

The Effect of Neuronal CoQ10 Deficiency and Mitochondrial Dysfunction on a Rotenone-Induced Neuronal Cell Model of Parkinson’s Disease
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Result
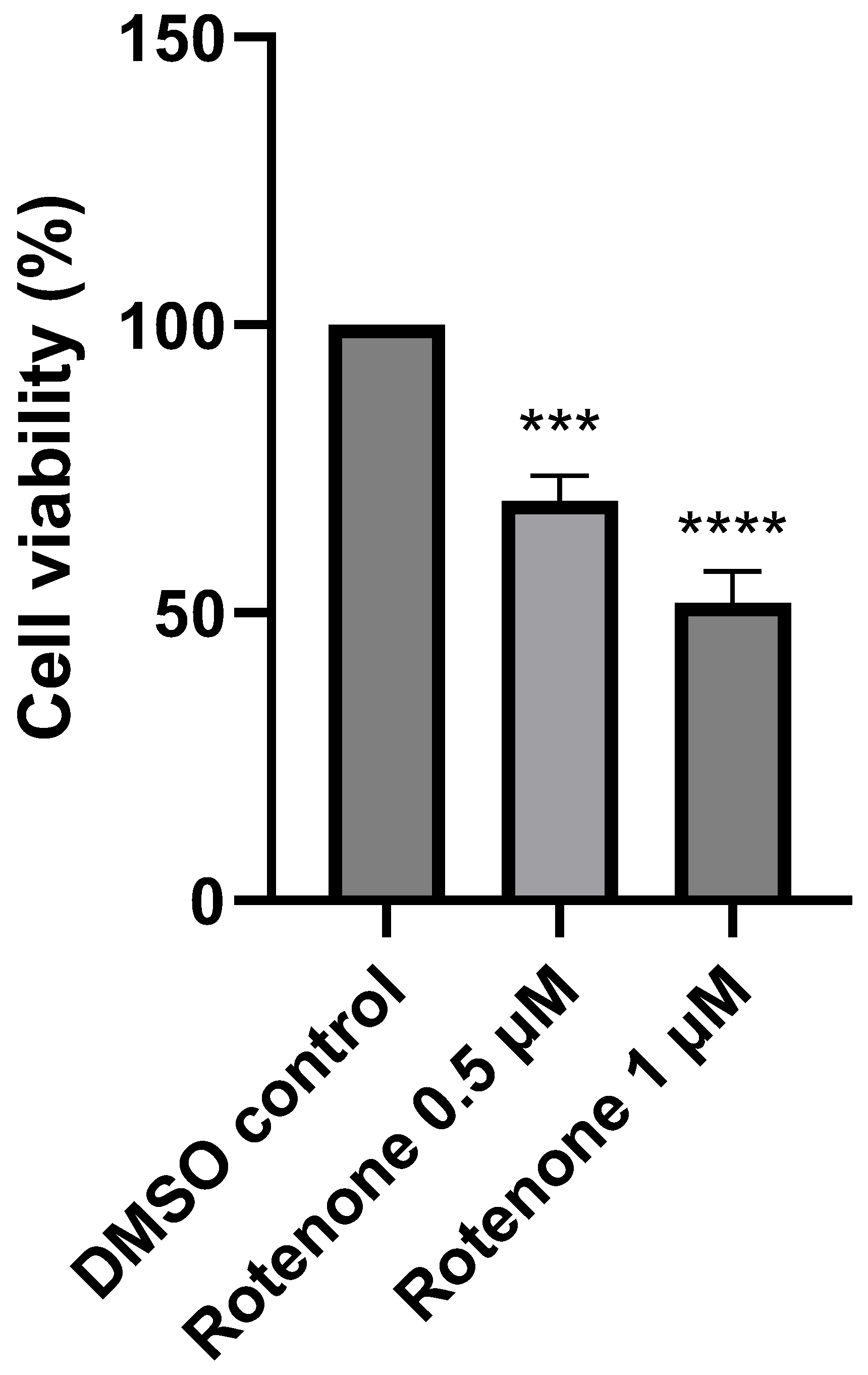
2.1. The Effect of Rotenone Treatment on Cell Viability
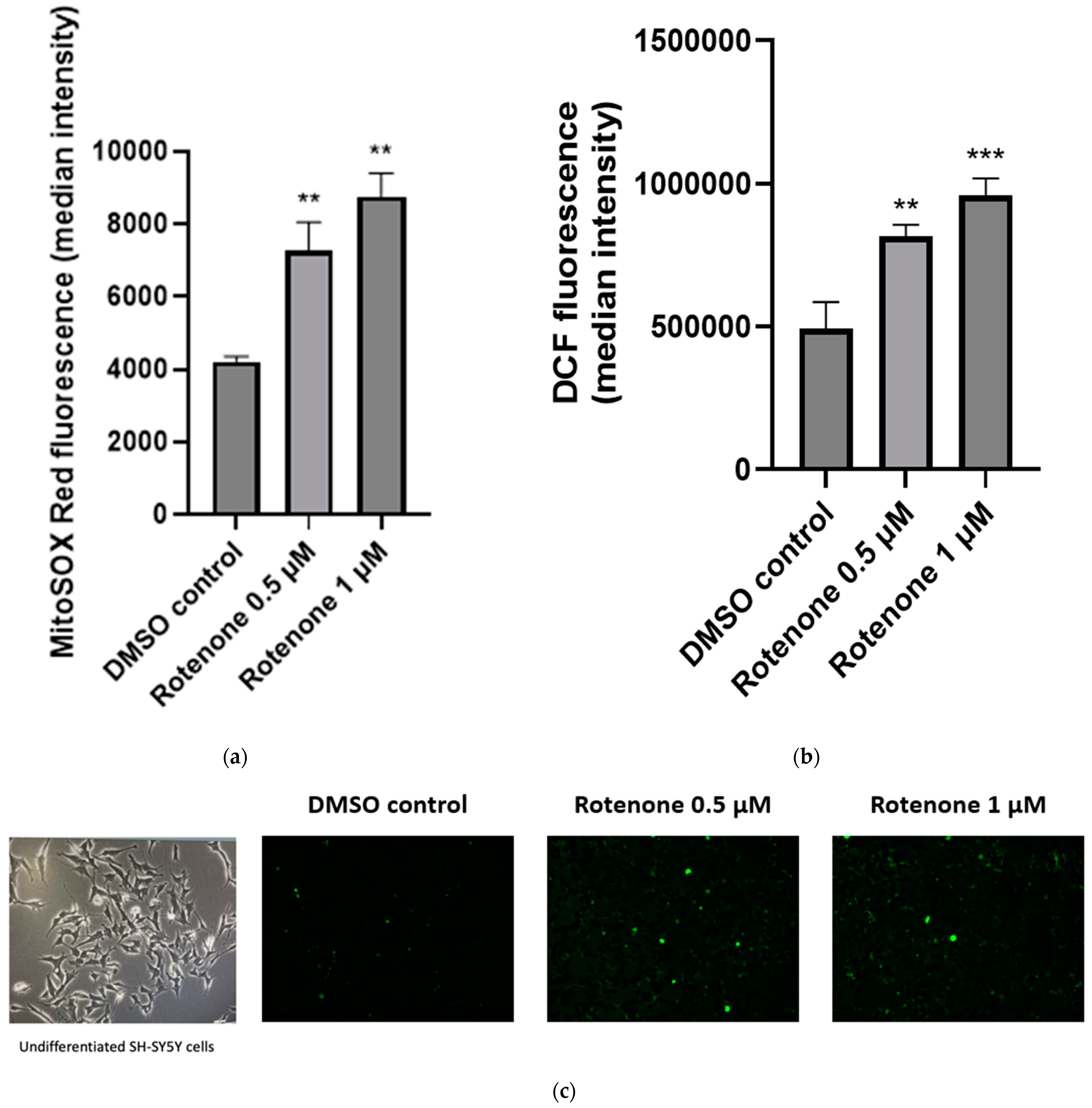
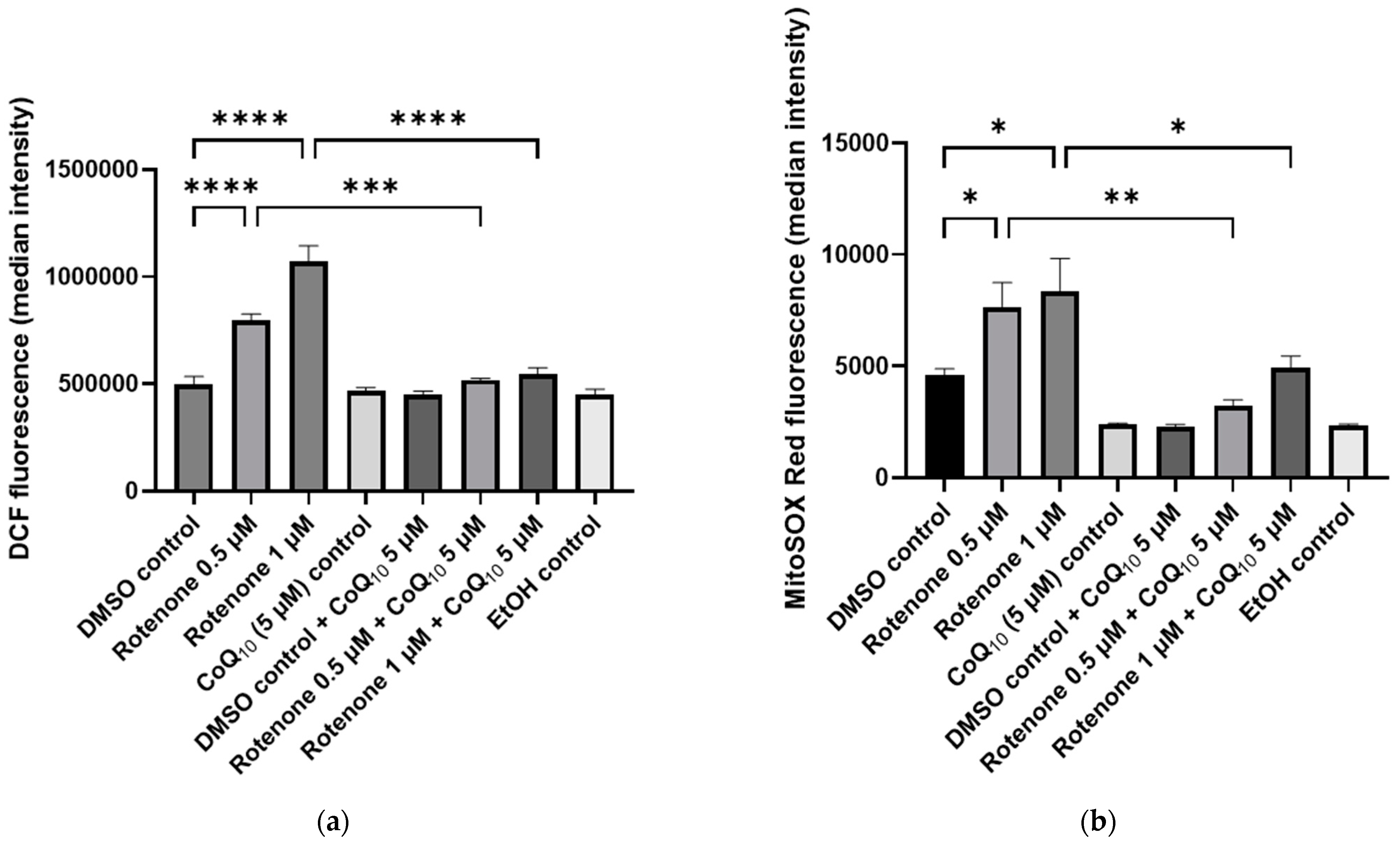
2.2. The Effect of Rotenone Treatment on Cellular ROS Production
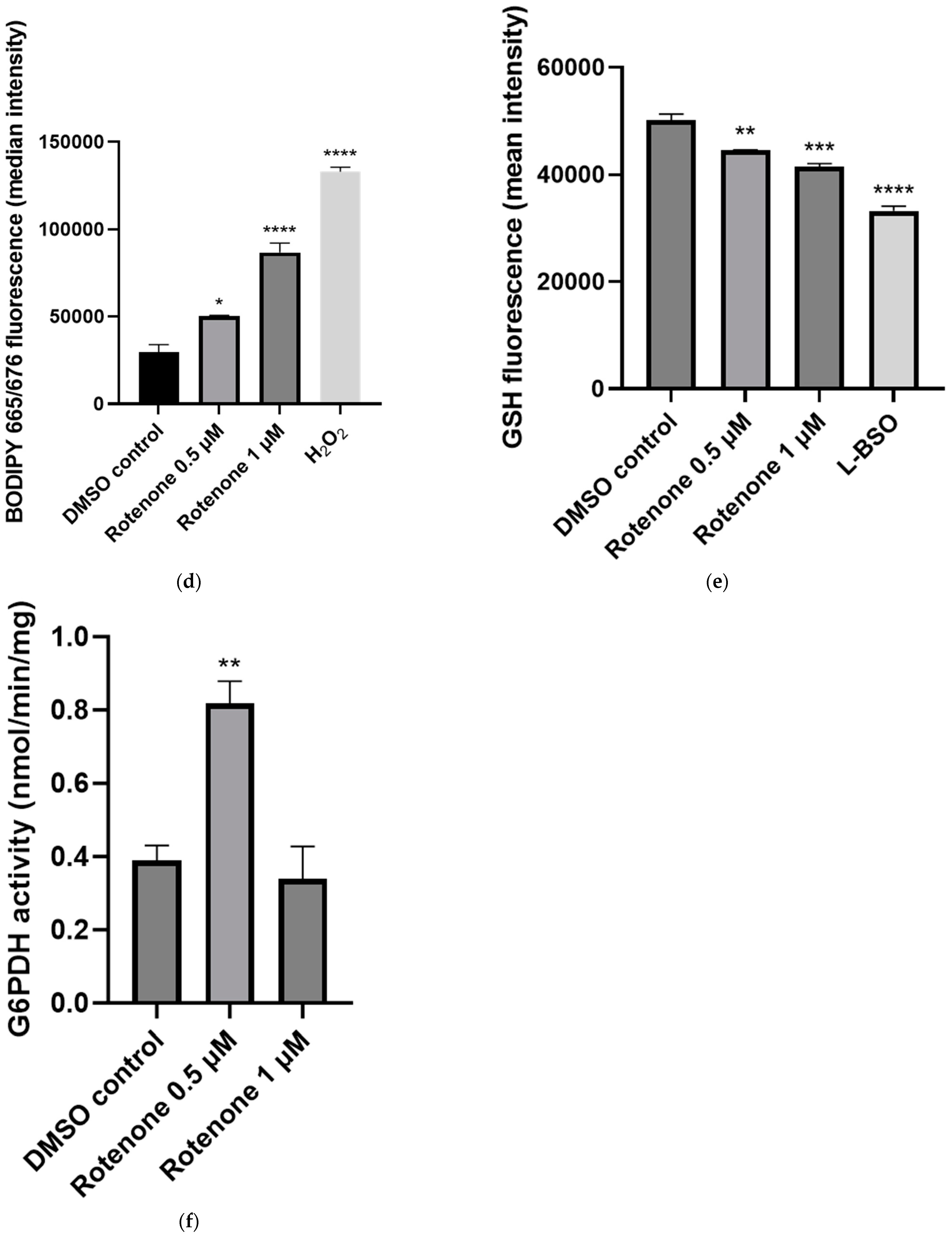
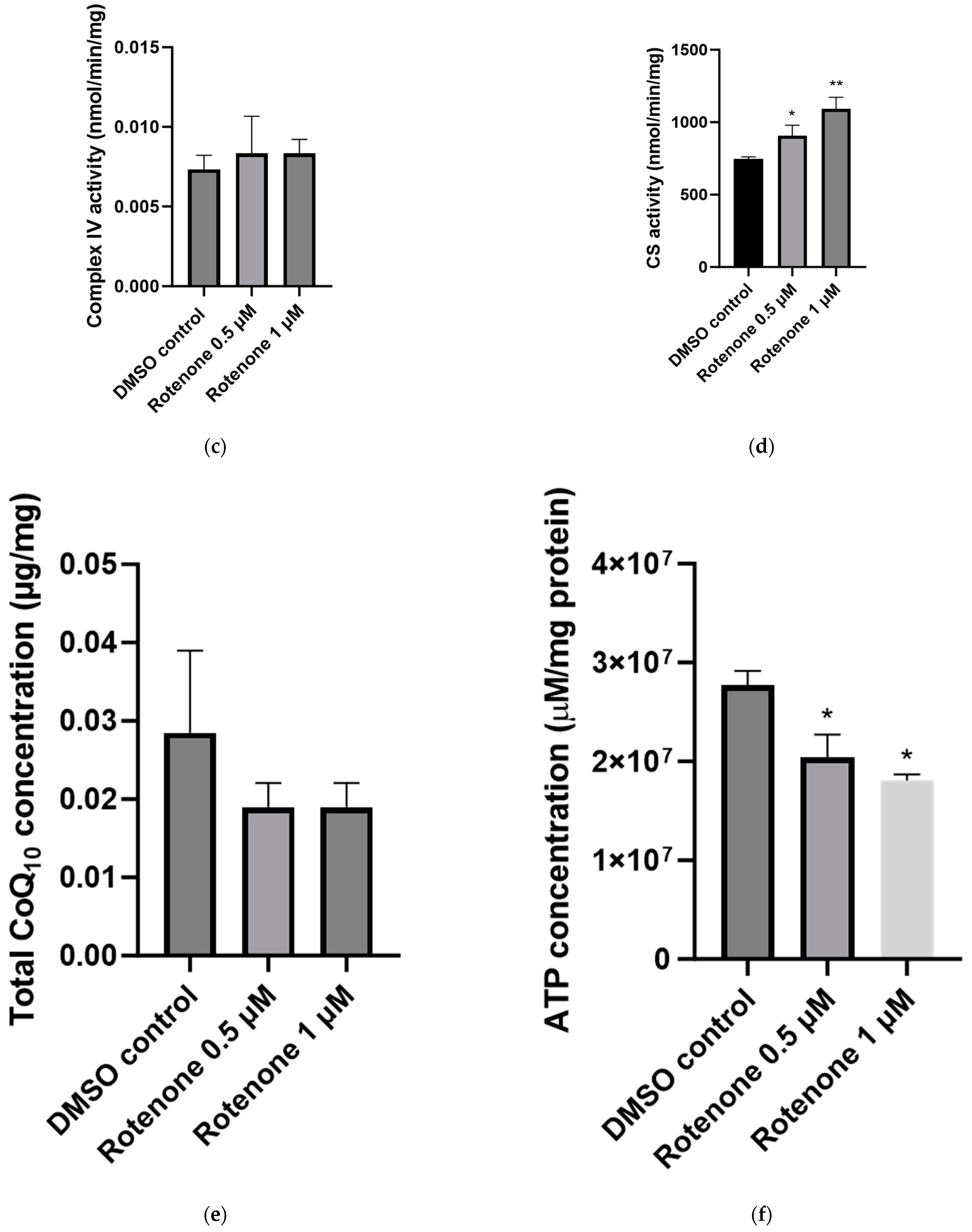
2.3. The Effect of Rotenone Treatment on Mitochondrial Function
2.4. The Effect of CoQ10 Supplementation on Rotenone-Treated Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals
All Reagents Were of Analytical Grade
4.2. Cell Culture
4.3. Rotenone Exposure
4.4. CoQ10 Supplementation
4.5. Cell Viability Assay
4.6. Intracellular ROS Measurement
4.7. Mitochondrial Superoxide Measurement
4.8. MRC Enzyme Activities
4.9. Lipid Peroxidation Measurement
4.10. Reduced Glutathione Measurement
4.11. Intracellular ATP Measurement
4.12. G6PDH Enzyme Activity
4.13. Quantification of Cellular CoQ10 Content
4.14. Total Protein Determination
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cuenca, L.; Gil-Martinez, A.L.; Cano-Fernandez, L.; Sanchez-Rodrigo, C.; Estrada, C.; Fernandez-Villalba, E.; Herrero, M.T. Parkinson’s disease: A short story of 200 years. Histol. Histopathol. 2019, 34, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 (Suppl. S1), 14–20. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Lim, K.L. Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr. Genom. 2013, 14, 486–501. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects [Internet]; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 1. [Google Scholar] [PubMed]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066, Erratum in Lancet 2009, 374, 684. [Google Scholar] [CrossRef] [PubMed]
- Odnokoz, O.; Nakatsuka, K.; Wright, C.; Castellanos, J.; Klichko, V.I.; Kretzschmar, D.; Orr, W.C.; Radyuk, S.N. Mitochondrial Redox Signaling Is Critical to the Normal Functioning of the Neuronal System. Front. Cell Dev. Biol. 2021, 9, 613036. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ramalingam, M.; Kim, S.J. Reactive oxygen/nitrogen species and their functional correlations in neurodegenerative diseases. J. Neural. Transm. 2012, 119, 891–910. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Zhu, X.; Wang, X.; Lee, H.G.; Nunomura, A.; Petersen, R.B.; Perry, G.; Smith, M.A. Mitochondria: A therapeutic target in neurodegeneration. Biochim. Biophys. Acta 2010, 1802, 212–220. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nicolson, G.L. Mitochondrial Dysfunction and Chronic Disease: Treatment with Natural Supplements. Integr. Med. 2014, 13, 35–43. [Google Scholar] [PubMed] [PubMed Central]
- Huang, C.W.; Lin, K.M.; Hung, T.Y.; Chuang, Y.C.; Wu, S.N. Multiple Actions of Rotenone, an Inhibitor of Mitochondrial Respiratory Chain, on Ionic Currents and Miniature End-Plate Potential in Mouse Hippocampal (mHippoE-14) Neurons. Cell Physiol. Biochem. 2018, 47, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Chernivec, E.; Cooper, J.; Naylor, K. Exploring the Effect of Rotenone-A Known Inducer of Parkinson’s Disease-On Mitochondrial Dynamics in Dictyostelium discoideum. Cells 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fato, R.; Bergamini, C.; Leoni, S.; Lenaz, G. Mitochondrial production of reactive oxygen species: Role of complex I and quinone analogues. Biofactors 2008, 32, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.; Allen, G.F.; Mamais, A.; Ling, H.; Li, A.; Duberley, K.E.; Hargreaves, I.P.; Pope, S.; Holton, J.L.; Lees, A.; et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson’s disease. Neurobiol. Aging 2014, 35, 1111–1115. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vos, M. Mitochondrial Complex I deficiency: Guilty in Parkinson’s disease. Signal Transduct. Target. Ther. 2022, 7, 136. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nandipati, S.; Litvan, I. Environmental Exposures and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2016, 13, 881. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barcelos, I.P.; Haas, R.H. CoQ10 and Aging. Biology 2019, 8, 28. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Manzar, H.; Abdulhussein, D.; Yap, T.E.; Cordeiro, M.F. Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. Int. J. Mol. Sci. 2020, 21, 9299. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neergheen, V.; Chalasani, A.; Wainwright, L.; Yubero, D.; Montero, R.; Artuch, R.; Hargreaves, I. Coenzyme Q10 in the Treatment of Mitochondrial Disease. J. Inborn Errors Metab. Screen. 2017, 5, e160063. [Google Scholar] [CrossRef]
- Onaolapo, O.J.; Odeniyi, A.O.; Jonathan, S.O.; Samuel, M.O.; Amadiegwu, D.; Olawale, A.; Tiamiyu, A.O.; Ojo, F.O.; Yahaya, H.A.; Ayeni, O.J.; et al. An Investigation of the Anti-Parkinsonism Potential of Co-enzyme Q10 and Co-enzyme Q10/Levodopa-carbidopa Combination in Mice. Curr. Aging Sci. 2021, 14, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Attia, H.N.; Maklad, Y.A. Neuroprotective effects of coenzyme Q10 on paraquat-induced Parkinson’s disease in experimental animals. Behav. Pharmacol. 2018, 29, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Rauchová, H. Coenzyme Q10 effects in neurological diseases. Physiol. Res. 2021, 70 (Suppl. S4), S683–S714. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xie, H.R.; Hu, L.S.; Li, G.Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sherer, T.B.; Richardson, J.R.; Testa, C.M.; Seo, B.B.; Panov, A.V.; Yagi, T.; Matsuno-Yagi, A.; Miller, G.W.; Greenamyre, J.T. Mechanism of toxicity of pesticides acting at complex I: Relevance to environmental etiologies of Parkinson’s disease. J. Neurochem. 2007, 100, 1469–1479. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Balakrishnan, R.; Elangovan, N.; Mohankumar, T.; Nataraj, J.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M.; Akbar, M.; Abdul Sattar Khan, M. Isolongifolene attenuates rotenone-induced mitochondrial dysfunction, oxidative stress and apoptosis. Front. Biosci. (Schol. Ed.) 2018, 10, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chinta, S.J.; Andersen, J.K. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: Implications for Parkinson’s disease. Free Radic. Biol. Med. 2006, 41, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.D.; Zhao, X.; Li, Y.; Li, G.R.; Liu, X.L. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci. Lett. 1986, 67, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Bharath, S.; Andersen, J.K. Glutathione depletion in a midbrain-derived immortalized dopaminergic cell line results in limited tyrosine nitration of mitochondrial complex I subunits: Implications for Parkinson’s disease. Antioxid. Redox Signal. 2005, 7, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Davey, G.P.; Peuchen, S.; Clark, J.B. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J. Biol. Chem. 1998, 273, 12753–12757. [Google Scholar] [CrossRef] [PubMed]
- Telford, J.E.; Kilbride, S.M.; Davey, G.P. Complex I is rate-limiting for oxygen consumption in the nerve terminal. J. Biol. Chem. 2009, 284, 9109–9114. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Duberley, K.E.; Heales, S.J.; Abramov, A.Y.; Chalasani, A.; Land, J.M.; Rahman, S.; Hargreaves, I.P. Effect of Coenzyme Q10 supplementation on mitochondrial electron transport chain activity and mitochondrial oxidative stress in Coenzyme Q10 deficient human neuronal cells. Int. J. Biochem. Cell Biol. 2014, 50, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.C.; Shaposhnykov, A.A.; Träger, S.; Engler, J.B.; Witte, M.E.; Roth, V.; Vieira, V.; Paauw, N.; Bauer, S.; Schwencke-Westphal, C.; et al. Enhancing mitochondrial activity in neurons protects against neurodegeneration in a mouse model of multiple sclerosis. eLife 2021, 10, e61798. [Google Scholar] [CrossRef] [PubMed]
- Poole, O.V.; Hanna, M.G.; Pitceathly, R.D. Mitochondrial disorders: Disease mechanisms and therapeutic approaches. Discov. Med. 2015, 20, 325–331. [Google Scholar] [PubMed]
- Selak, M.A.; de Chadarevian, J.P.; Melvin, J.J.; Grover, W.D.; Salganicoff, L.; Kaye, E.M. Mitochondrial activity in Pompe’s disease. Pediatr. Neurol. 2000, 23, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Hargreaves, I.; Clayton, P.; Heales, S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatr. 2001, 139, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Calabrò, V.; Mason, P.J.; Filosa, S.; Civitelli, D.; Cittadella, R.; Tagarelli, A.; Martini, G.; Brancati, C.; Luzzatto, L. Genetic heterogeneity of glucose-6-phosphate dehydrogenase deficiency revealed by single-strand conformation and sequence analysis. Am. J. Hum. Genet. 1993, 52, 527–536. [Google Scholar] [PubMed] [PubMed Central]
- Tiwari, M. Glucose 6 phosphatase dehydrogenase (G6PD) and neurodegenerative disorders: Mapping diagnostic and therapeutic opportunities. Genes. Dis. 2017, 4, 196–203. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, H.C.; Wu, Y.H.; Yen, W.C.; Liu, H.Y.; Hwang, T.L.; Stern, A.; Chiu, D.T. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meng, Q.; Zhang, Y.; Hao, S.; Sun, H.; Liu, B.; Zhou, H.; Wang, Y.; Xu, Z.X. Recent findings in the regulation of G6PD and its role in diseases. Front. Pharmacol. 2022, 13, 932154. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mejías, R.; Villadiego, J.; Pintado, C.O.; Vime, P.J.; Gao, L.; Toledo-Aral, J.J.; Echevarría, M.; López-Barneo, J. Neuroprotection by transgenic expression of glucose-6-phosphate dehydrogenase in dopaminergic nigrostriatal neurons of mice. J. Neurosci. 2006, 26, 4500–4508. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Filosa, S.; Fico, A.; Paglialunga, F.; Balestrieri, M.; Crooke, A.; Verde, P.; Abrescia, P.; Bautista, J.M.; Martini, G. Failure to increase glucose consumption through the pentose-phosphate pathway results in the death of glucose-6-phosphate dehydrogenase gene-deleted mouse embryonic stem cells subjected to oxidative stress. Biochem. J. 2003, 370 Pt. 3, 935–943. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pandolfi, P.P.; Sonati, F.; Rivi, R.; Mason, P.; Grosveld, F.; Luzzatto, L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995, 14, 5209–5215. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- López-Lluch, G.; Del Pozo-Cruz, J.; Sánchez-Cuesta, A.; Cortés-Rodríguez, A.B.; Navas, P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Raizner, A.E. Coenzyme Q10. Methodist DeBakey Cardiovasc. J. 2019, 15, 185–191. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Maroz, A.; Anderson, R.F.; Smith, R.A.; Murphy, M.P. Reactivity of ubiquinone and ubiquinol with superoxide and the hydroperoxyl radical: Implications for in vivo antioxidant activity. Free Radic. Biol. Med. 2009, 46, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; MacRae, C.L.; Broome, S.C.; D’souza, R.F.; Narang, R.; Wang, H.W.; Mori, T.A.; Hickey, A.J.R.; Mitchell, C.J.; Merry, T.L. MitoQ and CoQ10 supplementation mildly suppresses skeletal muscle mitochondrial hydrogen peroxide levels without impacting mitochondrial function in middle-aged men. Eur. J. Appl. Physiol. 2020, 120, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Cochemé, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Han, B.; Zhao, Y.; Li, G.; Wang, T.; He, J.; Du, W.; Cao, X.; Gan, J.; Wang, Z.; et al. Rosmarinic Acid Attenuates Rotenone-Induced Neurotoxicity in SH-SY5Y Parkinson’s Disease Cell Model through Abl Inhibition. Nutrients 2022, 14, 3508. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- López, L.C.; Quinzii, C.M.; Area, E.; Naini, A.; Rahman, S.; Schuelke, M.; Salviati, L.; Dimauro, S.; Hirano, M. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: Time- and compound-dependent effects. PLoS ONE 2010, 5, e11897. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sylvester, P.W. Optimization of the tetrazolium dye (MTT) colorimetric assay for cellular growth and viability. Methods Mol. Biol. 2011, 716, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Thermofisher. CM-H2DCFDA (General Oxidative Stress Indicator). 2023. Available online: https://www.thermofisher.cn/order/catalog/product/C6827 (accessed on 11 January 2024).
- Thermofisher. MitoSOX™ Mitochondrial Superoxide Indicators, for Live-Cell Imaging. 2005. Available online: https://tools.thermofisher.com/content/sfs/manuals/mp36008.pdf (accessed on 11 January 2024).
- Hargreaves, I.P. Biochemical Assessment and Monitoring of Mitochondrial Disease. J. Clin. Med. 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Thermofisher. BODIPY™ 665/676 (Lipid Peroxidation Sensor). 2023. Available online: https://www.thermofisher.cn/order/catalog/product/B3932 (accessed on 11 January 2024).
- Lange, C.; Lehmann, C.; Mahler, M.; Bednarski, P.J. Comparison of Cellular Death Pathways after mTHPC-mediated Photodynamic Therapy (PDT) in Five Human Cancer Cell Lines. Cancers 2019, 11, 702. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thermofisher. Monochlorobimane (mBCl). 2023. Available online: https://www.thermofisher.cn/order/catalog/product/M1381MP (accessed on 11 January 2024).
- Sebastià, J.; Cristòfol, R.; Martín, M.; Rodríguez-Farré, E.; Sanfeliu, C. Evaluation of fluorescent dyes for measuring intracellular glutathione content in primary cultures of human neurons and neuroblastoma SH-SY5Y. Cytom. Part A 2003, 51, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Drew, R.; Miners, J.O. The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochem. Pharmacol. 1984, 33, 2989–2994. [Google Scholar] [CrossRef] [PubMed]
- Promega, UK. CellTiter-Glo® Luminescent Cell Viability Assay. Technical Bulletin. 2023. Available online: https://www.promega.com.cn/resources/protocols/technical-bulletins/0/celltiter-glo-luminescent-cell-viability-assay-protocol/ (accessed on 11 January 2024).
- Crouch, S.P.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods. 1993, 160, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Abcam, UK. Glucose 6 Phopshate Dehydrogenase Assay Kit (Colorimetric) (ab102529). 2023. Available online: https://www.abcam.com/products/assay-kits/glucose-6-phosphate-dehydrogenase-assay-kit-colorimetric-ab102529.html (accessed on 11 January 2024).
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2021, 21, 363–381. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millichap, L.; Turton, N.; Damiani, E.; Marcheggiani, F.; Orlando, P.; Silvestri, S.; Tiano, L.; Hargreaves, I.P. The Effect of Neuronal CoQ10 Deficiency and Mitochondrial Dysfunction on a Rotenone-Induced Neuronal Cell Model of Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 6622. https://doi.org/10.3390/ijms25126622
Millichap L, Turton N, Damiani E, Marcheggiani F, Orlando P, Silvestri S, Tiano L, Hargreaves IP. The Effect of Neuronal CoQ10 Deficiency and Mitochondrial Dysfunction on a Rotenone-Induced Neuronal Cell Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(12):6622. https://doi.org/10.3390/ijms25126622
Chicago/Turabian StyleMillichap, Lauren, Nadia Turton, Elisabetta Damiani, Fabio Marcheggiani, Patrick Orlando, Sonia Silvestri, Luca Tiano, and Iain P. Hargreaves. 2024. "The Effect of Neuronal CoQ10 Deficiency and Mitochondrial Dysfunction on a Rotenone-Induced Neuronal Cell Model of Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 12: 6622. https://doi.org/10.3390/ijms25126622