
The Ubiquinone-Ubiquinol Redox Cycle and Its Clinical Consequences: An Overview
Abstract
:1. Introduction
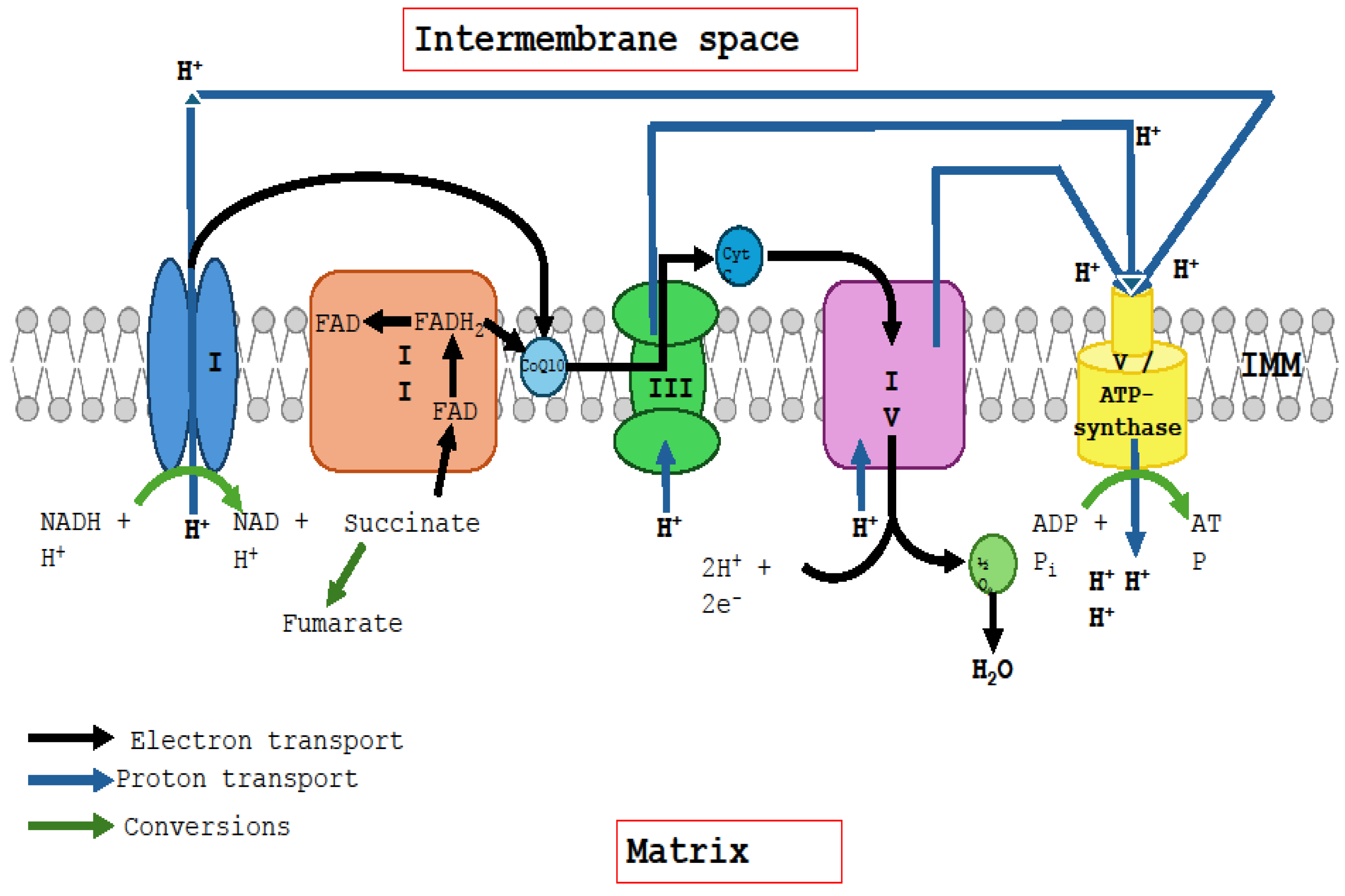
2. The Ubiquinone–Ubiquinol Redox Cycle
2.1. Interaction of Ubiquinone with Complexes I and II
2.2. The Q Cycle
3. Extramitochondrial Ubiquinone–Ubiquinol Redox: Role of Oxidoreductase Enzymes
4. Clinical Implications of the Ubiquinone–Ubiquinol Redox Cycle
4.1. Deficiencies of Redox Cycle Components: Complexes I, II, and III
4.2. Extramitochondrial Oxidoreductase Enzyme Deficiency
4.3. CoQ10 Deficiency
4.4. Clinical Benefits of CoQ10 and Selenium Co-Supplementation
5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Crane, F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of Human Coenzyme Q10 Metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef] [PubMed]
- Wikström, M.; Djurabekova, A.; Sharma, V. On the role of ubiquinone in the proton translocation mechanism of respiratory complex I. FEBS Lett. 2023, 597, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. Subcell. Biochem. 2018, 87, 167–227. [Google Scholar] [CrossRef] [PubMed]
- Luna-Sánchez, M.; Hidalgo-Gutiérrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, Á.; Romero, M.; Sayed, R.K.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2017, 9, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Crofts, A.R. The modified Q-cycle: A look back at its development and forward to a functional model. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148417. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Nordman, T.; Olsson, J.M.; Damdimopoulos, A.; Björkhem-Bergman, L.; Nalvarte, I.; Eriksson, L.C.; Arnér, E.S.; Spyrou, G.; Björnstedt, M. The mammalian cytosolic selenoenzyme thioredoxin reductase reduces ubiquinone. A novel mechanism for defense against oxidative stress. J. Biol. Chem. 2003, 278, 2141–2146. [Google Scholar] [CrossRef] [PubMed]
- Tinkov, A.A.; Bjørklund, G.; Skalny, A.V.; Holmgren, A.; Skalnaya, M.G.; Chirumbolo, S.; Aaseth, J. The role of the thioredoxin/thioredoxin reductase system in the metabolic syndrome: Towards a possible prognostic marker? Cell. Mol. Life Sci. 2018, 75, 1567–1586. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef]
- Nordman, T.; Xia, L.; Björkhem-Bergman, L.; Damdimopoulos, A.; Nalvarte, I.; Arnér, E.S.; Spyrou, G.; Eriksson, L.C.; Björnstedt, M.; Olsson, J.M. Regeneration of the antioxidant ubiquinol by lipoamide dehydrogenase, thioredoxin reductase and glutathione reductase. Biofactors 2003, 18, 45–50. [Google Scholar] [CrossRef]
- Beyer, R.E.; Segura-Aguilar, J.; di Bernardo, S.; Cavazzoni, M.; Fato, R.; Fiorentini, D.; Galli, M.C.; Setti, M.; Landi, L.; Lenaz, G. The two-electron quinone reductase DT-diaphorase generates and maintains the antioxidant (reduced) form of coenzyme Q in membranes. Mol. Asp. Med. 1997, 18 (Suppl. S1), S15–S23. [Google Scholar] [CrossRef]
- Takahashi, T.; Okamoto, T.; Kishi, T. Characterization of NADPH-dependent ubiquinone reductase activity in rat liver cytosol: Effect of various factors on ubiquinone-reducing activity and discrimination from other quinone reductases. J. Biochem. 1996, 119, 256–263. [Google Scholar] [CrossRef]
- Lind, C.; Cadenas, E.; Hochstein, P.; Ernster, L. DT-diaphorase: Purification, properties, and function. Methods Enzymol. 1990, 186, 287–301. [Google Scholar] [CrossRef]
- Swalwell, H.; Kirby, D.M.; Blakely, E.L.; Mitchell, A.; Salemi, R.; Sugiana, C.; Compton, A.G.; Tucker, E.J.; Ke, B.-X.; Lamont, P.J.; et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur. J. Hum. Genet. 2011, 19, 769–775. [Google Scholar] [CrossRef]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: Is riboflavin supplementation effective? Orphanet J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef]
- Bernsen, P.L.; Gabreëls, F.J.; Ruitenbeek, W.; Sengers, R.C.; Stadhouders, A.M.; Renier, W.O. Successful treatment of pure myopathy, associated with complex I deficiency, with riboflavin and carnitine. Arch. Neurol. 1991, 48, 334–338. [Google Scholar] [CrossRef]
- Bernsen, P.L.; Gabreëls, F.J.; Ruitenbeek, W.; Hamburger, H.L. Treatment of complex I deficiency with riboflavin. J. Neurol. Sci. 1993, 118, 181–187. [Google Scholar] [CrossRef]
- Scholte, H.R.; Busch, H.F.; Bakker, H.D.; Bogaard, J.M.; Luyt-Houwen, I.E.; Kuyt, L.P. Riboflavin-responsive complex I deficiency. Biochim. Biophys. Acta 1995, 1271, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Ogle, R.F.; Christodoulou, J.; Fagan, E.; Blok, R.B.; Kirby, D.M.; Seller, K.L.; Dahl, H.H.; Thorburn, D.R. Mitochondrial myopathy with tRNA(Leu(UUR)) mutation and complex I deficiency responsive to riboflavin. J. Pediatr. 1997, 130, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Artuch, R.; Brea-Calvo, G.; Briones, P.; Aracil, A.; Galván, M.; Espinós, C.; Corral, J.; Volpini, V.; Ribes, A.; Andreu, A.L.; et al. Cerebellar ataxia with coenzyme Q10 deficiency: Diagnosis and follow-up after coenzyme Q10 supplementation. Neurol. Sci. 2006, 246, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Curtabbi, A.; Guarás, A.; Cabrera-Alarcón, J.L.; Rivero, M.; Calvo, E.; Rosa-Moreno, M.; Vázquez, J.; Medina, M.; Enríquez, J.A. Regulation of respiratory complex I assembly by FMN cofactor targeting. Redox Biol. 2024, 69, 103001. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.; McFarland, R.; Taylor, R.W.; Alston, C.L. The genetic basis of isolated mitochondrial complex II deficiency. Mol. Genet. Metab. 2020, 131, 53–65. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; Zeviani, M. Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front. Genet. 2015, 6, 134. [Google Scholar] [CrossRef]
- Banerjee, R.; Purhonen, J.; Kallijärvi, J. The mitochondrial coenzyme Q junction and complex III: Biochemistry and pathophysiology. FEBS J. 2022, 289, 6936–6958. [Google Scholar] [CrossRef]
- Hill, K.E.; McCollum, G.W.; Boeglin, M.E.; Burk, R.F. Thioredoxin reductase activity is decreased by selenium deficiency. Biochem. Biophys. Res. Commun. 1997, 234, 293–295. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yang, G.; Hong, C.; Liu, J.; Holle, E.; Yu, X.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J. Clin. Investig. 2003, 112, 1395–1406. [Google Scholar] [CrossRef]
- Pei, J.; Fu, W.; Yang, L.; Zhang, Z.; Liu, Y. Oxidative stress is involved in the pathogenesis of Keshan disease (an endemic dilated cardiomyopathy) in China. Oxidative Med. Cell. Longev. 2013, 2013, 474203. [Google Scholar] [CrossRef]
- Szabó, E.; Ambrus, A. Lipoamide dehydrogenase (LADH) deficiency: Medical perspectives of the structural and functional characterization of LADH and its pathogenic variants. Biol. Futur. 2023, 74, 109–118. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free. Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Mantle, D.; Millichap, L.; Castro-Marrero, J.; Hargreaves, I.P. Primary Coenzyme Q10 Deficiency: An Update. Antioxidants 2023, 12, 1652. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Turton, N.; Hargreaves, I.P. Depletion and Supplementation of Coenzyme Q10 in Secondary Deficiency Disorders. Front. Biosci. (Landmark Ed.) 2022, 27, 322. [Google Scholar] [CrossRef]
- Alehagen, U.; Johansson, P.; Björnstedt, M.; Rosén, A.; Dahlström, U. Cardiovascular mortality and N-terminal-proBNP reduced after combined selenium and coenzyme Q10 supplementation: A 5-year prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. Int. J. Cardiol. 2013, 167, 1860–1866. [Google Scholar] [CrossRef]
- Johansson, P.; Dahlström, Ö.; Dahlström, U.; Alehagen, U. Improved Health-Related Quality of Life, and More Days out of Hospital with Supplementation with Selenium and Coenzyme Q10 Combined. Results from a Double Blind, Placebo-Controlled Prospective Study. J. Nutr. Health Aging 2015, 19, 870–877. [Google Scholar] [CrossRef]
- Johansson, P.; Dahlström, Ö.; Dahlström, U.; Alehagen, U. Effect of selenium and Q10 on the cardiac biomarker NT-proBNP. Scand. Cardiovasc. J. 2013, 47, 281–288. [Google Scholar] [CrossRef]
- Alehagen, U.; Lindahl, T.L.; Aaseth, J.; Svensson, E.; Johansson, P. Levels of sP-selectin and hs-CRP Decrease with Dietary Intervention with Selenium and Coenzyme Q10 Combined: A Secondary Analysis of a Randomized Clinical Trial. PLoS ONE 2015, 10, e0137680. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Aaseth, J.; Johansson, P. Less increase of copeptin and MR-proADM due to intervention with selenium and coenzyme Q10 combined: Results from a 4-year prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. Biofactors 2015, 41, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Aaseth, J.; Johansson, P. Reduced Cardiovascular Mortality 10 Years after Supplementation with Selenium and Coenzyme Q10 for Four Years: Follow-Up Results of a Prospective Randomized Double-Blind Placebo-Controlled Trial in Elderly Citizens. PLoS ONE 2015, 10, e0141641. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Aaseth, J.; Alexander, J.; Johansson, P. Still reduced cardiovascular mortality 12 years after supplementation with selenium and coenzyme Q10 for four years: A validation of previous 10-year follow-up results of a prospective randomized double-blind placebo-controlled trial in elderly. PLoS ONE 2018, 13, e0193120. [Google Scholar] [CrossRef]
- Kalén, A.; Appelkvist, E.L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef]
- Alehagen, U.; Johansson, P.; Björnstedt, M.; Rosén, A.; Post, C.; Aaseth, J. Relatively high mortality risk in elderly Swedish subjects with low selenium status. Eur. J. Clin. Nutr. 2016, 70, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Aaseth, J.; Alexander, J.; Johansson, P.; Larsson, A. Supplemental selenium and coenzyme Q10 reduce glycation along with cardiovascular mortality in an elderly population with low selenium status—A four-year, prospective, randomised, double-blind placebo-controlled trial. J. Trace Elem. Med. Biol. 2020, 61, 126541. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Aaseth, J.; Lindahl, T.L.; Larsson, A.; Alexander, J. Dietary Supplementation with Selenium and Coenzyme Q10 Prevents Increase in Plasma D-Dimer While Lowering Cardiovascular Mortality in an Elderly Swedish Population. Nutrients 2021, 13, 1344. [Google Scholar] [CrossRef] [PubMed]
- Opstad, T.B.; Alexander, J.; Aaseth, J.O.; Larsson, A.; Seljeflot, I.; Alehagen, U. Selenium and Coenzyme Q10 Intervention Prevents Telomere Attrition, with Association to Reduced Cardiovascular Mortality-Sub-Study of a Randomized Clinical Trial. Nutrients 2022, 14, 3346. [Google Scholar] [CrossRef] [PubMed]
- Opstad, T.B.; Alexander, J.; Aaseth, J.; Larsson, A.; Seljeflot, I.; Alehagen, U. Increased SIRT1 Concentration Following Four Years of Selenium and Q10 Intervention Associated with Reduced Cardiovascular Mortality at 10-Year Follow-Up-Sub-Study of a Previous Prospective Double-Blind Placebo-Controlled Randomized Clinical Trial. Antioxidants 2023, 12, 759. [Google Scholar] [CrossRef] [PubMed]
- Dunning, B.J.; Bourgonje, A.R.; Bulthuis, M.L.C.; Alexander, J.; Aaseth, J.O.; Larsson, A.; van Goor, H.; Alehagen, U. Selenium and coenzyme Q10 improve the systemic redox status while reducing cardiovascular mortality in elderly population-based individuals. Free. Radic. Biol. Med. 2023, 204, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Aaseth, J.; Larsson, A.; Alexander, J. Decreased Concentration of Fibroblast Growth Factor 23 (FGF-23) as a Result of Supplementation with Selenium and Coenzyme Q10 in an Elderly Swedish Population: A Sub-Analysis. Cells 2022, 11, 509. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Aaseth, J.; Alexander, J.; Brismar, K.; Larsson, A. Selenium and Coenzyme Q10 Supplementation Improves Renal Function in Elderly Deficient in Selenium: Observational Results and Results from a Subgroup Analysis of a Prospective Randomised Double-Blind Placebo-Controlled Trial. Nutrients 2020, 12, 3780. [Google Scholar] [CrossRef] [PubMed]
- Castro-Marrero, J.; Domingo, J.C.; Cordobilla, B.; Ferrer, R.; Giralt, M.; Sanmartín-Sentañes, R.; Alegre-Martín, J. Does Coenzyme Q10 Plus Selenium Supplementation Ameliorate Clinical Outcomes by Modulating Oxidative Stress and Inflammation in Individuals with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome? Antioxid. Redox Signal. 2022, 36, 729–739. [Google Scholar] [CrossRef]
- Alahmar, A.T.; Calogero, A.E.; Singh, R.; Cannarella, R.; Sengupta, P.; Dutta, S. Coenzyme Q10, oxidative stress, and male infertility: A review. Clin. Exp. Reprod. Med. 2021, 48, 97–104. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Mantle, D. Supplementation with selenium and coenzyme Q10 in critically ill patients. Br. J. Hosp. Med. 2019, 80, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, B.; Behl, C. Selenoproteins, cholesterol-lowering drugs, and the consequences: Revisiting of the mevalonate pathway. Trends Cardiovasc. Med. 2004, 14, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Park, S.J.; Lange, M.; Tseyang, T.; Doshi, M.B.; Kim, T.Y.; Song, Y.; Kim, D.I.; Greer, P.L.; Olzmann, J.A.; et al. Selenium reduction of ubiquinone via SQOR suppresses ferroptosis. Nat. Metab. 2024, 6, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Dybring, A. Bioavailability of Coenzyme Q10: An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants 2020, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- She, J.; Sheng, R.; Qin, Z.H. Pharmacology and Potential Implications of Nicotinamide Adenine Dinucleotide Precursors. Aging Dis. 2021, 12, 1879–1897. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid. Redox Signal. 2019, 30, 251–294. [Google Scholar] [CrossRef] [PubMed]
- Castro-Marrero, J.; Segundo, M.J.; Lacasa, M.; Martinez-Martinez, A.; Sentañes, R.S.; Alegre-Martin, J. Effect of Dietary Coenzyme Q10 Plus NADH Supplementation on Fatigue Perception and Health-Related Quality of Life in Individuals with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Prospective, Randomized, Double-Blind, Placebo-Controlled Trial. J. Nutr. 2021, 13, 2658. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Maier, A.B.; Tao, R.; Lin, Z.; Vaidya, A.; Pendse, S.; Thasma, S.; Andhalkar, N.; Avhad, G.; Kumbhar, V. The efficacy and safety of β-nicotinamide mononucleotide (NMN) supplementation in healthy middle-aged adults: A randomized, multicenter, double-blind, placebo-controlled, parallel-group, dose-dependent clinical trial. Geroscience 2023, 45, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Dallas, D.C.; Sanctuary, M.R.; Qu, Y.; Khajavi, S.H.; Van Zandt, A.E.; Dyandra, M.; Frese, S.A.; Barile, D.; German, J.B. Personalising protein nourishment. Crit. Rev. Food Sci. Nutr. 2017, 57, 3313–3331. [Google Scholar] [CrossRef]
- Gardner, M.L. Gastrointestinal absorption of intact proteins. Annu. Rev. Nutr. 1988, 8, 329–350. [Google Scholar] [CrossRef]
- Zieliński, Ł.P.; Smith, A.C.; Smith, A.G.; Robinson, A.J. Metabolic flexibility of mitochondrial respiratory chain disorders predicted by computer modelling. Mitochondrion 2016, 31, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Mráček, T.; Drahota, Z.; Houštěk, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Ralser, M.; Fan, J.; Rabinowitz, J.D. The pentose phosphate pathway in health and disease. Nat. Metab. 2023, 5, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Cantley, L.C. Toward a better understanding of folate metabolism in health and disease. J. Exp. Med. 2019, 216, 253–266. [Google Scholar] [CrossRef]
- Schirris, T.J.J.; Rossell, S.; de Haas, R.; Frambach, S.J.C.M.; Hoogstraten, C.A.; Renkema, G.H.; Beyrath, J.D.; Willems, P.H.G.M.; Huynen, M.A.; Smeitink, J.A.M.; et al. Stimulation of cholesterol biosynthesis in mitochondrial complex I-deficiency lowers reductive stress and improves motor function and survival in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166062. [Google Scholar] [CrossRef]

{kind=link}
| Supplement | Baseline Serum Level | End of Study Serum Level |
|---|---|---|
| Coenzyme Q10 | 0.82 mg/L | 2.17 mg/L |
| Selenium-enriched yeast | 67.1 μg/L | 210.3 μg/L |
| Reference | Change in Outcome Variable after 48 Months of Combined Supplementation | Statistical Significance |
|---|---|---|
| Alehagen et al. [33] | Reduced cardiovascular mortality in the active treatment group vs. the placebo group (5.9% vs. 12.6%) | p = 0.015 |
| Alehagen et al. [35] | Lower NT-proBNP level values in the active treatment group vs. the placebo group (mean values: 214 ng/L vs. 302 ng/L) | p = 0.014 |
| Alehagen et al. [33] | Better cardiac function score in echocardiography for the active treatment group vs. the placebo group | p = 0.03 |
| Alehagen et al. [36] | Improved CRP levels (from of 4.1 ng/mL down to 2.1 ng/mL) in the active treatment group vs. a slight rise (from 4.8 ng/mL up to 5.1 ng/mL) in the placebo group | p = 0.009 |
| Alehagen et al. [37] | Less increase in copeptin and MR-proADM levels in the active treatment group vs. the placebo group | p = 0.031 p = 0.026 |
| Johansson et al. [34] | More days out of hospital in the active treatment group vs. the placebo group (1779 compared with 1533) | p = 0.03 |
| Johansson et al. [34] | Less decline in health-related quality of life domains, including physical performance, vitality, and overall quality of life, in the active treatment group vs. the placebo group | p = 0.001 |
| Alehagen et al. [43] | Lower D-dimer concentrations in the active treatment group (0.22 mg/L) vs. the placebo group (0.34 mg/L) | p = 0.006 |
| Opstad et al. [44] | Less shortening of leukocyte telomere length in the active treatment group (+0.019) vs. the placebo group after 42 months (−0.129) | p = 0.02 |
| Opstad et al. [45] | Increased SIRT1 concentration in the active treatment group (from 252 to 469 ng/mL) vs. decreased SIRT1 concentration in the placebo group (from 269 down to 190 ng/mL) | p = 0.006 |
| Dunning et al. [46] | Increased serum sulphydryl levels, reduced cardiovascular disease risk | p = 0.002 |
| Alehagen et al. [47] | Reduced serum levels of fibroblast growth factor 23, reduced risk of cardiovascular disease | p = 0.01 |
| Alehagen et al. [48] | Serum markers of renal dysfunction (creatinine, cystatin-C) reduced | p = 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantle, D.; Dewsbury, M.; Hargreaves, I.P. The Ubiquinone-Ubiquinol Redox Cycle and Its Clinical Consequences: An Overview. Int. J. Mol. Sci. 2024, 25, 6765. https://doi.org/10.3390/ijms25126765
Mantle D, Dewsbury M, Hargreaves IP. The Ubiquinone-Ubiquinol Redox Cycle and Its Clinical Consequences: An Overview. International Journal of Molecular Sciences. 2024; 25(12):6765. https://doi.org/10.3390/ijms25126765
Chicago/Turabian StyleMantle, David, Mollie Dewsbury, and Iain P. Hargreaves. 2024. "The Ubiquinone-Ubiquinol Redox Cycle and Its Clinical Consequences: An Overview" International Journal of Molecular Sciences 25, no. 12: 6765. https://doi.org/10.3390/ijms25126765