
Association between Circulating T Cells and the Gut Microbiome in Healthy Individuals: Findings from a Pilot Study

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
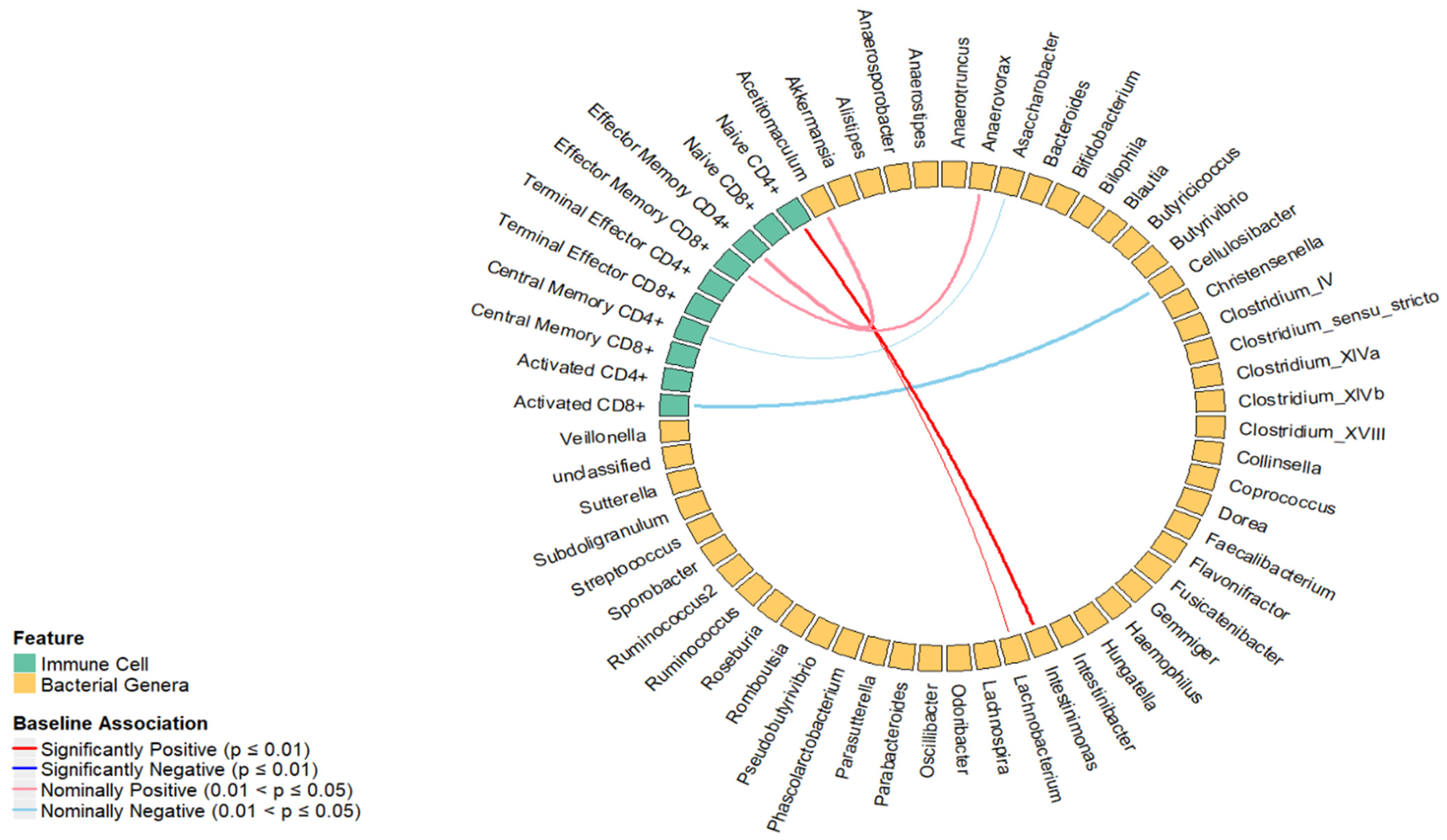
2.1. Cross-Sectional Association between the Percentage of T Cell Subsets and Relative Abundance of Bacterial Genera
2.2. Longitudinal Analysis Percentage of T Cell Subsets at the Baseline and Fold Change of Bacterial Genera over 6 Weeks
3. Discussion
Strengths and Limitations
4. Material and Methods
4.1. Study Design
4.2. Collection and Processing of the Stool Samples
4.3. Data Preprocessing
4.4. Collection of Blood Samples and Measurement of T Cell Subsets
4.5. Covariates
4.6. Statistical Analysis
4.7. Sparse Canonical Correlation Analysis (sCCA)
4.8. Linear Regression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosser, E.C.; Piper, C.J.; Matei, D.E.; Blair, P.A.; Rendeiro, A.F.; Orford, M.; Alber, D.G.; Krausgruber, T.; Catalan, D.; Klein, N.; et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metab. 2020, 31, 837–851.e10. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 283–297, Erratum in Semin. Immunopathol. 2019, 41, 299. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cai, X.; Zheng, Y.; Zhang, M.; Fei, W.; Sun, D.; Zhao, M.; Ye, Y.; Zheng, C. Short-chain fatty acids regulate B cells differentiation via the FFA2 receptor to alleviate rheumatoid arthritis. Br. J. Pharmacol. 2022, 179, 4315–4329. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.L.; Essigmann, H.T.; Hoffman, K.L.; Palm, N.W.; Gunter, S.M.; Sederstrom, J.M.; Petrosino, J.F.; Jun, G.; Aguilar, D.; Perkison, W.B.; et al. Impact of Diabetes on the Gut and Salivary IgA Microbiomes. Infect. Immun. 2020, 88, e00301-20. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A.; Ter Horst, R.; Jansen, T.; Jacobs, L.; Bonder, M.J.; et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 2016, 167, 1125–1136.e8. [Google Scholar] [CrossRef]
- Jung, Y.-J.; Kim, K.-H.; Ko, E.-J.; Lee, Y.; Kim, M.-C.; Lee, Y.-T.; Kim, C.-H.; Jeeva, S.; Park, B.R.; Kang, S.-M. Adjuvant effects of killed Lactobacillus casei DK128 on enhancing T helper type 1 immune responses and the efficacy of influenza vaccination in normal and CD4-deficient mice. Vaccine 2020, 38, 5783–5792. [Google Scholar] [CrossRef] [PubMed]
- Henrick, B.M.; Rodriguez, L.; Lakshmikanth, T.; Pou, C.; Henckel, E.; Arzoomand, A.; Olin, A.; Wang, J.; Mikes, J.; Tan, Z.; et al. Bifidobacteria-mediated immune system imprinting early in life. Cell 2021, 184, 3884–3898.e11. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Cantabrana, C.; Delgado, S.; Ruiz, L.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and Their Health-Promoting Effects. Microbiol. Spectr. 2017, 5, 73–98. [Google Scholar] [CrossRef]
- Avershina, E.; Storrø, O.; Øien, T.; Johnsen, R.; Wilson, R.; Egeland, T.; Rudi, K. Bifidobacterial Succession and Correlation Networks in a Large Unselected Cohort of Mothers and Their Children. Appl. Environ. Microbiol. 2013, 79, 497–507. [Google Scholar] [CrossRef]
- Sun, S.; Luo, L.; Liang, W.; Yin, Q.; Guo, J.; Rush, A.M.; Lv, Z.; Liang, Q.; Fischbach, M.A.; Sonnenburg, J.L.; et al. Bifidobacterium alters the gut microbiota and modulates the functional metabolism of T regulatory cells in the context of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2020, 117, 27509–27515. [Google Scholar] [CrossRef]
- Brennan, C.A.; Clay, S.L.; Lavoie, S.L.; Bae, S.; Lang, J.K.; Fonseca-Pereira, D.; Rosinski, K.G.; Ou, N.; Glickman, J.N.; Garrett, W.S. Fusobacterium nucleatum drives a pro-inflammatory intestinal microenvironment through metabolite receptor-dependent modulation of IL-17 expression. Gut Microbes 2021, 13, 1987780. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ruan, G.; Cheng, Y.; Yi, A.; Chen, D.; Wei, Y. The role of Th17 cells in inflammatory bowel disease and the research progress. Front. Immunol. 2023, 13, 1055914. [Google Scholar] [CrossRef] [PubMed]
- Hiippala, K.; Jouhten, H.; Ronkainen, A.; Hartikainen, A.; Kainulainen, V.; Jalanka, J.; Satokari, R. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients 2018, 10, 988. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, A.N.; West, N.R.; Stubbington, M.J.; Wendt, E.; Suijker, K.I.; Datsi, A.; This, S.; Danne, C.; Campion, S.; Duncan, S.H.; et al. Circulating and Tissue-Resident CD4+ T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology 2017, 153, 1320–1337.e16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sparks, J.B.; Karyala, S.V.; Settlage, R.; Luo, X.M. Host adaptive immunity alters gut microbiota. ISME J. 2014, 9, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Pagliari, D.; Piccirillo, C.A.; Larbi, A.; Cianci, R. The Interactions between Innate Immunity and Microbiota in Gastrointestinal Diseases. J. Immunol. Res. 2015, 2015, 1–3. [Google Scholar] [CrossRef]
- Yang, Y.; Palm, N.W. Immunoglobulin A and the microbiome. Curr. Opin. Microbiol. 2020, 56, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef]
- Dutta, D.; Sen, A.; Satagopan, J. Sparse canonical correlation to identify breast cancer related genes regulated by copy number aberrations. PLoS ONE 2022, 17, e0276886. [Google Scholar] [CrossRef]
- Du, L.; Liu, K.; Yao, X.; Risacher, S.L.; Han, J.; Saykin, A.J.; Guo, L.; Shen, L. Multi-Task Sparse Canonical Correlation Analysis with Application to Multi-Modal Brain Imaging Genetics. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Minnier, J.; Emmett, M.R.; Perez, R.; Ding, L.-H.; Barnette, B.L.; Larios, R.E.; Hong, C.; Hwang, T.H.; Yu, Y.; Fallgren, C.M.; et al. Associations between lipids in selected brain regions, plasma miRNA, and behavioral and cognitive measures following 28Si ion irradiation. Sci. Rep. 2021, 11, 14899. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qian, Y.; Wei, K.; Kong, W. Identifying Biomarkers of Alzheimer’s Disease via a Novel Structured Sparse Canonical Correlation Analysis Approach. J. Mol. Neurosci. 2021, 72, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Dong, W.; Yang, Q.; Yu, X.; Ma, J.; Gu, W.; Huang, Y. Altered Gut Microbiota Related to Inflammatory Responses in Patients With Huntington’s Disease. Front. Immunol. 2021, 11, 603594. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yan, L.; Chen, H.; Wu, N.; Wang, W.; Wang, D. Cyclocarya paliurus polysaccharides alleviate type 2 diabetic symptoms by modulating gut microbiota and short-chain fatty acids. Phytomedicine 2020, 77, 153268. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.L.; Hopkins, L.J.; Gangloff, M.; Bryant, C.E. The molecular basis for recognition of bacterial ligands at equine TLR2, TLR1 and TLR6. Vet. Res. 2013, 44, 50. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Beyaert, R. Role of Toll-Like Receptors in Pathogen Recognition. Clin. Microbiol. Rev. 2003, 16, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Ramasubramanian, R.; Meier, H.C.S.; Vivek, S.; Klopack, E.; Crimmins, E.M.; Faul, J.; Nikolich-Žugich, J.; Thyagarajan, B. Evaluation of T-cell aging-related immune phenotypes in the context of biological aging and multimorbidity in the Health and Retirement Study. Immun. Ageing 2022, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Klöß, S.; Dehmel, S.; Braun, A.; Parnham, M.J.; Köhl, U.; Schiffmann, S. From Cancer to Immune-Mediated Diseases and Tolerance Induction: Lessons Learned From Immune Oncology and Classical Anti-cancer Treatment. Front. Immunol. 2020, 11, 1423. [Google Scholar] [CrossRef]
- Bourque, J.; Hawiger, D. Variegated Outcomes of T Cell Activation by Dendritic Cells in the Steady State. J. Immunol. 2022, 208, 539–547. [Google Scholar] [CrossRef]
- Tian, T.; Zhang, X.; Luo, T.; Wang, D.; Sun, Y.; Dai, J. Effects of Short-Term Dietary Fiber Intervention on Gut Microbiota in Young Healthy People. Diabetes Metab. Syndr. Obes. 2020, 14, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Santarossa, S.; Sitarik, A.R.; Johnson, C.C.; Li, J.; Lynch, S.V.; Ownby, D.R.; Ramirez, A.; Yong, G.L.; Cassidy-Bushrow, A.E. Associations of physical activity with gut microbiota in pre-adolescent children. Phys. Act. Nutr. 2021, 25, 24–37. [Google Scholar] [CrossRef]
- Wei, M.; Li, C.; Dai, Y.; Zhou, H.; Cui, Y.; Zeng, Y.; Huang, Q.; Wang, Q. High-Throughput Absolute Quantification Sequencing Revealed Osteoporosis-Related Gut Microbiota Alterations in Han Chinese Elderly. Front. Cell. Infect. Microbiol. 2021, 11, 630372. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Jiang, W.; Zhang, X.; Qin, L.; Su, B.; Li, Z.; Sun, J.; Zhang, Y.; Zhang, T.; Lu, X.; et al. Elevated Level of CD4+ T Cell Immune Activation in Acutely HIV-1-Infected Stage Associates With Increased IL-2 Production and Cycling Expression, and Subsequent CD4+ T Cell Preservation. Front. Immunol. 2018, 9, 616. [Google Scholar] [CrossRef]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef]
- Kestens, L.; Vanham, G.; Gigase, P.; Young, G.; Hannet, I.; Vanlangendonck, F.; Hulstaert, F.; Bach, B.A. Expression of activation antigens, HLA-DR and CD38, on CD8 lymphocytes during HIV-1 infection. AIDS 1992, 6, 793–798. [Google Scholar] [CrossRef]
- Yang, Z.-H.; Liu, F.; Zhu, X.-R.; Suo, F.-Y.; Jia, Z.-J.; Yao, S.-K. Altered profiles of fecal bile acids correlate with gut microbiota and inflammatory responses in patients with ulcerative colitis. World J. Gastroenterol. 2021, 27, 3609–3629. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Zhang, Y.; He, W.; Tian, Z.; Lin, J.; Liu, Z.; Li, Y.; Chen, M.; Han, S.; Liang, J.; et al. Gut Microbiota and Metabolite Changes in Patients With Ulcerative Colitis and Clostridioides difficile Infection. Front. Microbiol. 2022, 13, 802823. [Google Scholar] [CrossRef]
- Du, Y.; Li, X.; An, Y.; Song, Y.; Lu, Y. Association of gut microbiota with sort-chain fatty acids and inflammatory cytokines in diabetic patients with cognitive impairment: A cross-sectional, non-controlled study. Front. Nutr. 2022, 9, 930626. [Google Scholar] [CrossRef]
- Qian, Y.; Yang, X.; Xu, S.; Wu, C.; Song, Y.; Qin, N.; Chen, S.-D.; Xiao, Q. Alteration of the fecal microbiota in Chinese patients with Parkinson’s disease. Brain Behav. Immun. 2018, 70, 194–202. [Google Scholar] [CrossRef]
- Eicher, T.P.; Mohajeri, M.H. Overlapping Mechanisms of Action of Brain-Active Bacteria and Bacterial Metabolites in the Pathogenesis of Common Brain Diseases. Nutrients 2022, 14, 2661. [Google Scholar] [CrossRef] [PubMed]
- Ogita, T.; Yamamoto, Y.; Mikami, A.; Shigemori, S.; Sato, T.; Shimosato, T. Oral Administration of Flavonifractor plautii Strongly Suppresses Th2 Immune Responses in Mice. Front. Immunol. 2020, 11, 379. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Bangs, D.J.; Sidney, J.; Kolla, R.V.; De Silva, A.D.; de Silva, A.M.; Crotty, S.; Peters, B.; Sette, A. Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc. Natl. Acad. Sci. USA 2015, 112, E4256–E4263. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Babor, M.; Lane, J.; Schulten, V.; Patil, V.S.; Seumois, G.; Rosales, S.L.; Fu, Z.; Picarda, G.; Burel, J.; et al. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nat. Commun. 2017, 8, 1473. [Google Scholar] [CrossRef] [PubMed]
- Noriega, B.S.; Sanchez-Gonzalez, M.A.; Salyakina, D.; Coffman, J. Understanding the Impact of Omega-3 Rich Diet on the Gut Microbiota. Case Rep. Med. 2016, 2016, 3089303. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; An, Y.; Hao, F.; Wang, Y.; Tang, H. Correlations of Fecal Metabonomic and Microbiomic Changes Induced by High-fat Diet in the Pre-Obesity State. Sci. Rep. 2016, 6, 21618. [Google Scholar] [CrossRef] [PubMed]
- Prizment, A.E.; Staley, C.; Onyeaghala, G.C.; Vivek, S.; Thyagarajan, B.; Straka, R.J.; Demmer, R.T.; Knights, D.; Meyer, K.A.; Shaukat, A.; et al. Randomised clinical study: Oral aspirin 325 mg daily vs placebo alters gut microbial composition and bacterial taxa associated with colorectal cancer risk. Aliment. Pharmacol. Ther. 2020, 52, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sarwar, K.A.; Kelly, R.S.; Lasky-Su, J.; Zeiger, R.S.; O‘Connor, G.T.; Sandel, M.T.; Bacharier, L.B.; Beigelman, A.; Laranjo, N.; Gold, D.R.; et al. Integrative analysis of the intestinal metabolome of childhood asthma. J. Allergy Clin. Immunol. 2019, 144, 442–454. [Google Scholar] [CrossRef]
- Witten, D.M.; Tibshirani, R.; Hastie, T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics 2009, 10, 515–534. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Immune Cell | Estimate | Standard Error | p-Value | Bacterial Genera (Phylum) |
|---|---|---|---|---|
| Naïve CD4+ | 0.03 | 0.01 | <0.01 | Intestinimonas (Bacillota) |
| Activated CD8+ | −0.11 | 0.05 | 0.03 | Cellulosibacter (Bacillota) |
| Immune Cell | Estimate | Standard Error | p-Value | Bacterial Genera (Phylum) |
|---|---|---|---|---|
| Activated CD4+ | −0.07 | 0.03 | 0.03 | Anaerovorax (Bacillota) |
| Terminal Effector CD4+ | 0.05 | 0.02 | 0.01 | Flavonifractor (Bacillota) |
| Naïve CD4+ | −0.03 | 0.03 | 0.02 | Clostridium_XlVb (Bacillota) |
| Microbiome | Immune Cells and Time Point | Estimate (p-Value) | |
|---|---|---|---|
| Intestinimonas | Naïve CD4+ | Baseline | 0.03 (<0.01) |
| Clostridium_XlVb | 6-weeks change | −0.03 (0.02) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vivek, S.; Shen, Y.S.; Guan, W.; Onyeaghala, G.; Oyenuga, M.; Staley, C.; Karger, A.B.; Prizment, A.E.; Thyagarajan, B. Association between Circulating T Cells and the Gut Microbiome in Healthy Individuals: Findings from a Pilot Study. Int. J. Mol. Sci. 2024, 25, 6831. https://doi.org/10.3390/ijms25136831
Vivek S, Shen YS, Guan W, Onyeaghala G, Oyenuga M, Staley C, Karger AB, Prizment AE, Thyagarajan B. Association between Circulating T Cells and the Gut Microbiome in Healthy Individuals: Findings from a Pilot Study. International Journal of Molecular Sciences. 2024; 25(13):6831. https://doi.org/10.3390/ijms25136831
Chicago/Turabian StyleVivek, Sithara, You Shan Shen, Weihua Guan, Guillaume Onyeaghala, Mosunmoluwa Oyenuga, Christopher Staley, Amy B. Karger, Anna E. Prizment, and Bharat Thyagarajan. 2024. "Association between Circulating T Cells and the Gut Microbiome in Healthy Individuals: Findings from a Pilot Study" International Journal of Molecular Sciences 25, no. 13: 6831. https://doi.org/10.3390/ijms25136831