigarette smoking-induced lung fibroblasts activation by regulating mitochondrial oxidative stress and lipid metabolism. J. Transl. Med. 2022, 20, 222. [Google Scholar] [CrossRef] [PubMed]
- Weidner, J.; Jarenbäck, L.; Åberg, I.; Westergren-Thorsson, G.; Ankerst, J.; Bjermer, L.; Tufvesson, E. Endoplasmic reticulum, Golgi, and lysosomes are disorganized in lung fibroblasts from chronic obstructive pulmonary disease patients. Physiol. Rep. 2018, 6, e13584. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.-R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, M.O.; Kim, V.; Cornwell, W.D.; Rogers, T.J.; Kosmider, B.; Bahmed, K.; Barrero, C.; Merali, S.; Shetty, N.; Kelsen, S.G. Secretion of the endoplasmic reticulum stress protein, GRP78, into the BALF is increased in cigarette smokers. Respir. Res. 2017, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Peng, H.; Guo, W.; Luo, M.; Duan, W.; Chen, P.; Zhou, Y. Cigarette Smoke Extract Promotes Human Lung Myofibroblast Differentiation by the Induction of Endoplasmic Reticulum Stress. In Proceedings of the American Thoracic Society 2019 International Conference, Dallas, TX, USA, 17–22 May 2019. [Google Scholar]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Dombroski, B.A.; Nayak, R.R.; Ewens, K.G.; Ankener, W.; Cheung, V.G.; Spielman, R.S. Gene Expression and Genetic Variation in Response to Endoplasmic Reticulum Stress in Human Cells. Am. J. Hum. Genet. 2010, 86, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Kelsen, S.G. The Unfolded Protein Response in Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S2), S138–S145. [Google Scholar] [PubMed]
- Garcia-Ryde, M.; van der Burg, N.M.; Larsson, C.E.; Larsson-Callerfelt, A.-K.; Westergren-Thorsson, G.; Bjermer, L.; Tufvesson, E. Lung Fibroblasts from Chronic Obstructive Pulmonary Disease Subjects Have a Deficient Gene Expression Response to Cigarette Smoke Extract Compared to Healthy. Int. J. Chronic Obstr. Pulm. Dis. 2023, 18, 2999–3014. [Google Scholar] [CrossRef]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.-M.; Seeger, W.; et al. Epithelial Endoplasmic Reticulum Stress and Apoptosis in Sporadic Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef]
- Wrench, C.L.; Baker, J.R.; Monkley, S.; Fenwick, P.S.; Murray, L.; Donnelly, L.E.; Barnes, P.J. Small airway fibroblasts from patients with chronic obstructive pulmonary disease exhibit cellular senescence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2024, 326, L266–L279. [Google Scholar] [CrossRef] [PubMed]
- Min, T.; Bodas, M.; Mazur, S.; Vij, N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. 2011, 89, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Lozon, T.I.; Eastman, A.J.; Matute-Bello, G.; Chen, P.; Hallstrand, T.S.; Altemeier, W.A.; You, K.; Parikh, P.; Khandalavala, K.; Wicher, S.A.; et al. PKR-dependent CHOP induction limits hyperoxia-induced lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L422–L429. [Google Scholar] [CrossRef]
- Brandsma, C.-A.; Timens, W.; Jonker, M.R.; Rutgers, B.; Noordhoek, J.A.; Postma, D.S. Differential effects of fluticasone on extracellular matrix production by airway and parenchymal fibroblasts in severe COPD. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L582–L589. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Zhai, W.; Lu, X.; Wang, G. The Cross-Links of Endoplasmic Reticulum Stress, Autophagy, and Neurodegeneration in Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 691881. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Yamashita, Y.; Tanaka, T.; Takaki, M.; Le, M.N.; Yoshida, L.-M.; Morimoto, K. Cigarette smoke induces endoplasmic reticulum stress and suppresses efferocytosis through the activation of RhoA. Sci. Rep. 2020, 10, 12620. [Google Scholar] [CrossRef] [PubMed]
- Yeap, J.W.; Ali, I.A.H.; Ibrahim, B.; Tan, M.L. Chronic obstructive pulmonary disease and emerging ER stress-related therapeutic targets. Pulm. Pharmacol. Ther. 2023, 81, 102218. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, W.Y. Protein synthesis rhythm. J. Theor. Biol. 1975, 55, 167–200. [Google Scholar] [CrossRef]
- Okuda, T.; Wakaguri, H.; Suzuki, Y.; Sugano, S. Monitoring endoplasmic reticulum stress responsive mRNAs by RNA sequencing. Gene 2012, 500, 63–72. [Google Scholar] [CrossRef]
- Park, J.-H.; Shin, J.-M.; Yang, H.-W.; Kim, T.H.; Lee, S.H.; Lee, H.-M.; Cho, J.-G.; Park, I.-H. Cigarette Smoke Extract Stimulates MMP-2 Production in Nasal Fibroblasts via ROS/PI3K, Akt, and NF-κB Signaling Pathways. Antioxidants 2020, 9, 739. [Google Scholar] [CrossRef]
- Farid, M.; Kanaji, N.; Michalski, J.; Nakanishi, M.; Sato, T.; Wang, X.; Basma, H.; Nelson, A.; Liu, X.; Rennard, S.I. Cigarette Smoke Extract (CSE) Stimulates The Production of Vascular Endothelial Growth Factor (VEGF) by Human Lung Fibroblasts. Am. J. Respir. Crit. Care Med. 2011, 183, A3473. [Google Scholar]
- Krimmer, D.I.; Burgess, J.K.; Wooi, T.K.; Black, J.L.; Oliver, B.G.G. Matrix Proteins from Smoke-Exposed Fibroblasts Are Pro-proliferative. Am. J. Respir. Cell Mol. Biol. 2012, 46, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kanaji, N.; Basma, H.; Nelson, A.; Farid, M.; Sato, T.; Nakanishi, M.; Wang, X.; Michalski, J.; Li, Y.; Gunji, Y.; et al. Fibroblasts that resist cigarette smoke-induced senescence acquire profibrotic phenotypes. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L364–L373. [Google Scholar] [CrossRef] [PubMed]
- Malmström, J.; Larsen, K.; Hansson, L.; Löfdahl, C.-G.; Nörregard-Jensen, O.; Marko-Varga, G.; Westergren-Thorsson, G. Proteoglycan and proteome profiling of central human pulmonary fibrotic tissue utilizing miniaturized sample preparation: A feasibility study. Proteomics 2002, 2, 394–404. [Google Scholar] [CrossRef]
- Tufvesson, E.; Nihlberg, K.; Westergren-Thorsson, G.; Bjermer, L. Leukotriene receptors are differently expressed in fibroblast from peripheral versus central airways in asthmatics and healthy controls. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 67–73. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BAL Cells | Lung Fibroblasts | |||
|---|---|---|---|---|
| Healthy n = 35 | COPD n = 7 | Healthy n = 24 | COPD n = 19 | |
| Age, years a | 57 (20–73) | 58 (57–68) | 66 (20–74) | 68 (52–77) |
| Male/Female, n (%) | 13/22 (37%/63%) | 3/4 (43%/57%) | 8/16 (33%/67%) | 11/8 (58%/42%) |
| Smoking status: | ||||
| Never-smokers, n | 20 | 0 | 7 | 0 |
| Ex-smokers, n | 2 | 1 | 12 | 14 |
| Current-smokers, n | 13 | 6 | 5 | 5 |
| GOLD stage 1/2/3/4, n | - | 2/4/1/0 | - | 2/13/3/1 |
| Packyears a | 0 (0–56) | 43 (12–50) | 1 (0–70) | 43 (12–100) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Ryde, M.; van der Burg, N.M.D.; Berlin, F.; Westergren-Thorsson, G.; Bjermer, L.; Ankerst, J.; Larsson-Callerfelt, A.-K.; Andersson, C.K.; Tufvesson, E. Expression of Stress-Induced Genes in Bronchoalveolar Lavage Cells and Lung Fibroblasts from Healthy and COPD Subjects. Int. J. Mol. Sci. 2024, 25, 6600. https://doi.org/10.3390/ijms25126600
Garcia-Ryde M, van der Burg NMD, Berlin F, Westergren-Thorsson G, Bjermer L, Ankerst J, Larsson-Callerfelt A-K, Andersson CK, Tufvesson E. Expression of Stress-Induced Genes in Bronchoalveolar Lavage Cells and Lung Fibroblasts from Healthy and COPD Subjects. International Journal of Molecular Sciences. 2024; 25(12):6600. https://doi.org/10.3390/ijms25126600
Chicago/Turabian StyleGarcia-Ryde, Martin, Nicole M. D. van der Burg, Frida Berlin, Gunilla Westergren-Thorsson, Leif Bjermer, Jaro Ankerst, Anna-Karin Larsson-Callerfelt, Cecilia K. Andersson, and Ellen Tufvesson. 2024. "Expression of Stress-Induced Genes in Bronchoalveolar Lavage Cells and Lung Fibroblasts from Healthy and COPD Subjects" International Journal of Molecular Sciences 25, no. 12: 6600. https://doi.org/10.3390/ijms25126600