
The Anti-Atherosclerotic Effects of Endothelin Receptor Antagonist, Bosentan, in Combination with Atorvastatin—An Experimental Study

Abstract
:1. Introduction
2. Results
2.1. Body Weight and Biochemical Analysis
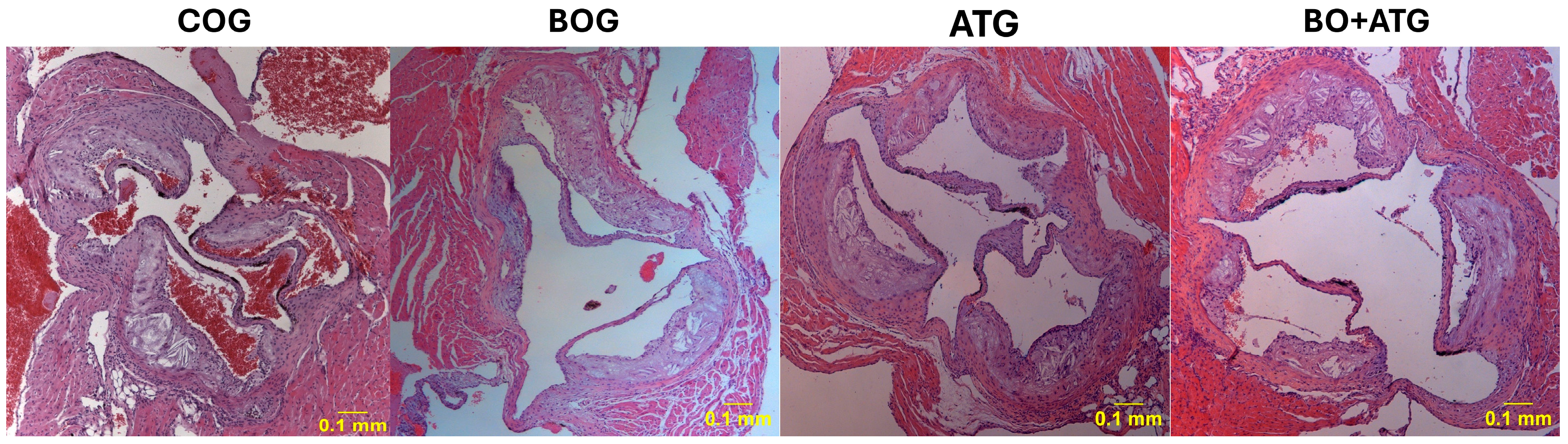
2.2. Morphometry, Collagen, and Elastin
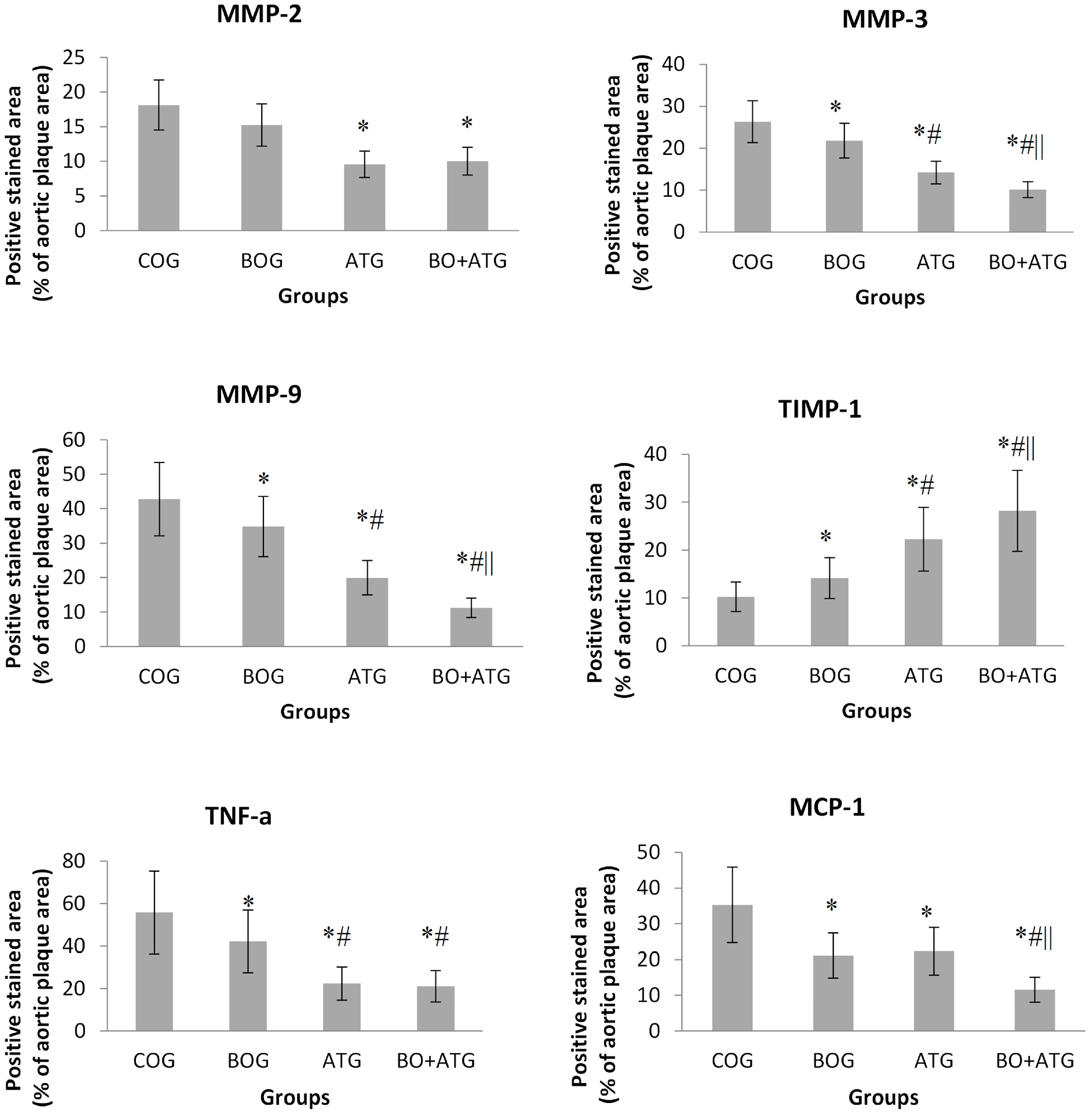
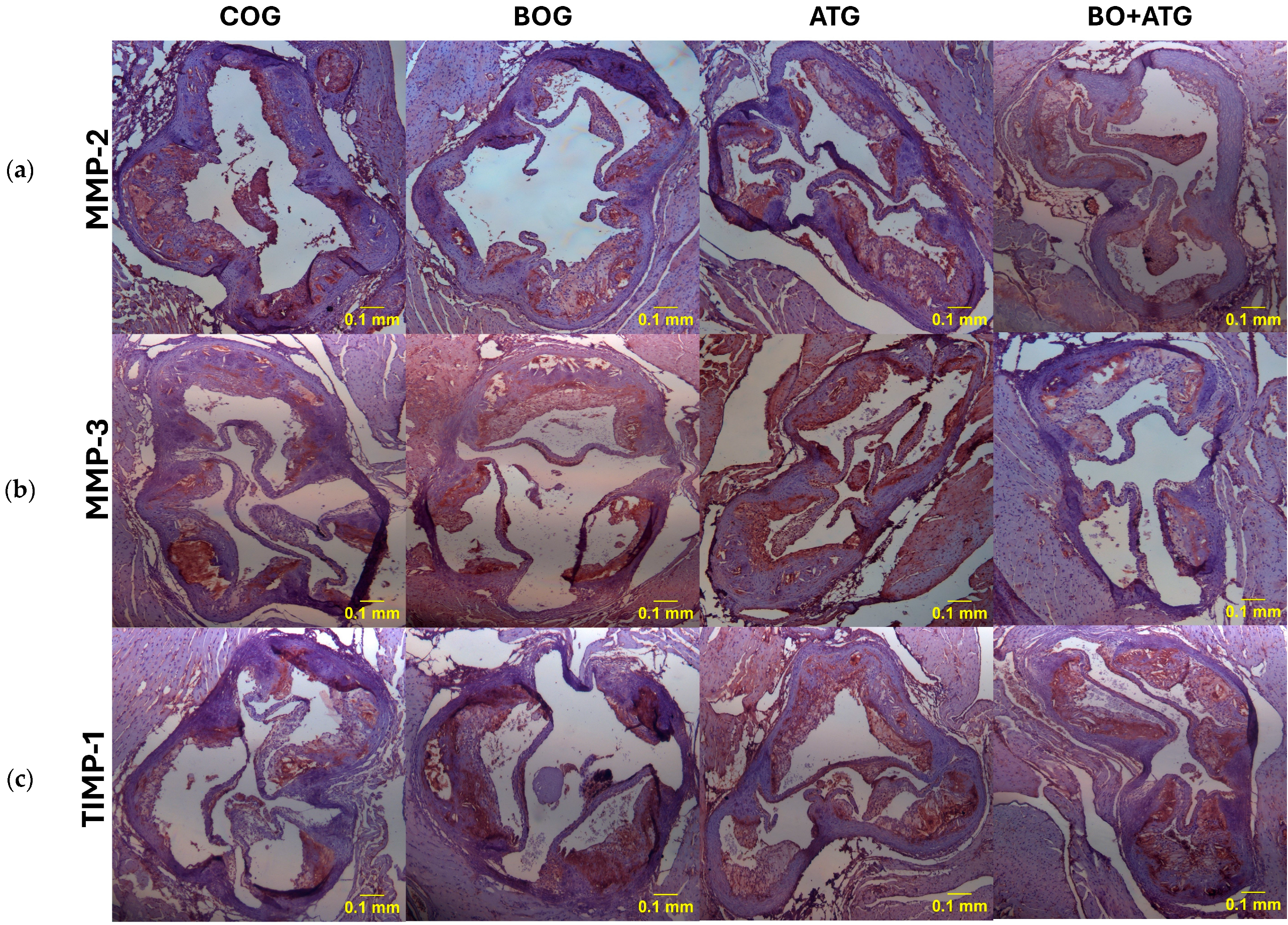
2.3. Immunohistochemistry
3. Discussion
4. Materials and Methods
4.1. Animal Model and Experimental Design
- (1)
- Control group (COG): normal saline was administered every day by esophageal gavage to make all interventions comparable between groups.
- (2)
- Bosentan group (BOG): bosentan (Actelion Pharmaceuticals LTD, Allschwil, Switzerland) was administered by esophageal gavage (100 mg/kg/day).
- (3)
- Atorvastatin group (ATG): mice were treated with atorvastatin (20 mg/kg/day) that was administered by esophageal gavage. The detailed protocol has been described in previous publication [8].
- (4)
- Bosentan and atorvastatin (BO + ATG): concomitant bosentan and atorvastatin administration by esophageal gavage, as described previously, for 6 weeks.
4.2. Histological Parameters
4.3. Histomorphometry
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Libby, P.; Nahrendorf, M.; Swirski, F.K. Monocyte Heterogeneity in Cardiovascular Disease. Semin. Immunopathol. 2013, 35, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Schönbeck, U.; Yan, Z.Q. Innate and Adaptive Immunity in the Pathogenesis of Atherosclerosis. Circ. Res. 2002, 91, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis--an Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of Atherosclerosis Plaque Progression. Heart Lung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Bornfeldt, K.E.; Goldberg, I.J. Cardiovascular Disease in Diabetes, beyond Glucose. Cell Metab. 2021, 33, 1519–1545. [Google Scholar] [CrossRef] [PubMed]
- Pleus, S.; Tytko, A.; Landgraf, R.; Heinemann, L.; Werner, C.; Müller-Wieland, D.; Ziegler, A.G.; Müller, U.A.; Freckmann, G.; Kleinwechter, H.; et al. Definition, Classification, Diagnosis and Differential Diagnosis of Diabetes Mellitus: Update 2023. Exp. Clin. Endocrinol. Diabetes 2024, 132, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Howard-Alpe, G.; Foëx, P.; Biccard, B. Cardiovascular Protection by Anti-Inflammatory Statin Therapy. Best Pract. Res. Clin. Anaesthesiol. 2008, 22, 111–133. [Google Scholar] [CrossRef] [PubMed]
- Stasinopoulou, M.; Kadoglou, N.P.E.; Christodoulou, E.; Paronis, E.; Kostomitsopoulos, N.G.; Valsami, G.; Liapis, C.D.; Kakisis, J. Statins’ Withdrawal Induces Atherosclerotic Plaque Destabilization in Animal Model-A “Rebound” Stimulation of Inflammation. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 377–386. [Google Scholar] [CrossRef]
- Wierzbicki, A.S.; Poston, R.; Ferro, A. The Lipid and Non-Lipid Effects of Statins. Pharmacol. Ther. 2003, 99, 95–112. [Google Scholar] [CrossRef]
- Gallo, G.; Savoia, C. New Insights into Endothelial Dysfunction in Cardiometabolic Diseases: Potential Mechanisms and Clinical Implications. Int. J. Mol. Sci. 2024, 25, 2973. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A Novel Potent Vasoconstrictor Peptide Produced by Vascular Endothelial Cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Böhm, F.; Pernow, J. The Importance of Endothelin-1 for Vascular Dysfunction in Cardiovascular Disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Miwa, S.; Sawamura, T.; Ninomiya, H.; Okamoto, Y. Subcellular Mechanisms of Endothelin Action in Vascular System. Eur. J. Pharmacol. 1999, 375, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Peacock, A.J.; Dawes, K.E.; Shock, A.; Gray, A.J.; Reeves, J.T.; Laurent, G.J. Endothelin-1 and Endothelin-3 Induce Chemotaxis and Replication of Pulmonary Artery Fibroblasts. Am. J. Respir. Cell Mol. Biol. 1992, 7, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Hirata, Y.; Hiroe, M.; Tsujino, M.; Adachi, S.; Takamoto, T.; Nitta, M.; Taniguchi, K.; Marumo, F. Endothelin-1 Induces Hypertrophy with Enhanced Expression of Muscle-Specific Genes in Cultured Neonatal Rat Cardiomyocytes. Circ. Res. 1991, 69, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, L.; Gray, M.; Honbo, N.Y.; Chentoufi, J.; Bergman, M.; Karliner, J.S. Endothelin-1 Stimulates Cardiac Fibroblast Proliferation through Activation of Protein Kinase C. J. Mol. Cell Cardiol. 2000, 32, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Ghatei, M.A.; Lam, H.C.; O’Halloran, D.J.; Bloom, S.R. Elevated Plasma Endothelin in Patients with Diabetes Mellitus. Diabetologia 1990, 33, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Settergren, M.; Pernow, J.; Brismar, K.; Jörneskog, G.; Kalani, M. Endothelin-A Receptor Blockade Increases Nutritive Skin Capillary Circulation in Patients with Type 2 Diabetes and Microangiopathy. J. Vasc. Res. 2008, 45, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Mather, K.J.; Mirzamohammadi, B.; Lteif, A.; Steinberg, H.O.; Baron, A.D. Endothelin Contributes to Basal Vascular Tone and Endothelial Dysfunction in Human Obesity and Type 2 Diabetes. Diabetes 2002, 51, 3517–3523. [Google Scholar] [CrossRef]
- Galiè, N.; Beghetti, M.; Gatzoulis, M.A.; Granton, J.; Berger, R.M.; Lauer, A.; Chiossi, E.; Landzberg, B. Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 (BREATHE-5) Investigators Bosentan Therapy in Patients with Eisenmenger Syndrome: A Multicenter, Double-Blind, Randomized, Placebo-Controlled Study. Circulation 2006, 114, 48–54. [Google Scholar] [CrossRef]
- Gatzoulis, M.A.; Beghetti, M.; Galiè, N.; Granton, J.; Berger, R.M.; Lauer, A.; Chiossi, E.; Landzberg, M. BREATHE-5 Investigators Longer-Term Bosentan Therapy Improves Functional Capacity in Eisenmenger Syndrome: Results of the BREATHE-5 Open-Label Extension Study. Int. J. Cardiol. 2008, 127, 27–32. [Google Scholar] [CrossRef]
- De Haro, J.; Bleda, S.; Varela, C.; Esparza, L.; Acin, F. Bosentan population-based randomized trial for clinical and endothelial function assessment on endothelin antagonist therapy in patients with intermittent claudication CLAU Investigators. Effect of Bosentan on claudication distance and endothelium-dependent vasodilation in Hispanic patients with peripheral arterial disease. Am. J. Cardiol. 2016, 117, 295–301. [Google Scholar] [CrossRef]
- De Haro, J.; Bleda, S.; Gonzalez-Hidalgo, C.; Michel, I.; Acin, F. Long-Term Effects of Bosentan on Cardiovascular Events in Hispanic Patients with Intermittent Claudication: Four-Year Follow-up of the CLAU Trial: The CLAU Randomized Trial Long-Term Outcome. Am. J. Cardiovasc. Drugs 2019, 19, 203–209. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, Z.; Li, G. The Protective Effect of Bosentan against Atherosclerosis in Apolipoprotein E-Deficient Mice Is Mediated by miRNA-21. Biomed. Res. Int. 2019, 2019, 8348430. [Google Scholar] [CrossRef]
- Mulder, P.; Richard, V.; Derumeaux, G.; Hogie, M.; Henry, J.P.; Lallemand, F.; Compagnon, P.; Macé, B.; Comoy, E.; Letac, B.; et al. Role of Endogenous Endothelin in Chronic Heart Failure: Effect of Long-Term Treatment with an Endothelin Antagonist on Survival, Hemodynamics, and Cardiac Remodeling. Circulation 1997, 96, 1976–1982. [Google Scholar] [CrossRef]
- Kedzierski, R.M.; Yanagisawa, M. Endothelin System: The Double-Edged Sword in Health and Disease. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 851–876. [Google Scholar] [CrossRef]
- Ihling, C.; Szombathy, T.; Bohrmann, B.; Brockhaus, M.; Schaefer, H.E.; Loeffler, B.M. Coexpression of Endothelin-Converting Enzyme-1 and Endothelin-1 in Different Stages of Human Atherosclerosis. Circulation 2001, 104, 864–869. [Google Scholar] [CrossRef]
- Kowala, M.C.; Rose, P.M.; Stein, P.D.; Goller, N.; Recce, R.; Beyer, S.; Valentine, M.; Barton, D.; Durham, S.K. Selective Blockade of the Endothelin Subtype A Receptor Decreases Early Atherosclerosis in Hamsters Fed Cholesterol. Am. J. Pathol. 1995, 146, 819–826. [Google Scholar]
- Best, P.J.; Lerman, A. Endothelin in Cardiovascular Disease: From Atherosclerosis to Heart Failure. J. Cardiovasc. Pharmacol. Ther. 2000, 35, S61–S63. [Google Scholar] [CrossRef]
- Jabarpour, M.; Rashtchizadeh, N.; Argani, H.; Ghorbanihaghjo, A.; Ranjbarzadhag, M.; Sanajou, D.; Panah, F.; Alirezaei, A. The Impact of Dyslipidemia and Oxidative Stress on Vasoactive Mediators in Patients with Renal Dysfunction. Int. Urol. Nephrol. 2019, 51, 2235–2242. [Google Scholar] [CrossRef]
- Rivera-Gonzalez, O.; Wilson, N.A.; Coats, L.E.; Taylor, E.B.; Speed, J.S. Endothelin Receptor Antagonism Improves Glucose Handling, Dyslipidemia, and Adipose Tissue Inflammation in Obese Mice. Clin. Sci. 2021, 135, 1773–1789. [Google Scholar] [CrossRef]
- Moustardas, P.; Kadoglou, N.P.; Katsimpoulas, M.; Kapelouzou, A.; Kostomitsopoulos, N.; Karayannacos, P.E.; Kostakis, A.; Liapis, C.D. The Complementary Effects of Atorvastatin and Exercise Treatment on the Composition and Stability of the Atherosclerotic Plaques in ApoE Knockout Mice. PLoS ONE 2014, 9, e108240. [Google Scholar] [CrossRef]
- Lee, S.G.; Lee, S.J.; Thuy, N.V.P.; Kim, J.S.; Lee, J.J.; Lee, O.H.; Kim, C.K.; Oh, J.; Park, S.; Lee, O.H.; et al. Synergistic Protective Effects of a Statin and an Angiotensin Receptor Blocker for Initiation and Progression of Atherosclerosis. PLoS ONE 2019, 14, e0215604. [Google Scholar] [CrossRef]
- Li, D.Q.; Lv, F.F.; Li, Z.C.; Dai, Z.Y.; Wang, H.X.; Han, Y. Anti-Atherosclerotic Effects between a Combined Treatment with Simvastatin plus Hirudin and Single Simvastatin Therapy in Patients with Early Type 2 Diabetes Mellitus. Ann. Transl. Med. 2019, 7, 302. [Google Scholar] [CrossRef]
- Omarjee, L.; Fontaine, C.; Mahe, G.; Jaquinandi, V. Improvement of peripheral artery disease with Sildenafil and Bosentan combined therapy in a patient with limited cutaneous systemic sclerosis: A case report. Medicine 2017, 96, e6988. [Google Scholar] [CrossRef]
- van der Vorst, E.P.C.; Weber, C.; Donners, M.M.P.C. A Disintegrin and Metalloproteases (ADAMs) in Cardiovascular, Metabolic and Inflammatory Diseases: Aspects for Theranostic Approaches. Thromb. Haemost. 2018, 118, 1167–1175. [Google Scholar] [CrossRef]
- Sharifi, M.A.; Wierer, M.; Dang, T.A.; Milic, J.; Moggio, A.; Sachs, N.; von Scheidt, M.; Hinterdobler, J.; Müller, P.; Werner, J.; et al. ADAMTS-7 Modulates Atherosclerotic Plaque Formation by Degradation of TIMP-1. Circ. Res. 2023, 133, 674–686. [Google Scholar] [CrossRef]
- Lorentzen, L.G.; Yeung, K.; Eldrup, N.; Eiberg, J.P.; Sillesen, H.H.; Davies, M.J. Proteomic Analysis of the Extracellular Matrix of Human Atherosclerotic Plaques Shows Marked Changes between Plaque Types. Matrix Biol. Plus 2024, 21, 100141. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Daskalopoulou, S.S.; Perrea, D.; Liapis, C.D. Matrix Metalloproteinases and Diabetic Vascular Complications. Angiology 2005, 56, 173–189. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Zamilpa, R. Temporal and Spatial Expression of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases Following Myocardial Infarction. Cardiovasc. Ther. 2012, 30, 31–41. [Google Scholar] [CrossRef]
- Wang, Y.; Johnson, J.A.; Fulp, A.; Sutton, M.A.; Lessner, S.M. Adhesive Strength of Atherosclerotic Plaque in a Mouse Model Depends on Local Collagen Content and Elastin Fragmentation. J. Biomech. 2013, 46, 716–722. [Google Scholar] [CrossRef]
- Kadoglou, N.P. The Beneficial Effects of a Direct Thrombin Inhibitor, Dabigatran Etexilate, on the Development and Stability of Atherosclerotic Lesions in Apolipoprotein E-Deficient Mice: Dabigatran Etexilate and Atherosclerosis. Cardiovasc. Drugs Ther. 2012, 26, 367–374. [Google Scholar] [CrossRef]
- Basiak, M.; Hachula, M.; Kosowski, M.; Machnik, G.; Maliglowka, M.; Dziubinska-Basiak, M.; Krysiak, R.; Okopien, B. The Effect of PCSK9 Inhibition on the Stabilization of Atherosclerotic Plaque Determined by Biochemical and Diagnostic Imaging Methods. Molecules 2023, 28, 5928. [Google Scholar] [CrossRef]
- Basiak, M. Impact of PCSK9 Inhibition on Proinflammatory Cytokines and Matrix Metalloproteinases Release in Patients with Mixed Hyperlipidemia and Vulnerable Atherosclerotic Plaque. Pharmaceuticals 2022, 15, 802. [Google Scholar] [CrossRef]
- Kadoglou, N.P.E.; Velidakis, N.; Khattab, E.; Kassimis, G.; Patsourakos, N. The Interplay between Statins and Adipokines. Is This Another Explanation of Statins’ “pleiotropic” Effects? Cytokine 2021, 148, 155698. [Google Scholar] [CrossRef]
- Umebashi, K.; Yamamoto, M.; Tokito, A.; Sudou, K.; Takenoshita, Y.; Jougasaki, M. Inhibitory Effects of Simvastatin on IL-33-Induced MCP-1 via the Suppression of the JNK Pathway in Human Vascular Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 13015. [Google Scholar] [CrossRef]
- Vieceli Dalla Sega, F.; Cimaglia, P.; Manfrini, M.; Fortini, F.; Marracino, L.; Bernucci, D.; Pompei, G.; Scala, A.; Trichilo, M.; De Carolis, B.; et al. Circulating Biomarkers of Endothelial Dysfunction and Inflammation in Predicting Clinical Outcomes in Diabetic Patients with Critical Limb Ischemia. Int. J. Mol. Sci. 2022, 23, 10641. [Google Scholar] [CrossRef]
- Sun, J.; Pan, S.; Yu, H.; Hu, H.; Sun, Y.U.; Yang, Z.; Hoffman, R.M.; Yuan, H. Anti-Inflammatory and Anti-Thrombotic Efficacy of Targeted Ultrasound Microbubbles on LPS-Induced HUVEC Cells. Anticancer Res. 2021, 41, 4761–4769. [Google Scholar] [CrossRef]
- Verma, S.; Li, S.H.; Badiwala, M.V.; Weisel, R.D.; Fedak, P.W.; Li, R.K.; Dhillon, B.; Mickle, D.A. Endothelin Antagonism and Interleukin-6 Inhibition Attenuate the Proatherogenic Effects of C-Reactive Protein. Circulation 2002, 105, 1890–1896. [Google Scholar] [CrossRef]
- Yao, E.H.; Wang, H.J.; Xu, C.S. Effects of Tongxinluo on the Neointima Formation and Expression of Inflammatory Cytokines in Rats after Carotid Artery Balloon Injury. Indian J. Pharmacol. 2014, 46, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhuang, S.; Jiang, S. Muscone inhibits the progression of atherosclerotic plaques in mice aorta by inhibiting the NF-κB/p65 pathway. Biochem. Biophys. Res. Commun. 2024, 702, 149628. [Google Scholar] [CrossRef] [PubMed]
- Dede, E.; Liapis, D.; Davos, C.; Katsimpoulas, M.; Varela, A.; Mpotis, I.; Kostomitsopoulos, N.; Kadoglou, N.P.E. The Effects of Exercise Training on Cardiac Matrix Metalloproteinases Activity and Cardiac Function in Mice with Diabetic Cardiomyopathy. Biochem. Biophys. Res. Commun. 2022, 586, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P. The Anti-Inflammatory Effects of Exercise Training Promote Atherosclerotic Plaque Stabilization in Apolipoprotein E Knockout Mice with Diabetic Atherosclerosis. Eur. J. Histochem. 2013, 57, e3. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| COG (n = 12) | BOG (n = 12) | ATG (n = 12) | BO + ATG (n = 12) | p | |
|---|---|---|---|---|---|
| Weight (g) | 34.4 ± 4.2 | 33.3 ± 4.8 | 32.9 ± 3.9 | 33.5 ± 4.2 | 0.512 |
| FPG (mg/dL) | 281 ± 29 | 289 ± 55 | 295 ± 45 | 309 ± 49 | 0.794 |
| TC (mg/dL) | 759 ± 131 | 758 ± 182 | 511 ± 131 * | 502 ± 191 * | <0.001 |
| TG (mg/dl) | 155 ± 41 | 145 ± 34 | 129 ± 30 | 131 ± 33 | 0.400 |
| COG (n = 12) | BOG (n = 12) | ATG (n = 12) | BO + ATG (n = 12) | p | |
|---|---|---|---|---|---|
| Lumen stenosis (%) | 24.6 ± 4.8 | 19.5 ± 2.2 a,c,d | 12.8 ± 4.8 a,b,d | 9.1 ± 2.7 a,b,c | <0.001 |
| Elastin (%) plaque | 8.12 ± 2.10 | 10.62 ± 6.52 c,d | 25.17 ± 6.91 a | 31.02 ± 5.23 a,b | <0.001 |
| Collagen (%) plaque | 14.21 ± 4.21 | 22.83 ± 4.79 a,c,d | 31.88 ± 5.97 a,b | 40.33 ± 8.72 a,b,c | <0.001 |
| Fibrous cap thickness (μm) | 9.12 ± 3.10 | 13.12 ± 3.23 a,c,d | 23.12 ± 5.44 a,b,d | 48.12 ± 6.21 a,b,c | <0.001 |
| a-actin (VSMCs) (%) plaque | 17.13 ± 3.21 | 26.88 ± 6.06 a | 20.53 ± 6.97 | 28.10 ± 6.82 a | 0.005 |
| Mac-3 (macrophages) (%) plaque | 34.56 ± 10.25 | 26.46 ± 6.82 a,c,d | 15.09 ± 3.22 a,b,c | 10.12 ± 3.78 a,b,c | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stasinopoulou, M.; Kostomitsopoulos, N.; Kadoglou, N.P.E. The Anti-Atherosclerotic Effects of Endothelin Receptor Antagonist, Bosentan, in Combination with Atorvastatin—An Experimental Study. Int. J. Mol. Sci. 2024, 25, 6614. https://doi.org/10.3390/ijms25126614
Stasinopoulou M, Kostomitsopoulos N, Kadoglou NPE. The Anti-Atherosclerotic Effects of Endothelin Receptor Antagonist, Bosentan, in Combination with Atorvastatin—An Experimental Study. International Journal of Molecular Sciences. 2024; 25(12):6614. https://doi.org/10.3390/ijms25126614
Chicago/Turabian StyleStasinopoulou, Marianna, Nikolaos Kostomitsopoulos, and Nikolaos P. E. Kadoglou. 2024. "The Anti-Atherosclerotic Effects of Endothelin Receptor Antagonist, Bosentan, in Combination with Atorvastatin—An Experimental Study" International Journal of Molecular Sciences 25, no. 12: 6614. https://doi.org/10.3390/ijms25126614