
Inhibition of Toll-like Receptors Alters Macrophage Cholesterol Efflux and Foam Cell Formation

 , , ,
, , ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
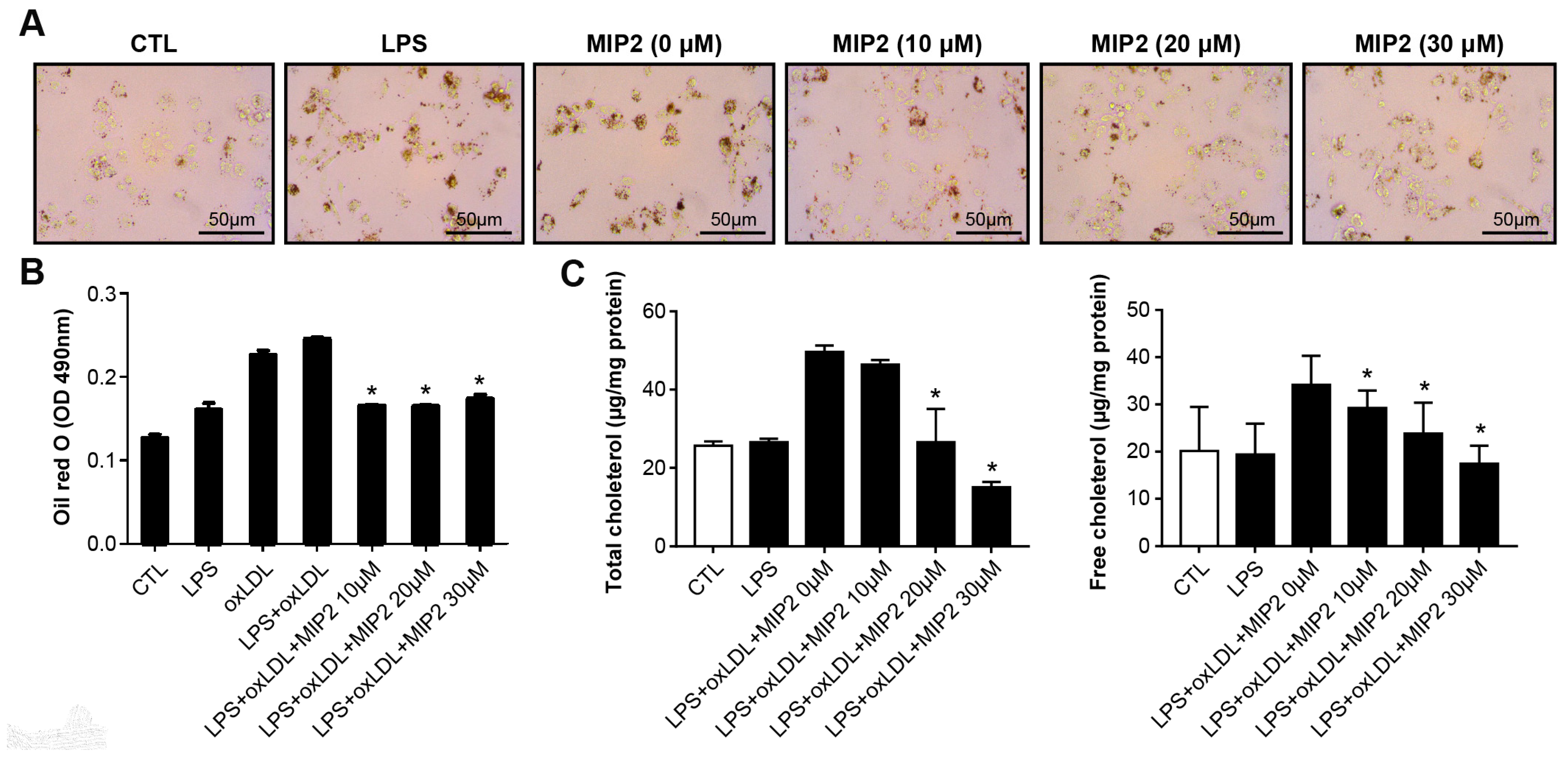
2.1. Foam Cell Formation and Cholesterol Assay after MIP2 Administration
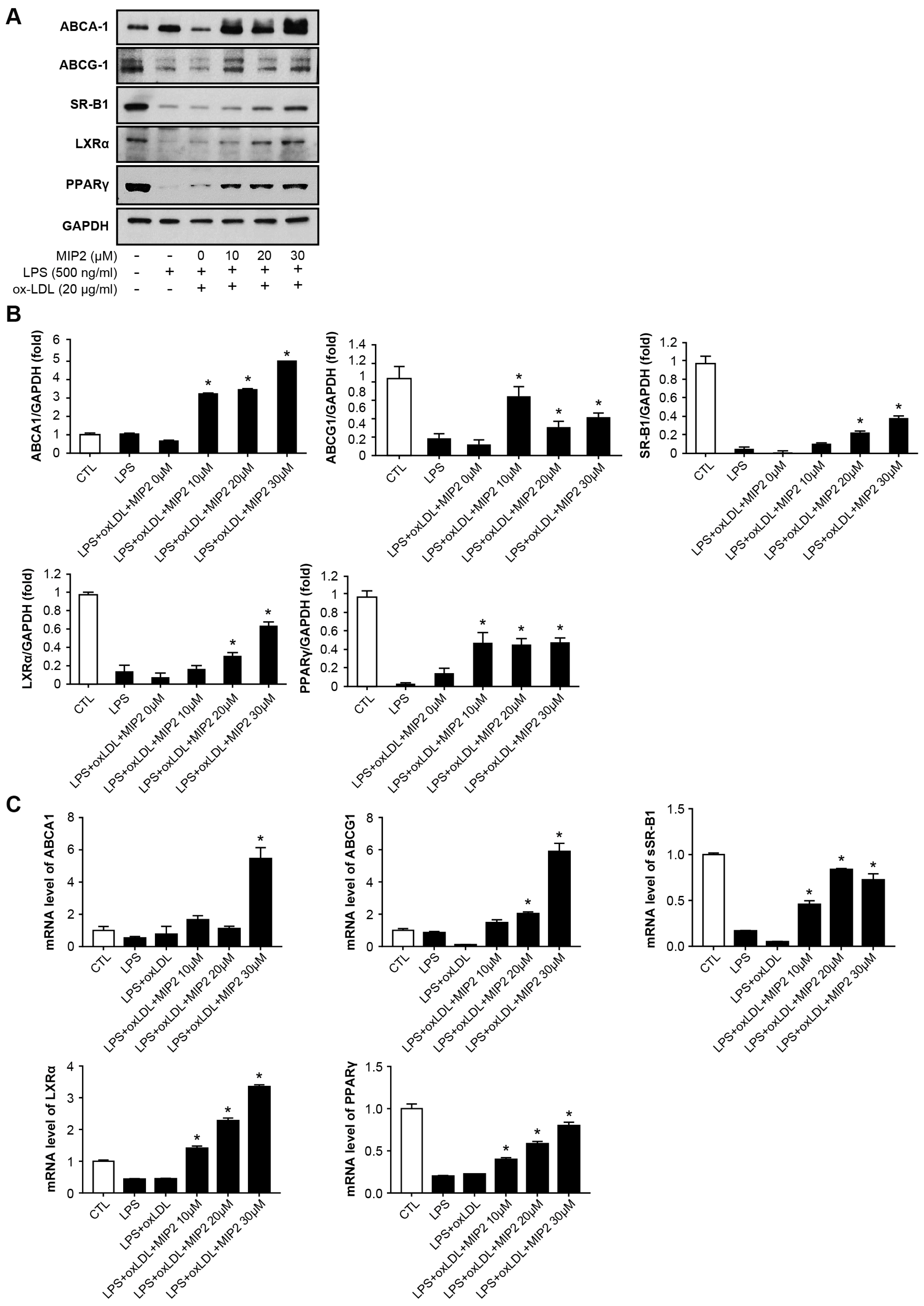
2.2. Expression of Cholesterol Efflux Transporters after MIP2 Administration
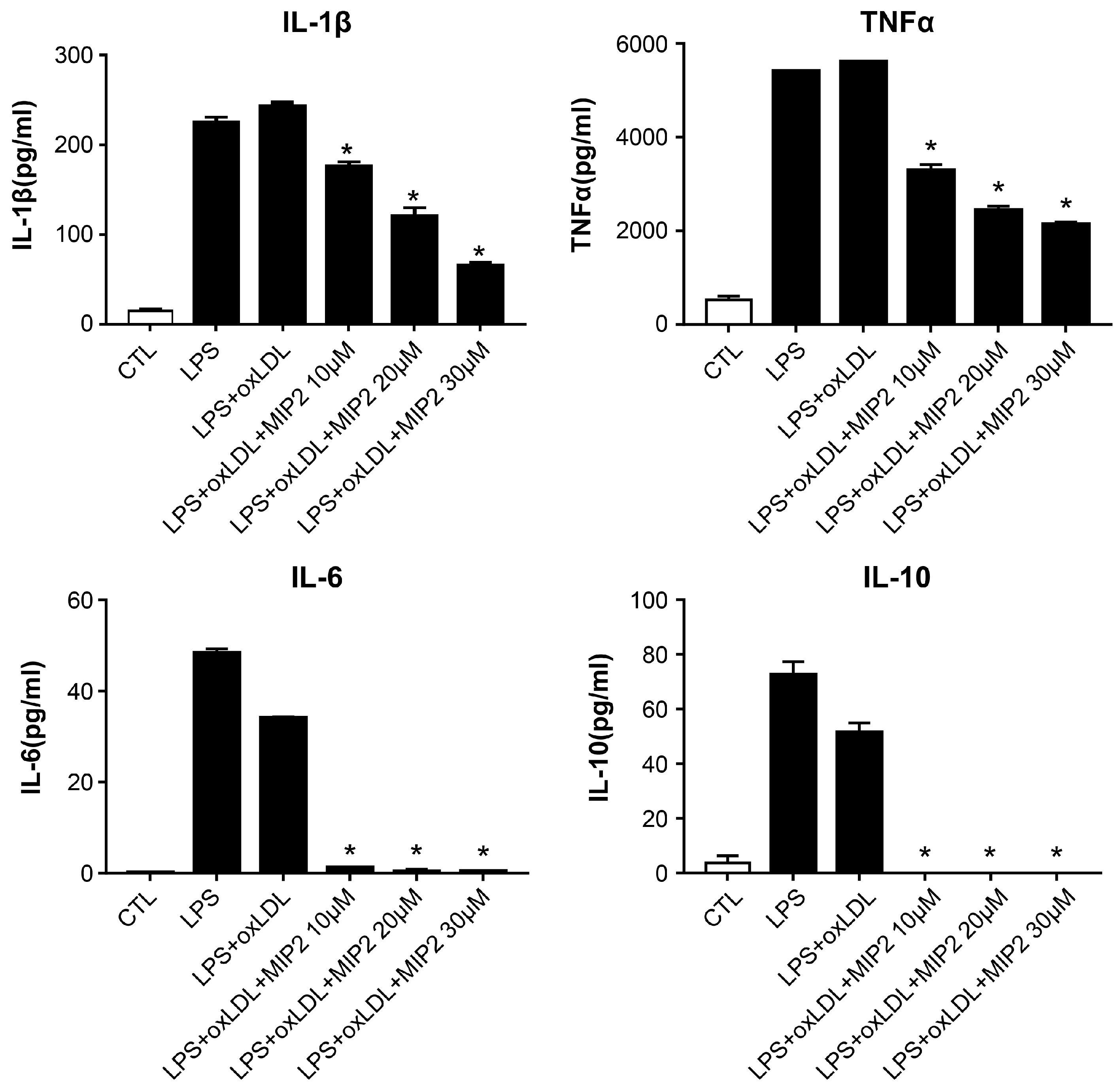
2.3. Change in Inflammatory Cytokines after MIP2 Administration
2.4. Change in Phosphorylation of Intracellular Transcription after MIP2 Administration
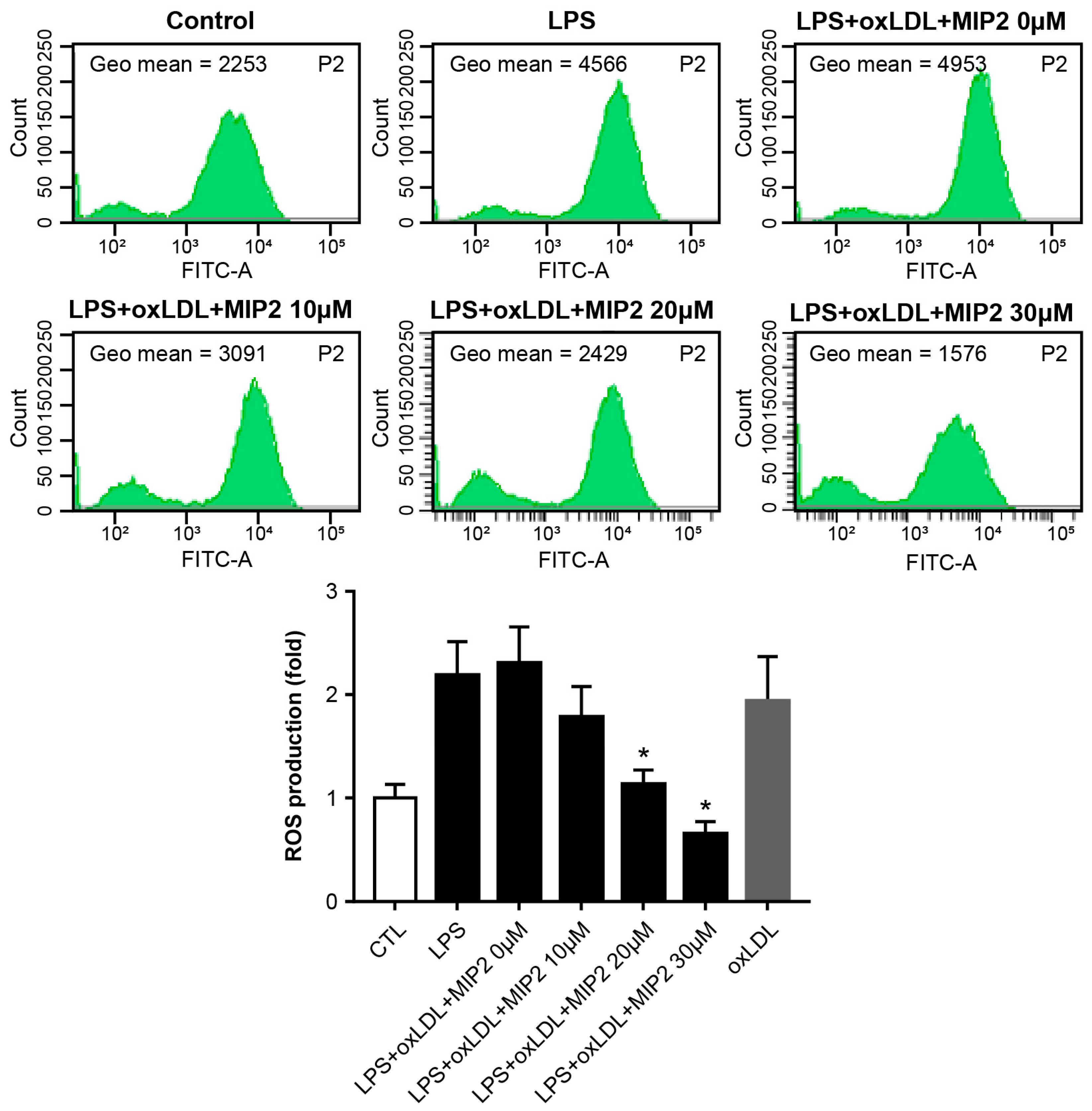
2.5. ROS Production after MIP2 Administration
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Oil Red-O Staining
4.3. Cholesterol Assay
4.4. Western Blotting
4.5. Quantitative Real-Time PCR
4.6. Enzyme-Linked Immunosobent Assay (ELISA)
4.7. ROS Production
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Berenice Martinez-Shio, E.; Martin Cardenas-Hernandez, A.; Jimenez-Suarez, V.; Marín-Jáuregui, L.S.; del Campo, C.C.-M.; González-Amaro, R.; Escobedo-Uribe, C.D.; Monsiváis-Urenda, A.E. Differentiation of circulating monocytes into macrophages with metabolically activated phenotype regulates inflammation in dyslipidemia patients. Clin. Exp. Immunol. 2022, 208, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Chen, H.; Qu, P.; Qiao, X.; Guo, L.; Liu, L. Novel insight on the role of Macrophages in atherosclerosis: Focus on polarization, apoptosis and efferocytosis. Int. Immunopharmacol. 2022, 113, 109260. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Pownall, H.J.; Rosales, C.; Gillard, B.K.; Gotto, A.M., Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat. Rev. Cardiol. 2021, 18, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Dergunov, A.D.; Baserova, V.B. Different Pathways of Cellular Cholesterol Efflux. Cell Biochem. Biophys. 2022, 80, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef]
- Hayakawa, S.; Tamura, A.; Nikiforov, N.; Koike, H.; Kudo, F.; Cheng, Y.; Miyazaki, T.; Kubekina, M.; Kirichenko, T.V.; Orekhov, A.N.; et al. Activated cholesterol metabolism is integral for innate macrophage responses by amplifying Myd88 signaling. JCI Insight 2022, 7, e138539. [Google Scholar] [CrossRef]
- Olejarz, W.; Łacheta, D.; Głuszko, A.; Migacz, E.; Kukwa, W.; Szczepański, M.J.; Tomaszewski, P.; Nowicka, G. RAGE and TLRs as Key Targets for Antiatherosclerotic Therapy. Biomed. Res. Int. 2018, 2018, 7675286. [Google Scholar] [CrossRef]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef]
- Wang, X.; Smith, C.; Yin, H. Targeting Toll-like receptors with small molecule agents. Chem. Soc. Rev. 2013, 42, 4859–4866. [Google Scholar] [CrossRef]
- Shah, M.; Kim, G.Y.; Achek, A.; Cho, E.-Y.; Baek, W.-Y.; Choi, Y.S.; Lee, W.H.; Kim, D.-J.; Lee, S.H.; Kim, W.; et al. The αC helix of TIRAP holds therapeutic potential in TLR-mediated autoimmune diseases. Biomaterials 2020, 245, 119974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, Q.; Shi, L.; Li, S.-W.; Wang, W.-H.; Wang, Q.-Q.; Lu, L.-X.; Xiao, H.; Wang, J.-H.; Li, F.-Y.; et al. Soluble CD4 effectively prevents excessive TLR activation of resident macrophages in the onset of sepsis. Signal Transduct. Target Ther. 2023, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Srivastava, M.; Saqib, U.; Liu, D.; Faisal, S.M.; Sugathan, S.; Bishnoi, S.; Baig, M.S. Potential therapeutic targets for inflammation in toll-like receptor 4 (TLR4)-mediated signaling pathways. Int. Immunopharmacol. 2016, 40, 79–89. [Google Scholar] [CrossRef]
- Flacher, V.; Neuberg, P.; Point, F.; Daubeuf, F.; Muller, Q.; Sigwalt, D.; Fauny, J.-D.; Remy, J.-S.; Frossard, N.; Wagner, A. Mannoside Glycolipid Conjugates Display Anti-inflammatory Activity by Inhibition of Toll-like Receptor-4 Mediated Cell Activation. ACS Chem. Biol. 2015, 10, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Kayani, T.; Jones, D.M.; Salmon, J.E.; Whirledge, S.; Chamley, L.W.; Abrahams, V.M. Antiphospholipid Antibodies Increase Endometrial Stromal Cell Decidualization, Senescence, and Inflammation via Toll-like Receptor 4, Reactive Oxygen Species, and p38 MAPK Signaling. Arthritis Rheumatol. 2022, 74, 1001–1012. [Google Scholar] [CrossRef]
- Li, Y.; Shen, S.; Ding, S.; Wang, L. LincRNA DYN-LRB2-2 upregulates cholesterol efflux by decreasing TLR2 expression in macrophages. J. Cell Biochem. 2018, 119, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Køllgaard, T.; Enevold, C.; Bendtzen, K.; Hansen, P.R.; Givskov, M.; Holmstrup, P.; Nielsen, C.H. Cholesterol crystals enhance TLR2- and TLR4-mediated pro-inflammatory cytokine responses of monocytes to the proatherogenic oral bacterium Porphyromonas gingivalis. PLoS ONE 2017, 12, e0172773. [Google Scholar] [CrossRef] [PubMed]
- Higashimori, M.; Tatro, J.B.; Moore, K.J.; Mendelsohn, M.E.; Galper, J.B.; Beasley, D. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 50–57. [Google Scholar] [CrossRef]
- Li, Y.; Shen, S.; Ding, S.; Wang, L. Toll-like receptor 2 downregulates the cholesterol efflux by activating the nuclear factor-κB pathway in macrophages and may be a potential therapeutic target for the prevention of atherosclerosis. Exp. Ther. Med. 2018, 15, 198–204. [Google Scholar] [CrossRef]
- Mullick, A.E.; Soldau, K.; Kiosses, W.B.; Bell, T.A., 3rd; Tobias, P.S.; Curtiss, L.K. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J. Exp. Med. 2008, 205, 373–383. [Google Scholar] [CrossRef]
- Liu, X.; Ukai, T.; Yumoto, H.; Davey, M.; Goswami, S.; Gibson, F.C., 3rd; Genco, C.A. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis 2008, 196, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Harkewicz, R.; Lee, J.H.; Boullier, A.; Almazan, F.; Li, A.C.; Witztum, J.L.; Bae, Y.S.; Miller, Y.I. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ. Res. 2009, 104, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.W.; Meng, X.; Fullerton, D.A.; Jin, C.; Reece, T.B.; Cleveland, J.C., Jr. Toll-like receptor 4 mediates oxidized LDL-induced macrophage differentiation to foam cells. J. Surg. Res. 2011, 171, e27–e31. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Boisvert, W.A.; Lee, C.H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Schuster, G.U.; Parini, P.; Wang, L.; Alberti, S.; Steffensen, K.R.; Hansson, G.K.; Angelin, B.; Gustafsson, J.-A. Accumulation of foam cells in liver X receptor-deficient mice. Circulation 2002, 106, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Moser, A.H.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. Downregulation of liver X receptor-alpha in mouse kidney and HK-2 proximal tubular cells by LPS and cytokines. J. Lipid Res. 2005, 46, 2377–2387. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, S.F.; Qin, Z.S.; Ye, J.; Zhao, Z.L.; Fang, H.H.; Yao, Z.W.; Gu, M.N.; Hu, Y.W. Propofol up-regulates expression of ABCA1, ABCG1, and SR-B1 through the PPARγ/LXRα signaling pathway in THP-1 macrophage-derived foam cells. Cardiovasc. Pathol. 2015, 24, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Endo-Umeda, K.; Kim, E.; Thomas, D.G.; Liu, W.; Dou, H.; Yalcinkaya, M.; Abramowicz, S.; Xiao, T.; Antonson, P.; Gustafsson, J.; et al. Myeloid LXR (Liver X Receptor) Deficiency Induces Inflammatory Gene Expression in Foamy Macrophages and Accelerates Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 719–731. [Google Scholar] [CrossRef]
- Awasthi, D.; Nagarkoti, S.; Kumar, A.; Dubey, M.; Singh, A.K.; Pathak, P.; Chandra, T.; Barthwal, M.K.; Dikshit, M. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-oxidase activation. Free Radic. Biol. Med. 2016, 93, 190–203. [Google Scholar] [CrossRef]
- Maeba, R.; Maruyama, A.; Tarutani, O.; Ueta, N.; Shimasaki, H. Oxidized low-density lipoprotein induces the production of superoxide by neutrophils. FEBS Lett. 1995, 377, 309–312. [Google Scholar] [PubMed]
- Feng, G.; Yang, X.; Li, Y.; Wang, X.; Tan, S.; Chen, F. LPS enhances platelets aggregation via TLR4, which is related to mitochondria damage caused by intracellular ROS, but not extracellular ROS. Cell Immunol. 2018, 328, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Singh, V.; Tiwari, R.L.; Chandra, T.; Kumar, A.; Dikshit, M.; Barthwal, M.K. The IRAK-ERK-p67phox-Nox-2 axis mediates TLR4, 2-induced ROS production for IL-1β transcription and processing in monocytes. Cell Mol. Immunol. 2016, 13, 745–763. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Kim, J.-Y.; Byeon, H.-E.; Kim, J.-W.; Kim, H.-A.; Suh, C.-H.; Choi, S.; Linton, M.F.; Jung, J.-Y. Inhibition of Toll-like Receptors Alters Macrophage Cholesterol Efflux and Foam Cell Formation. Int. J. Mol. Sci. 2024, 25, 6808. https://doi.org/10.3390/ijms25126808
Kim J, Kim J-Y, Byeon H-E, Kim J-W, Kim H-A, Suh C-H, Choi S, Linton MF, Jung J-Y. Inhibition of Toll-like Receptors Alters Macrophage Cholesterol Efflux and Foam Cell Formation. International Journal of Molecular Sciences. 2024; 25(12):6808. https://doi.org/10.3390/ijms25126808
Chicago/Turabian StyleKim, Jaemi, Ji-Yun Kim, Hye-Eun Byeon, Ji-Won Kim, Hyoun-Ah Kim, Chang-Hee Suh, Sangdun Choi, MacRae F. Linton, and Ju-Yang Jung. 2024. "Inhibition of Toll-like Receptors Alters Macrophage Cholesterol Efflux and Foam Cell Formation" International Journal of Molecular Sciences 25, no. 12: 6808. https://doi.org/10.3390/ijms25126808
APA StyleKim, J., Kim, J.-Y., Byeon, H.-E., Kim, J.-W., Kim, H.-A., Suh, C.-H., Choi, S., Linton, M. F., & Jung, J.-Y. (2024). Inhibition of Toll-like Receptors Alters Macrophage Cholesterol Efflux and Foam Cell Formation. International Journal of Molecular Sciences, 25(12), 6808. https://doi.org/10.3390/ijms25126808