
Synthesis, Molecular Electron Density Theory Study, Molecular Docking, and Pharmacological Evaluation of New Coumarin–Sulfonamide–Nitroindazolyl–Triazole Hybrids as Monoamine Oxidase Inhibitors
 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Nitration of Coumarin
2.2. MEDT Study of the Nitration Reaction of Coumarin in Sulfuric Acid
2.2.1. Analysis of the CDFT Reactivity Indices
2.2.2. Analysis of the Electronic Structure of Coumarin 1 and Coumarin: SO4H2 Complex 6
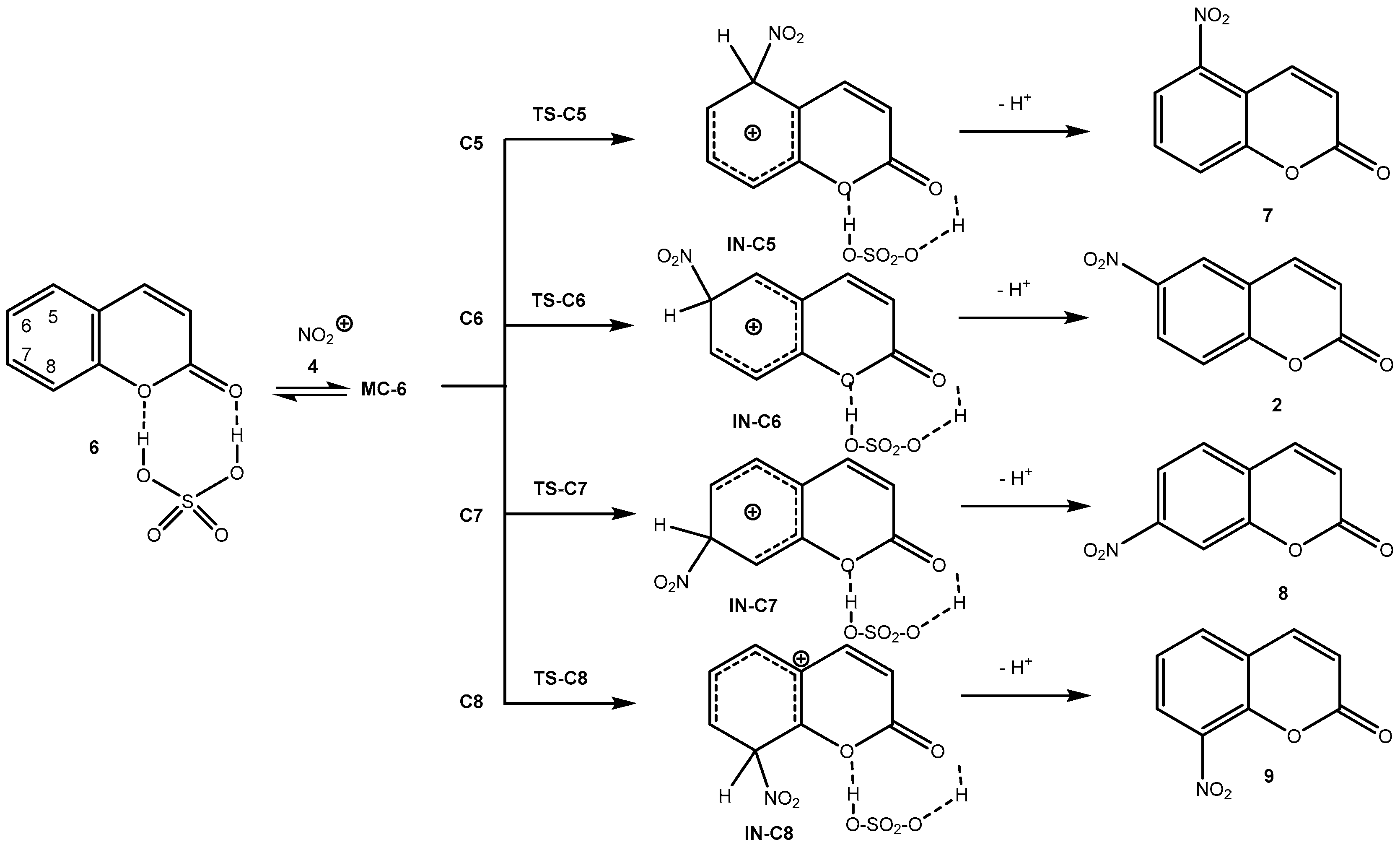
2.2.3. Study of the Reaction Mechanism and Regioselectivity Associated to the EAS Nitration Reaction of Coumarin: SO4H2 complex 6 with Nitronium NO2+ ion 4

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔH | ΔS | ΔG | |
|---|---|---|---|
| MC-6 | −0.40 | −22.30 | 6.25 |
| TS-C5 | 4.36 | −34.24 | 14.57 |
| TS-C6 | 2.81 | −34.08 | 12.97 |
| TS-C7 | 5.51 | −35.10 | 15.97 |
| TS-C8 | 4.30 | −39.94 | 16.20 |
| IN-C5 | −1.33 | −36.57 | 9.58 |
| IN-C6 | −8.50 | −34.14 | 1.68 |
| IN-C7 | −1.88 | −33.35 | 8.06 |
| IN-C8 | −8.19 | −38.55 | 3.30 |
| 7 | −104.64 | 10.73 | −107.84 |
| 2 | −99.76 | 10.57 | −102.91 |
| 8 | −100.76 | 8.90 | −103.41 |
| 9 | −101.93 | 14.73 | −106.32 |
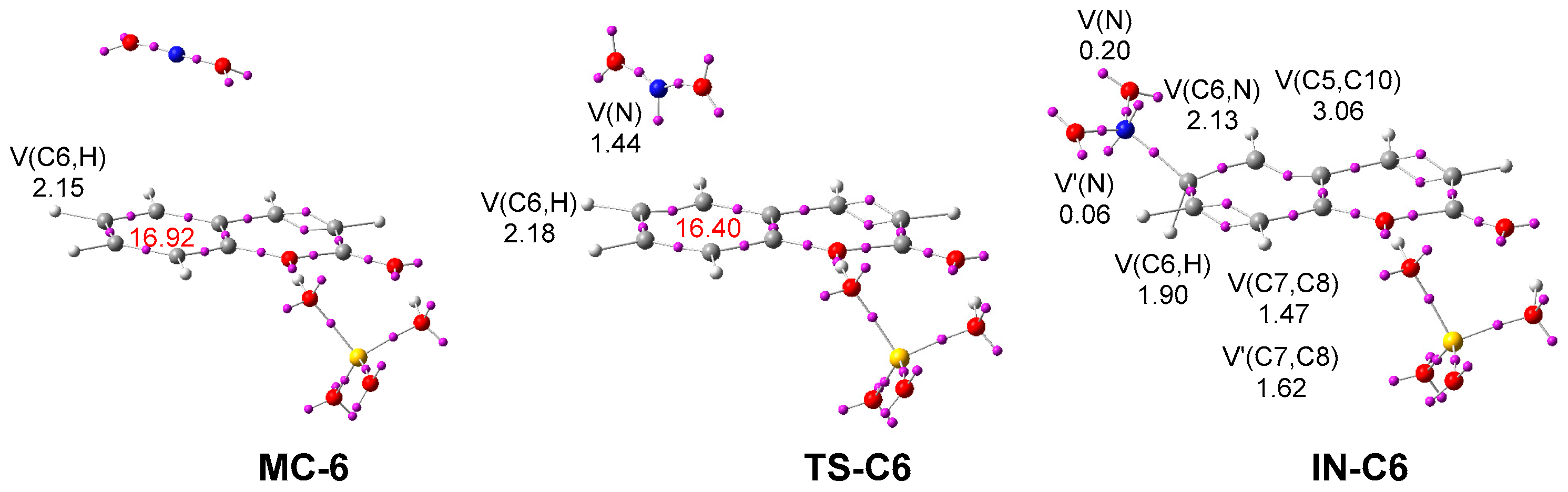
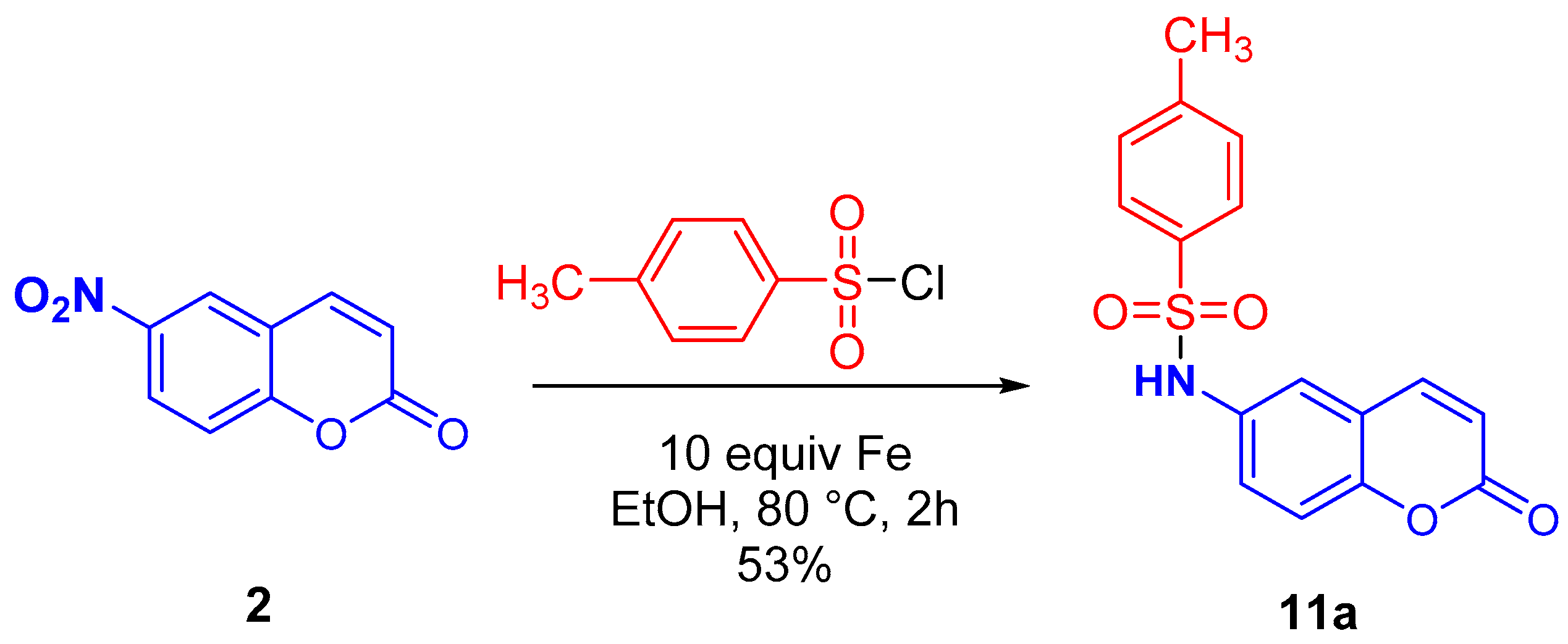
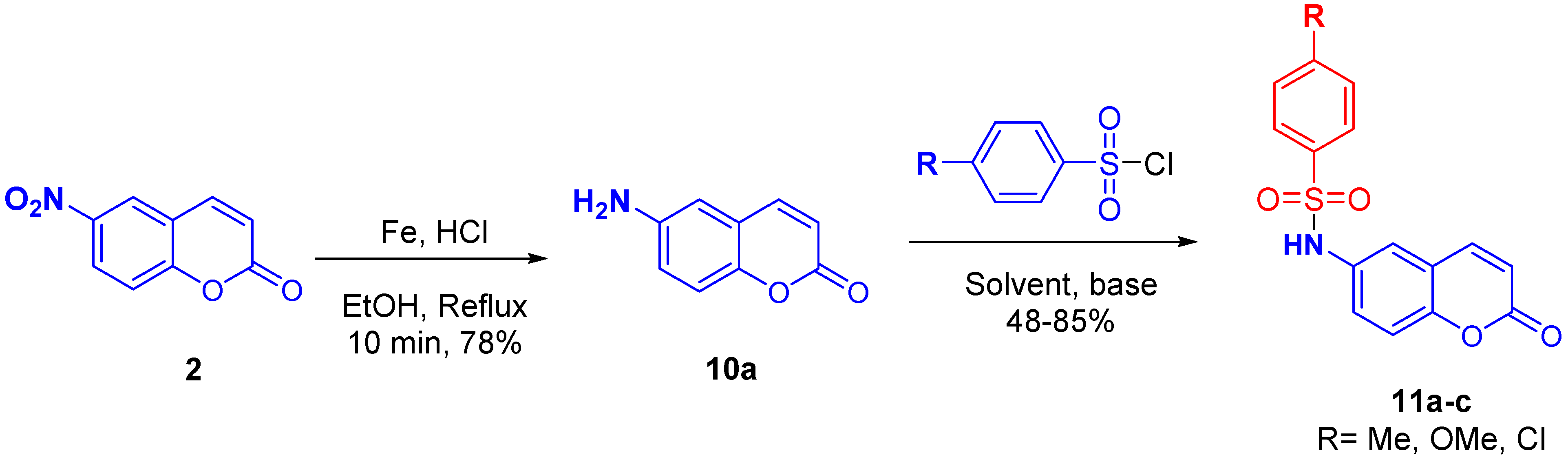
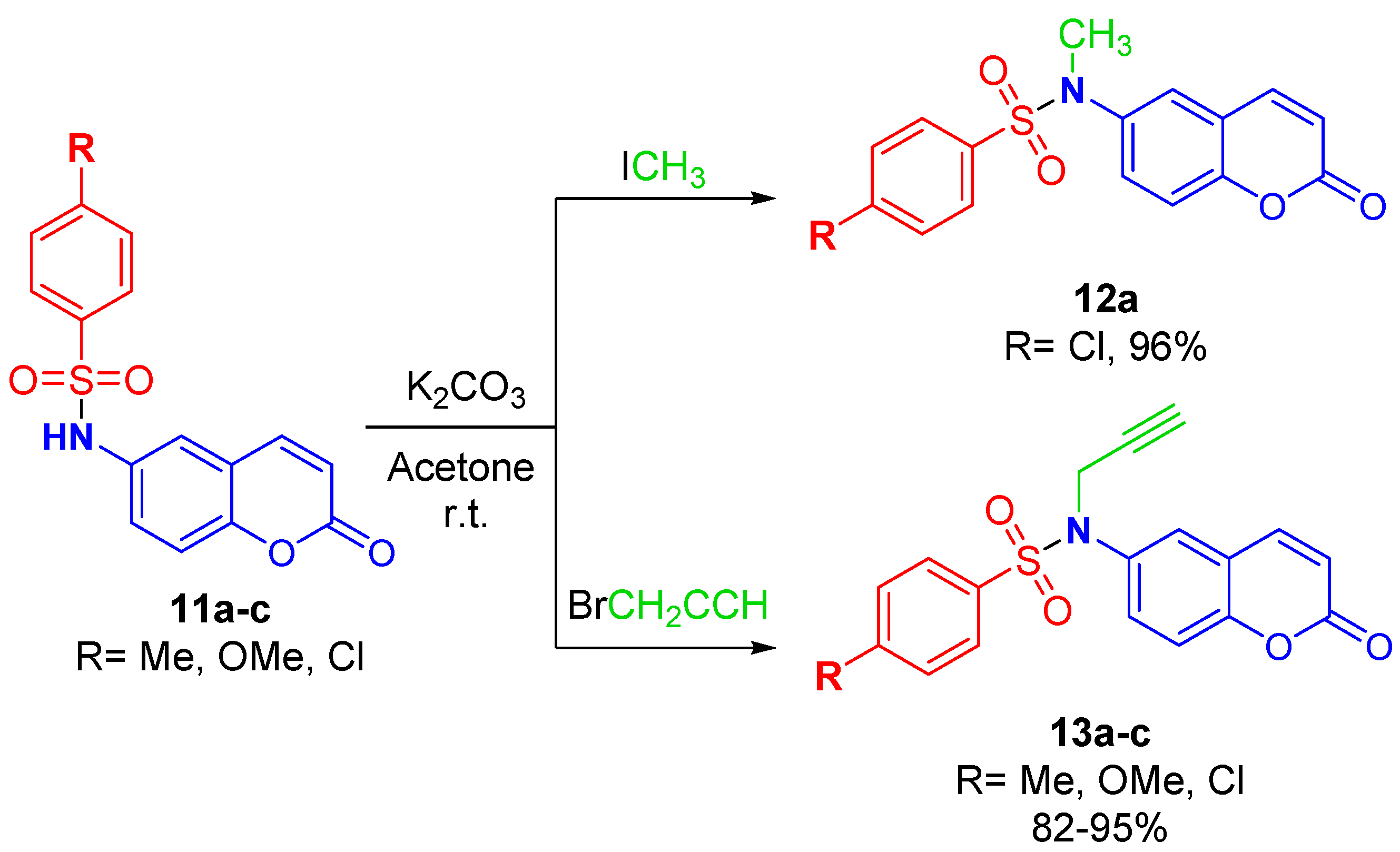
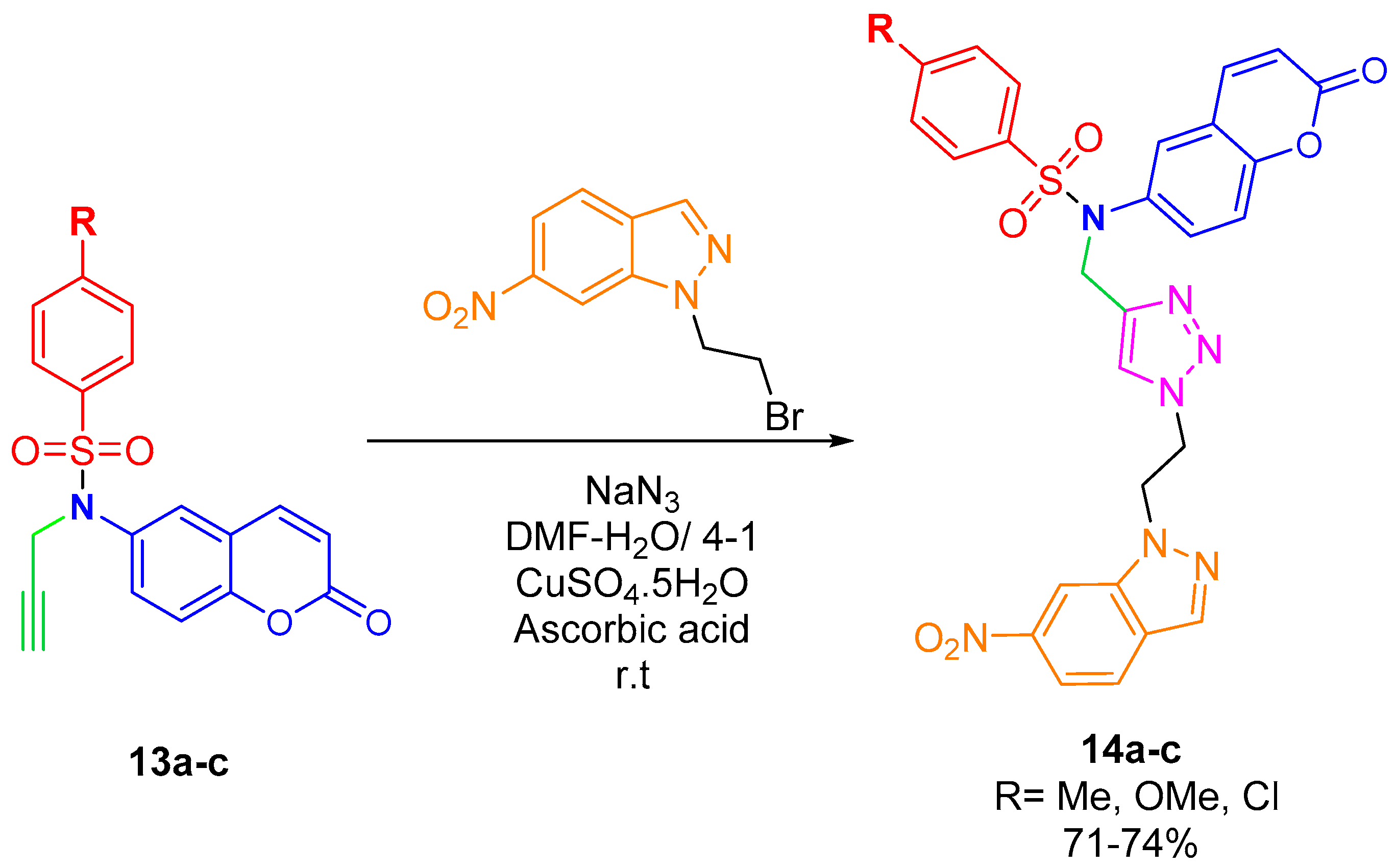
2.3. Synthesis of N-(2-oxo-2H-Chromen-6-yl)benzenesulfonamides
2.4. CuAAC Reaction of N-(2-oxo-2H-Chromen-6-yl)-N-(prop-2-ynyl)benzenesulfonamides
2.5. Biological Studies
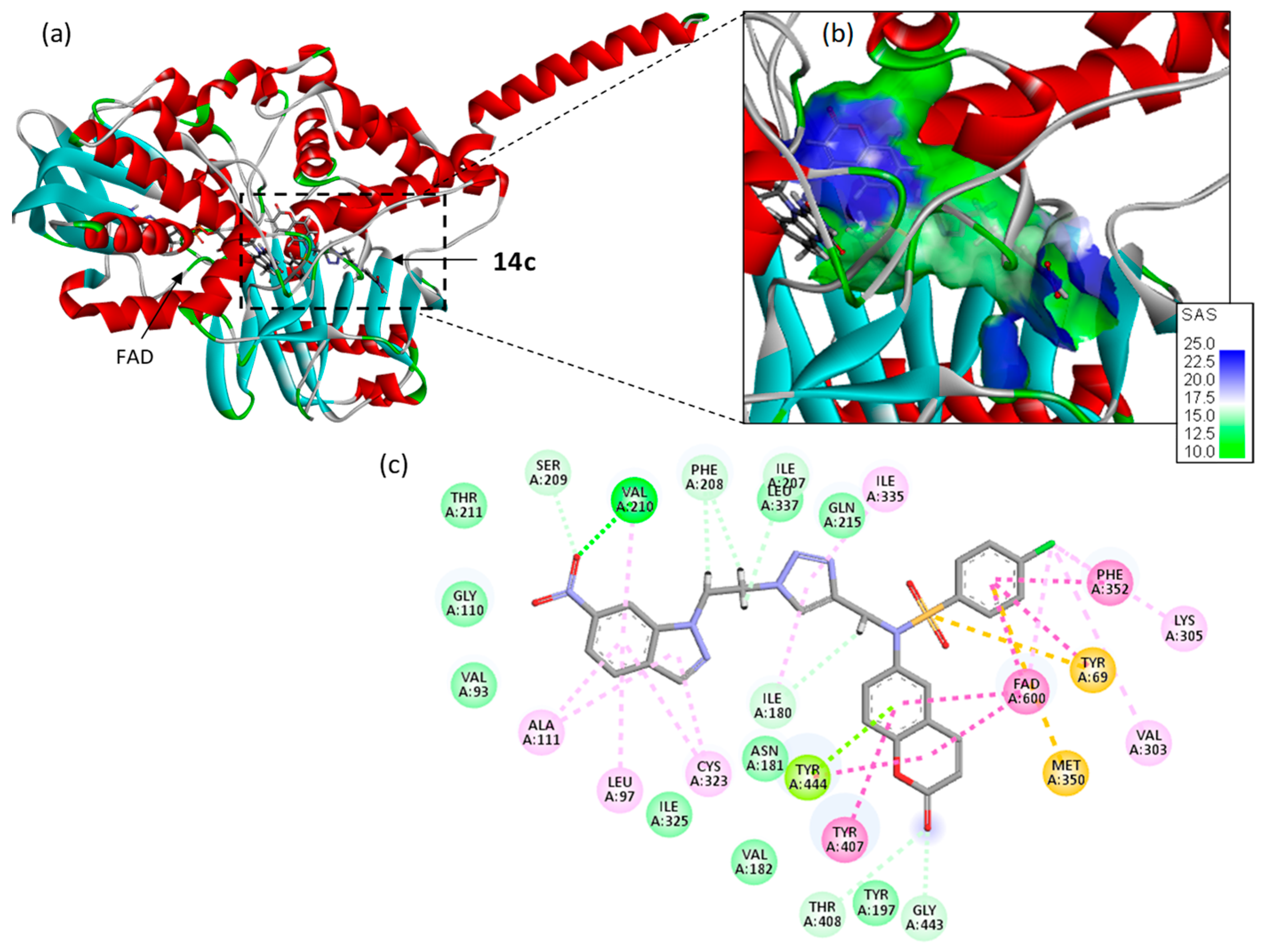
2.6. Docking Studies
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of 6-Nitro-2H-chromen-2-one (2)
3.3. Synthesis of 6-Amino-2H-chromen-2-one (10a)
3.4. Procedure for the Preparation of N-(2-oxo-2H-Chromen-6-yl)benzenesulfonamides (11a–c)
3.5. N-Alkylation of (11a–c)
3.5.1. General Procedure for Synthesis of 4-Chloro-N-methyl-N-(2-oxo-2H-chromen-6-yl)benzenesulfonamide (12a)
3.5.2. General Procedure for Synthesis of N-(2-oxo-2H-Chromen-6-yl)-N-(prop-2-ynyl)benzenesulfonamides (13a–c)
3.6. General Procedure for 1,3-Dipolar Cycloaddition of Azides with Terminal Alkynes
3.7. Computational Details
3.8. Biological Methods
3.9. Molecular Docking Procedure
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouhaoui, A.; Eddahmi, M.; Dib, M.; Khouili, M.; Aires, A.; Catto, M.; Bouissane, L. Synthesis and Biological Properties of Coumarin Derivatives. A Review. ChemistrySelect 2021, 6, 5848–5870. [Google Scholar] [CrossRef]
- Balewski, Ł.; Szulta, S.; Jalińska, A.; Kornicka, A. Recent Advances in Coumarin-Metal Complexes with Biological Properties. Front. Chem. 2021, 9, 781779. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.L.; Ke, X.; Liu, M. Coumarin-triazole Hybrids and Their Biological Activities. J. Heterocycl. Chem. 2018, 55, 791–802. [Google Scholar] [CrossRef]
- Tanvi, S.; Sunita, S.; Diksha, V.; Pardeep, K. Overview of Coumarins and its Derivatives: Synthesis and Biological Activity. Lett. Org. Chem. 2021, 18, 880–902. [Google Scholar]
- Xia, D.; Liu, H.; Cheng, X.; Maraswami, M.; Chen, Y.; Lv, X. Recent Developments of Coumarin-based Hybrids in Drug Discovery. Curr. Top. Med. Chem. 2022, 22, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Vega-Granados, K.; Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Martinez, A.; Alvarez de Cienfuegos, L.; Lupiañez, J.A.; Parra, A. Synthesis and Biological Activity of Triterpene–Coumarin Conjugates. J. Nat. Prod. 2021, 84, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Kaur, G.; Tandon, N.; Tandon, R.; Singh, I. Current advancements in synthesis, anticancer activity, and structure–activity relationship (SAR) of coumarin derivatives. Inorg. Chem. Commun. 2024, 167, 112605. [Google Scholar] [CrossRef]
- Al-Majedy, Y.K.; Al-Duhaidahawi, D.L.; Al-Azawi, K.F.; Al-Amiery, A.A.; Kadhum, A.A.H.; Mohamad, A.B. Coumarins as Potential Antioxidant Agents Complemented with Suggested Mechanisms and Approved by Molecular Modeling Studies. Molecules 2016, 21, 135. [Google Scholar] [CrossRef]
- Angelova, V.T.; Vassilev, N.G.; Nikolova-Mladenova, B.; Vitas, J.; Malbaša, R.; Momekov, G.; Djukic, M.; Saso, L. Antiproliferative and Antioxidative Effects of Novel Hydrazone Derivatives Bearing Coumarin and Chromene Moiety. Med. Chem. Res. 2016, 25, 2082–2092. [Google Scholar] [CrossRef]
- Hussain, M.K.; Ansari, M.I.; Yadav, N.; Gupta, P.K.; Gupta, A.K.; Saxena, R.; Fatima, I.; Manohar, M.; Kushwaha, P.; Khedgikar, V.; et al. Design and Synthesis of Erα/Erβ Selective Coumarin and Chromene Derivatives as Potential Anti-Breast Cancer and Anti-Osteoporotic Agents. RSC. Adv. 2014, 4, 8828–8845. [Google Scholar] [CrossRef]
- Ballazhi, L.; Popovski, E.; Jashari, A.; Imeri, F.; Ibrahimi, I.; Mikhova, B.; Mladenovska, K. Potential Antiproliferative Effect of Isoxazolo- and Thiazolo Coumarin Derivatives o Breast Cancer Mediated Bone and Lung Metastases. Acta Pharm. 2015, 65, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Saidu, N.E.B.; Valente, S.; Bana, E.; Kirsch, G.; Bagrel, D.; Montenarh, M. Coumarin Polysulfides Inhibit Cell Growth and Induce Apoptosis in HCT116 Colon Cancer Cells. Bioorg. Med. Chem. 2012, 20, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Luo, J.; Wu, F.; Hou, X.; Yan, G.; Zhou, M.; Li, R. Synthesis and Biological Evaluation of Novel 2,3-Dihydrochromeno[3,4-d]Imidazol-4(1H)-One Derivatives as Potent Anticancer Cell Proliferation and Migration Agents. Eur. J. Med. Chem. 2016, 114, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Ibrar, A.; Shehzadi, S.A.; Saeed, F.; Khan, I. Developing Hybrid Molecule Therapeutics for Diverse Enzyme Inhibitory Action: Active Role of Coumarin-Based Structural Leads in Drug Discovery. Bioorg. Med. Chem. 2018, 26, 3731–3762. [Google Scholar] [CrossRef] [PubMed]
- Bana, E.; Sibille, E.; Valente, S.; Cerella, C.; Chaimbault, P.; Kirsch, G.; Dicato, M.; Diederich, M.; Bagrel, D. A Novel Coumarin-Quinone Derivative SV37 Inhibits CDC25 Phosphatases, Impairs Proliferation, and Induces Cell Death. Mol. Carcinog. 2015, 54, 229–241. [Google Scholar] [CrossRef]
- Kumbar, S.S.; Hosamani, K.M.; Gouripur, G.C.; Joshi, S.D. Functionalization of 3-Chloroformylcoumarin to Coumarin Schiff Bases Using Reusable Catalyst: An Approach to Molecular Docking and Biological Studies. R. Soc. Open Sci. 2018, 5, 172416. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; Subhedar, D.D.; Shingate, B.B.; Khan, F.A.K.; Sangshetti, J.N.; Khedkar, V.M.; Nawale, L.; Sarkar, D.; Nayale, G.R.; Shinde, S.S. Synthesis, Biological Evaluation and Molecular Docking of Novel Coumarin Incorporated Triazoles as Antitubercular, Antioxidant and Antimicrobial Agents. Med. Chem. Res. 2016, 25, 790–804. [Google Scholar] [CrossRef]
- Papadopoulos, J.; Merkens, K.; Müller, T.J.J. Three-Component Synthesis and Photophysical Properties of Novel Coumarin-based Merocyanines. Chem. Eur. J. 2018, 24, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, J.; Müller, T.J.J. Rapid Synthesis of 4-Alkynyl Coumarins and Tunable Electronic Properties of Emission Solvatochromic Fluorophores. Dyes Pigm. 2019, 166, 357–366. [Google Scholar] [CrossRef]
- Geenen, S.R.; Presser, L.; Hölzel, T.; Ganter, C.; Müller, T.J.J. Electronic Finetuning of 8-Methoxy Psoralens by Palladium-Catalyzed Coupling—Acidochromicity and Solvatochromicity. Chem. Eur. J. 2020, 26, 8064–8075. [Google Scholar] [CrossRef]
- Geenen, S.R.; Schumann, T.; Müller, T.J.J. Fluorescent Donor-Acceptor-Psoralen Cruciforms by Consecutive Suzuki-Suzuki and Sonogashira-Sonogashira One-pot Syntheses. J. Org. Chem. 2020, 85, 9737–9750. [Google Scholar] [CrossRef] [PubMed]
- Bertling, J.; Thom, K.A.; Geenen, S.; Jeuken, H.; Presser, L.; Müller, T.J.J.; Gilch, P. Synthesis and Photophysics of Water-Soluble Psoralens with Red-Shifted Absorption. Photochem. Photobiol. 2021, 97, 1534–1547. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, C.F.; Yang, S.H.; Yang, W.C.; Yang, G.F. A Coumarin-Based Fluorescent Probe for Selective and Sensitive Detection of Thiophenols and its Application. Anal. Chem. 2014, 86, 3037–3042. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Wang, Q.; Teng, P.; Jiao, J.; Li, Y.; Xia, Q.; Zhang, W. Design, Synthesis, Antifungal Activity, and 3D-QASR of Novel Oxime Ether-Containing Coumarin Derivatives as Potential Fungicides. J. Agric. Food Chem. 2024, 72, 5983–5992. [Google Scholar] [CrossRef] [PubMed]
- Stokes, S.S.; Albert, R.; Buurman, E.T.; Andrews, B.A.; Shapiro, B.; Green, O.M.; McKenzie, A.R.; Otterbein, L.R. Inhibitors of the Acetyltransferase Domain of N-Acetylglucosamine-1-Phosphate-Uridylyltransferase/Glucosamine-1-Phosphate-Acetyltransferase (Glmu). Part 2: Optimization of Physical Properties Leading to Antibacterial Aryl Sulfonamides. Bioorg. Med. Chem. Lett. 2012, 22, 7019–7023. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y.; Kawanishi, Y.; Oda, K.; Sakata, T.; Mihara, S.; Asakura, K.; Konoike, T. Synthesis and Structure-Activity Relationships of Potent and Orally Active Sulfonamide ETB Selective Antagonists. Bioorg. Med. Chem. 2001, 9, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.F.; Thorley, M.; Kleeman, A.; Engel, J.; Kutscher, B.; Reichert George, D. Pharmaceutical Substances, 3rd Ed, A. Kleeman, J. Engel, B. Kutscher & D. Reichert George Thieme Verlag, Stuttgart/New York, 1999, 2286 pp., ISBN 3-13-558403-8/0-86577-817-5.[0pt] [Electronic Version. ISBN 3-13-115133-1/0-86577-818-3]. Bioseparation 1999, 8, 336. [Google Scholar]
- Ezabadi, I.R.; Camoutsis, C.; Zoumpoulakis, P.; Geronikaki, A.; Soković, M.; Glamočilija, J.; Ćirić, A. Sulfonamide-1,2,4-Triazole Derivatives as Antifungal and Antibacterial Agents: Synthesis, Biological Evaluation, Lipophilicity, and Conformational Studies. Bioorg. Med. Chem. 2008, 16, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Chibale, K.; Haupt, H.; Kendrick, H.; Yardley, V.; Saravanamuthu, A.; Fairlamb, A.H.; Croft, S.L. Antiprotozoal and Cytotoxicity Evaluation of Sulfonamide and Urea Analogues of Quinacrine. Bioorg. Med. Chem. Lett. 2001, 11, 2655–2657. [Google Scholar] [CrossRef]
- Lv, Q.; Zhang, J.; Cai, J.; Chen, L.; Liang, J.; Zhang, T.; Lin, J.; Chen, R.; Zhang, Z.; Guo, P.; et al. Design, synthesis and mechanism study of coumarin-sulfonamide derivatives as carbonic anhydrase IX inhibitors with anticancer activity. Chem. Biol. Interact. 2024, 393, 110947. [Google Scholar] [CrossRef]
- Bouissane, L.; El Kazzouli, S.; Léonce, S.; Pfeiffer, B.; Rakib, E.M.; Khouili, M.; Guillaumet, G. Synthesis and Biological Evaluation of N-(7-Indazolyl)benzenesulfonamide Derivatives as Potent Cell Cycle Inhibitors. Bioorg. Med. Chem. 2006, 14, 1078–1088. [Google Scholar] [CrossRef]
- Bouissane, L.; El Kazzouli, S.; Léger, J.M.; Christian, J.; Rakib, E.M.; Khouili, M.; Guillaumet, G. New and Efficient Synthesis of Bi- and Trisubstituted Indazoles. Tetrahedron 2005, 61, 8218–8225. [Google Scholar] [CrossRef]
- Talebi, M.R.; Nematollahi, D. Electrochemical Synthesis of Sulfonamide Derivatives: Electrosynthesis Conditions and Reaction Pathways. ChemElectroChem 2024, 11, e202300728. [Google Scholar] [CrossRef]
- Jiang, J.; Zeng, S.; Chen, D.; Cheng, C.; Deng, W.; Xiang, J. Synthesis of N-Arylsulfonamides via Fe-Promoted Reaction of Sulfonyl Halides with Nitroarenes in an Aqueous Medium. Org. Biomol. Chem. 2018, 16, 5016–5020. [Google Scholar] [CrossRef]
- Zhang, W.; Xie, J.; Rao, B.; Luo, M. Iron-Catalyzed N-Arylsulfonamide Formation through Directly Using Nitroarenes as Nitrogen Sources. J. Org. Chem. 2015, 80, 3504–3511. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lian, C.; Yue, G.; Liu, D.; Wei, L.; Ding, Y.; Zheng, X.; Lu, K.; Qiu, D.; Zhao, X. Synthesis of N-Arylsulfonamides through a Pd-Catalyzed Reduction Coupling Reaction of Nitroarenes with Sodium Arylsulfinates. Org. Biomol. Chem. 2018, 16, 8150–8154. [Google Scholar] [CrossRef]
- Stea, C.; Petzer, A.; Crou, C.; Petzer, J.P. Indazole derivatives as novel inhibitors of monoamine oxidase and D-amino acid oxidase. Med. Chem. Res. 2024, 33, 164–176. [Google Scholar] [CrossRef]
- Najafi, Z.; Mahdavi, M.; Saeedi, M.; Karimpour-Razkenari, E.; Asatouri, R.; Vafadarnejad, F.; Moghadam, F.H.; Khanavi, M.; Sharifzadeh, M.; Akbarzadeh, T. Novel Tacrine-1,2,3-Triazole Hybrids: In Vitro, In Vivo Biological Evaluation and Docking Study of Cholinesterase Inhibitors. Eur. J. Med. Chem. 2017, 125, 1200–1212. [Google Scholar] [CrossRef]
- Rohman, N.; Ardiansah, B.; Wukirsari, T.; Judeh, Z. Recent Trends in the Synthesis and Bioactivity of Coumarin, Coumarin–Chalcone, and Coumarin–Triazole Molecular Hybrids. Molecules 2024, 29, 1026. [Google Scholar] [CrossRef]
- Zeki, N.M.; Mustafa, Y.F. Coumarin hybrids: A sighting of their roles in drug targeting. Chem. Pap. 2024, 1–20. [Google Scholar] [CrossRef]
- Shetnev, A.; Shlenev, R.; Efimova, J.; Ivanovskii, S.; Tarasov, A.; Petzer, A.; Petzer, J.P. 1,3,4-Oxadiazol-2-ylbenzenesulfonamides as Privileged Structures for the Inhibition of Monoamine Oxidase B. Bioorg. Med. Chem Lett. 2019, 29, 126677. [Google Scholar] [CrossRef] [PubMed]
- Jalil, S.; Hussain, Z.; Ali Abid, S.M.; Hameed, A.; Iqbal, J. Quinoline–sulfonamides as a multi-targeting neurotherapeutic for cognitive decline: In vitro, in silico studies and ADME evaluation of monoamine oxidases and cholinesterases inhibitors. RSC Adv. 2024, 14, 8905–8920. [Google Scholar] [CrossRef] [PubMed]
- Chahardoli, A.; Mavaei, M.; Shokoohinia, Y.; Fattahi, A. Galbanic Acid, A Sesquiterpene Coumarin as a Novel Candidate for the Biosynthesis of Silver Nanoparticles: In Vitro Hemocompatibility, Antiproliferative, Antibacterial, Antioxidant, and Anti-Inflammatory Properties. Adv. Powder Technol. 2023, 24, 103928. [Google Scholar] [CrossRef]
- Bouissane, L.; Khouili, M.; Coudert, G.; Pujol, M.D.; Guillaumet, G. New and Promising Type of Leukotriene B4 (LTB4) Antagonists Based on the 1,4-Benzodioxine Structure. Eur. J. Med. Chem. 2023, 254, 115332. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Cyrille, N.N.; Idrice, A.A.; Maraf, M.B.; Charles, F.A.; Ibrahim, M.N.; Ríos-Gutiérrez, M.; Domingo, L.R. Understanding the Mechanism of Nitrobenzene Nitration with Nitronium Ion. A Molecular Electron Density Theory Study. Chem. Select. 2019, 4, 13313–13319. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Aurell, M.J. Unveiling the Regioselectivity in Electrophilic Aromatic Substitution Reactions of Deactivated Benzenes through Molecular Electron Density Theory. New J. Chem. 2021, 45, 13626–13638. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M. Application of Reactivity Indices in the Study of Polar Diels–Alder Reactions. In Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory; Liu, S., Ed.; WILEY-VCH GmbH: Weinheim, German, 2022; pp. 481–502. [Google Scholar]
- Moumad, A.; Bouhaoui, A.; Eddahmi, M.; Hafid, A.; Domingo, L.R.; Bouissane, L. Study of N-Methyl-5-nitroindazolylacrylonitriles as a Function of Quantum Parameters Employing Density Function Theory Methods: Comparative Theoretical Study and Nonlinear Optical Properties. Chem. Select. 2023, 8, e202300669. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Domingo, L.R. A New C–C Bond Formation Model Based on the Quantum Chemical Topology of Electron Density. RSC. Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular-Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of Chemical Bonds Based on Topological Analysis of Electron Localization Functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Enhanced Reactivity of Aza Aromatic Compounds Participating in Diels-Alder Reactions. Org. Biomol. Chem. 2020, 18, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Some Applications of the Transition State Method to the Calculation of Reaction Velocities, Especially In Solution. Trans. Faraday Soc. 1935, 31, 875–894. [Google Scholar] [CrossRef]
- Eddahmi, M.; Moura, N.M.M.; Bouissane, L.; Amiri, O.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Mendes, R.F.; Paz, F.A.A.; Neves, M.G.P.M.S.; Rakib, E.M. A Suitable Functionalization of Nitroindazoles with Triazolyl and Pyrazolyl Moieties via Cycloaddition Reactions. Molecules 2020, 25, 126. [Google Scholar] [CrossRef]
- Rullo, M.; Cipolloni, M.; Catto, M.; Colliva, C.; Miniero, D.V.; Latronico, T.; de Candia, M.; Benicchi, T.; Linusson, A.; Giacchè, N.; et al. Probing Fluorinated Motifs onto Dual AChE–MAO B Inhibitors: Rational Design, Synthesis, Biological Evaluation and Early-ADME Studies. J. Med. Chem. 2022, 65, 3962–3977. [Google Scholar] [CrossRef] [PubMed]
- Eddahmi, M.; La Spada, G.; Hafid, A.; Khouili, M.; Catto, M.; Bouissane, L. Towards Alzheimer’s Disease-Related Targets: One-Pot Cu(I)-Mediated Synthesis of New Nitroindazolyltriazoles. Bioorg. Chem. 2023, 130, 106261. [Google Scholar] [CrossRef] [PubMed]
- Rullo, M.; La Spada, G.; Miniero, D.V.; Gottinger, A.; Catto, M.; Delre, P.; Mastromarino, M.; Latronico, T.; Marchese, S.; Mangiatordi, G.F.; et al. Bioisosteric replacement based on 1,2,4-oxadiazoles in the discovery of 1H-indazole-bearing neuroprotective MAO B inhibitors. Eur. J. Med. Chem. 2023, 255, 115352. [Google Scholar] [CrossRef] [PubMed]
- Knez, D.; Sova, M.; Košak, U.; Gobec, S. Dual inhibitors of cholinesterases and monoamine oxidases for Alzheimer’s disease. Future Med. Chem. 2017, 9, 811–832. [Google Scholar] [CrossRef] [PubMed]
- Catto, M.; Nicolotti, O.; Leonetti, F.; Carotti, A.; Favia, A.D.; Soto-Otero, R.; Méndez-Álvarez, E.; Carotti, A. Structural Insights into MAO Inhibitory Potency and Selectivity of 7-substituted Coumarins from Ligand- and Target-based Approaches. J. Med. Chem. 2006, 49, 4912–4925. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Hehre, M.J.; Radom, L.; Schleyer, P.V.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Tomasi, J.; Persico, M. Molecular interactions in solution: And overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions–Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, 6th ed.; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Paolino, M.; de Candia, M.; Rosa Purgatorio, R.; Catto, M.; Saletti, M.; Tondo, A.R.; Nicolotti, O.; Cappelli, A.; Brizzi, A.; Mugnaini, C.; et al. Investigation on Novel E/Z 2-Benzylideneindan-1-one-based 2 Photoswitches with AChE and MAO-B Dual Inhibitory Activity. Molecules 2023, 28, 5857. [Google Scholar] [CrossRef]
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-A resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem. 2005, 26, 1689–1700. [Google Scholar] [CrossRef]
- Bailly, C.; Vergoten, G. Interaction of Camptothecin Anticancer Drugs with Ribosomal Proteins L15 and L11: A Molecular Docking Study. Molecules 2023, 28, 1828. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Vergoten, G. Binding of Vialinin A and p-Terphenyl Derivatives to Ubiquitin-Specific Protease 4 (USP4): A Molecular Docking Study. Molecules 2022, 27, 590. [Google Scholar] [CrossRef] [PubMed]
- Alcaro, S.; Gaspar, A.; Ortuso, F.; Milhazes, N.; Orallo, F.; Uriarte, E.; Yáñez, M.; Borges, F. Chromone-2- and -3-carboxylic acids inhibit differently monoamine oxidases A and B. Bioorg. Med. Chem. Lett. 2010, 20, 2709–2712. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, D.; Singh, T.G.; Kaur, R.; Kumar, B. Pyrazoline Derivatives as Promising MAO-A Targeting Antidepressants: An Update. Curr. Top. Med. Chem. 2024, 24, 401–415. [Google Scholar] [CrossRef]
- Olayinka, J.N.; Akawa, O.B.; Ogbu, E.K.; Eduviere, A.T.; Ozolua, R.I.; Soliman, M. Apigenin attenuates depressive-like behavior via modulating monoamine oxidase A enzyme activity in chronically stressed mice. Curr. Res. Pharmacol. Drug Discov. 2023, 11, 100161. [Google Scholar] [CrossRef]
- Benny, F.; Kumar, S.; Jayan, J.; Abdelgawad, M.A.; Ghoneim, M.M.; Kumar, A.; Manoharan, A.; Susan, R.; Sudevan, S.T.; Mathew, B. Review of beta-carboline and its derivatives as selective MAO-A inhibitors. Arch Pharm 2023, 356, e2300091. [Google Scholar] [CrossRef]



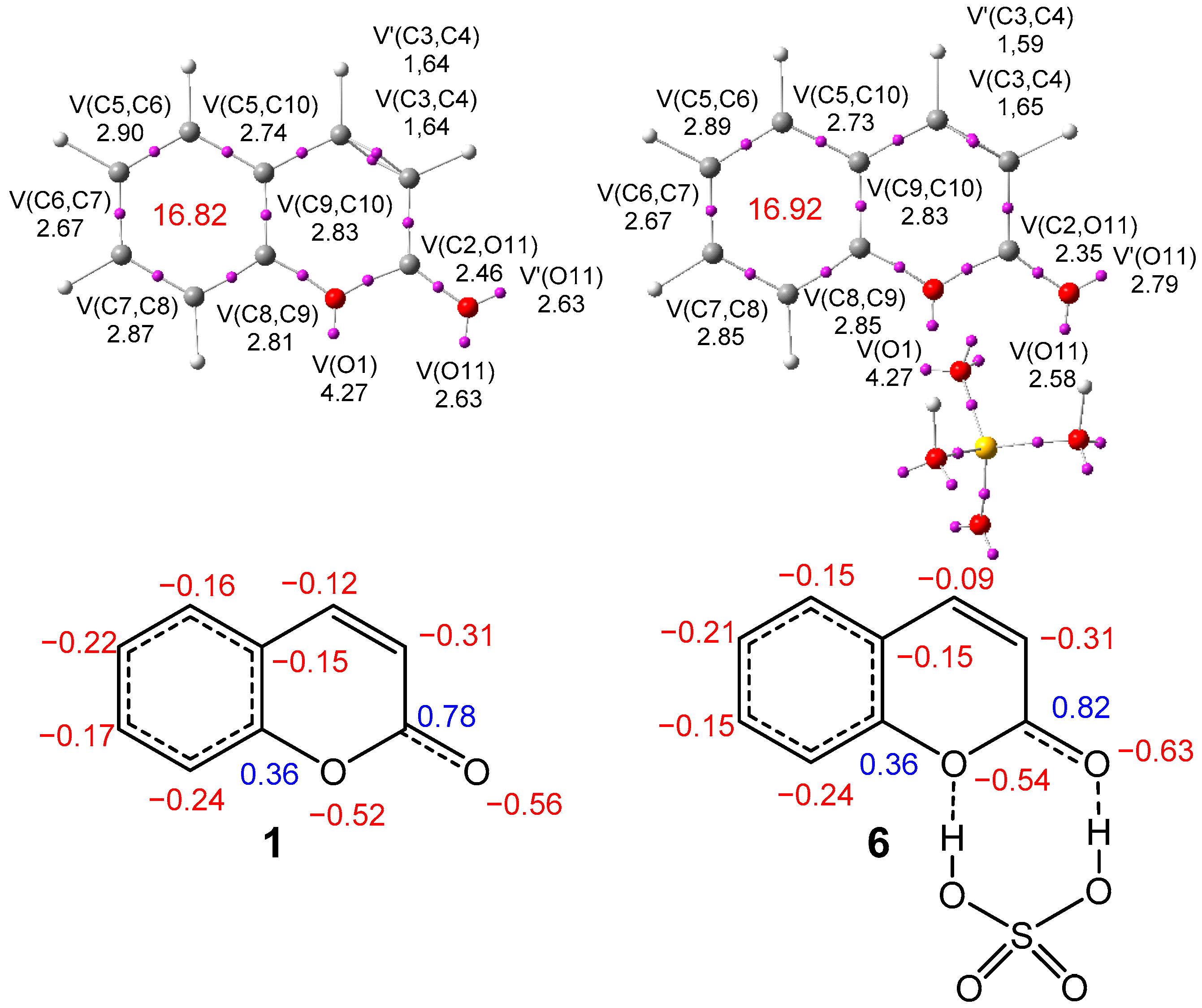
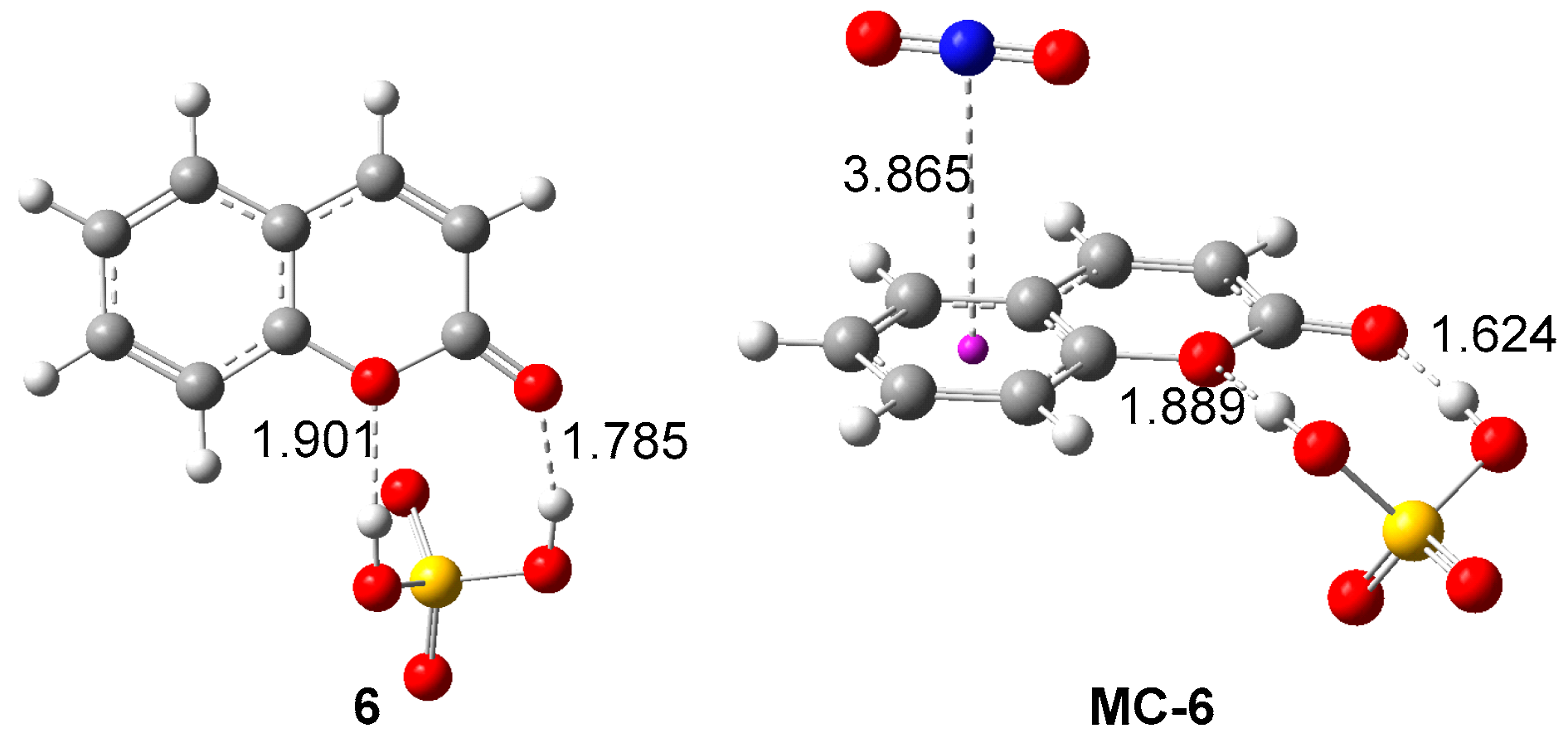
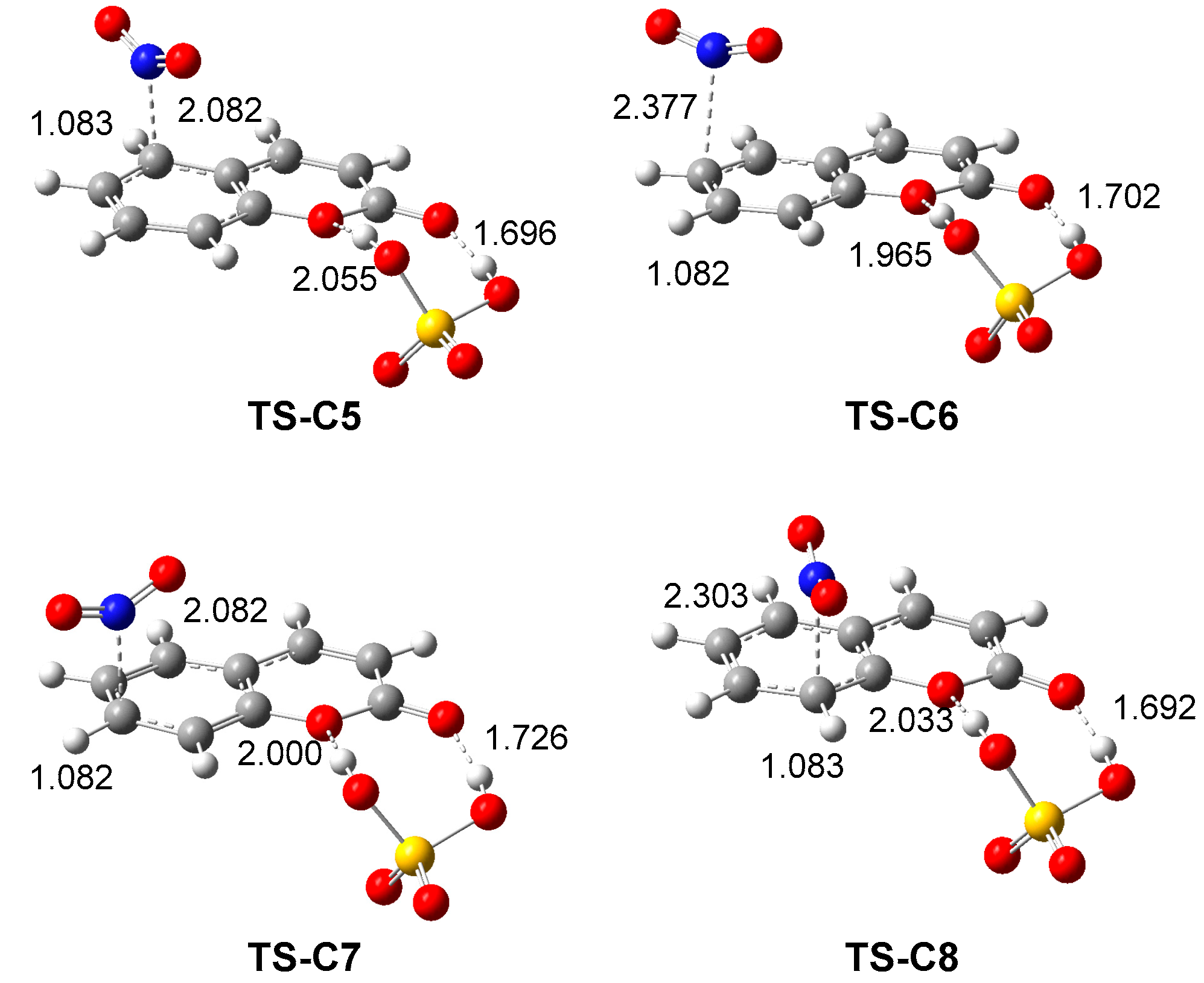

| µ | ɳ | ω | N | |
| Nitronium NO2+ ion 4 | −16.18 | 9.35 | 14.01 | −11.74 |
| Coumarin: SO4H2 complex 6 | −5.09 | 4.62 | 2.81 | 1.72 |
| Coumarin 1 | −4.19 | 4.62 | 1.90 | 2.62 |
| Benzene 3 | −3.30 | 6.80 | 0.80 | 2.42 |
| Entry | Product | Solvent | Base | T °C | Time (h) | Yield % |
|---|---|---|---|---|---|---|
| 1 | 11a | H2O | K2CO3 | r.t. | 24 | 51 |
| 2 | 11a | EtOH | K2CO3 | r.t. | 6 | 73 |
| 3 | 11a | EtOH | K2CO3 | 80 °C | 4 | 48 |
| 4 | 11a | - | Pyridine | r.t. | 1 | 85 |
| 5 | 11b | - | Pyridine | r.t. | 1 | 71 |
| 6 | 11c | - | Pyridine | r.t. | 1 | 82 |
| hAChE | hBChE | hMAO-A | hMAO-B | |||
|---|---|---|---|---|---|---|
| % Inhibition | IC50 µM | % Inhibition | % Inhibition | IC50 µM | % Inhibition | |
| 11a | 55 ± 3 | ni | 76 ± 3 | 2.46 ± 0.67 | 45 ± 2 | |
| 11b | 57 ± 4 | ni | 26 ± 3 | 34 ± 3 | ||
| 11c | 55 ± 3 | ni | 9 ± 4 | 26 ± 4 | ||
| 13a | 60 ± 3 | 3.73 ± 0.37 | ni | 13 ± 5 | 12 ± 1 | |
| 13b | 49 ± 3 | ni | 22 ± 7 | 28 ± 2 | ||
| 13c | 48 ± 2 | ni | 37 ± 5 | 20 ± 2 | ||
| 14a | 51 ± 2 | 48 ± 4 | 76 ± 5 | 14.8 ± 1.6 | 39 ± 3 | |
| 14b | 44 ± 3 | 31 ± 5 | 81 ± 5 | 16.9 ± 1.9 | 23 ± 4 | |
| 14c | 42 ± 1 | ni | 67 ± 5 | 28.0 ± 2.2 | 45 ± 1 | |
| Galantamine | 0.721 ± 0.152 | 8.78 ± 0.36 (b) | ||||
| Pargyline | 10.9 ± 0.6 | 2.69 ± 0.48 (b) | ||||
| Compound | Monoamine Oxidase A (MAO-A) | Monoamine Oxidase B (MAO-B) | ||
|---|---|---|---|---|
| ΔE (kcal/mol) | ΔG (kcal/mol) | ΔE (kcal/mol) | ΔG (kcal/mol) | |
| 11a | −52.30 | −22.20 | −53.85 | −15.00 |
| 11b | −56.65 | −18.40 | −59.70 | −17.65 |
| 11c | −56.65 | −24.30 | −58.40 | −18.50 |
| 13a | −56.80 | −17.20 | −58.70 | −14.80 |
| 13b | −59.40 | −14.50 | −66.60 | −15.50 |
| 13c | −56.10 | −18.75 | −60.55 | −16.75 |
| 14a | −89.60 | −24.70 | −94.30 | −26.60 |
| 14b | −89.65 | −36.30 | −94.40 | −20.25 |
| 14c | −91.75 | −21.90 | −97.40 | −30.00 |
| Harmine | −32.35 | −9.00 | - | - |
| Safinamide | - | - | −53.80 | −21.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eddahmi, M.; La Spada, G.; Domingo, L.R.; Vergoten, G.; Bailly, C.; Catto, M.; Bouissane, L. Synthesis, Molecular Electron Density Theory Study, Molecular Docking, and Pharmacological Evaluation of New Coumarin–Sulfonamide–Nitroindazolyl–Triazole Hybrids as Monoamine Oxidase Inhibitors. Int. J. Mol. Sci. 2024, 25, 6803. https://doi.org/10.3390/ijms25126803
Eddahmi M, La Spada G, Domingo LR, Vergoten G, Bailly C, Catto M, Bouissane L. Synthesis, Molecular Electron Density Theory Study, Molecular Docking, and Pharmacological Evaluation of New Coumarin–Sulfonamide–Nitroindazolyl–Triazole Hybrids as Monoamine Oxidase Inhibitors. International Journal of Molecular Sciences. 2024; 25(12):6803. https://doi.org/10.3390/ijms25126803
Chicago/Turabian StyleEddahmi, Mohammed, Gabriella La Spada, Luis R. Domingo, Gérard Vergoten, Christian Bailly, Marco Catto, and Latifa Bouissane. 2024. "Synthesis, Molecular Electron Density Theory Study, Molecular Docking, and Pharmacological Evaluation of New Coumarin–Sulfonamide–Nitroindazolyl–Triazole Hybrids as Monoamine Oxidase Inhibitors" International Journal of Molecular Sciences 25, no. 12: 6803. https://doi.org/10.3390/ijms25126803
APA StyleEddahmi, M., La Spada, G., Domingo, L. R., Vergoten, G., Bailly, C., Catto, M., & Bouissane, L. (2024). Synthesis, Molecular Electron Density Theory Study, Molecular Docking, and Pharmacological Evaluation of New Coumarin–Sulfonamide–Nitroindazolyl–Triazole Hybrids as Monoamine Oxidase Inhibitors. International Journal of Molecular Sciences, 25(12), 6803. https://doi.org/10.3390/ijms25126803