
Effects of High-Mobility Group Box-1 on Mucosal Immunity and Epithelial Differentiation in Colitic Carcinoma


Abstract
:1. Introduction
2. Results
2.1. Effects of HMGB1 in the Azoxymethane (AOM)-Induced Mouse Colon Carcinogenesis Model
2.2. Effects of HMGB1 in the Mouse Dextran Sulfate Sodium (DSS) Colitis-Associated Colon Carcinogenesis Model
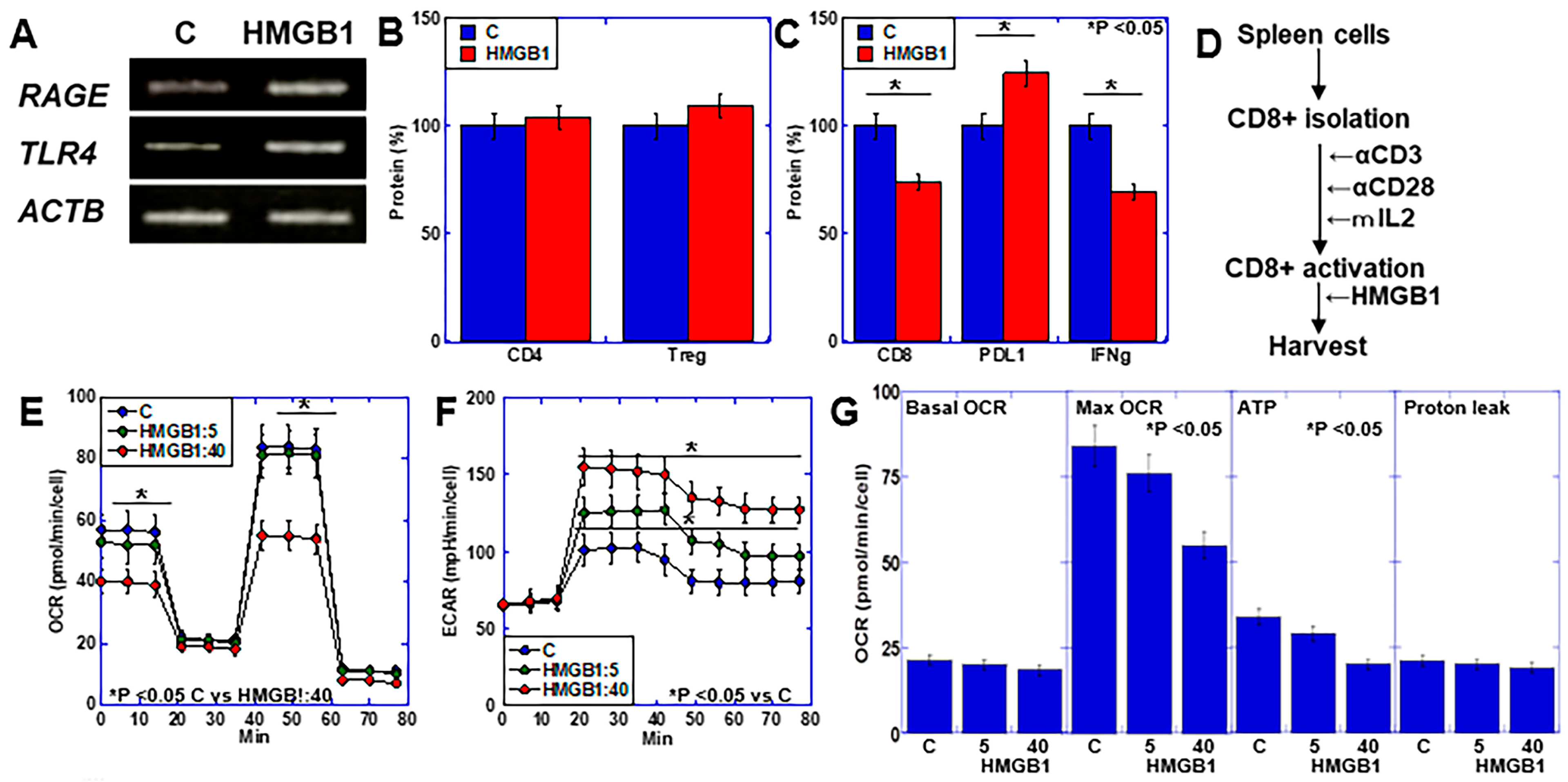
2.3. Effects of HMGB1 on Lymphocytes Using In Vitro CD8+ Cell Activation Assay
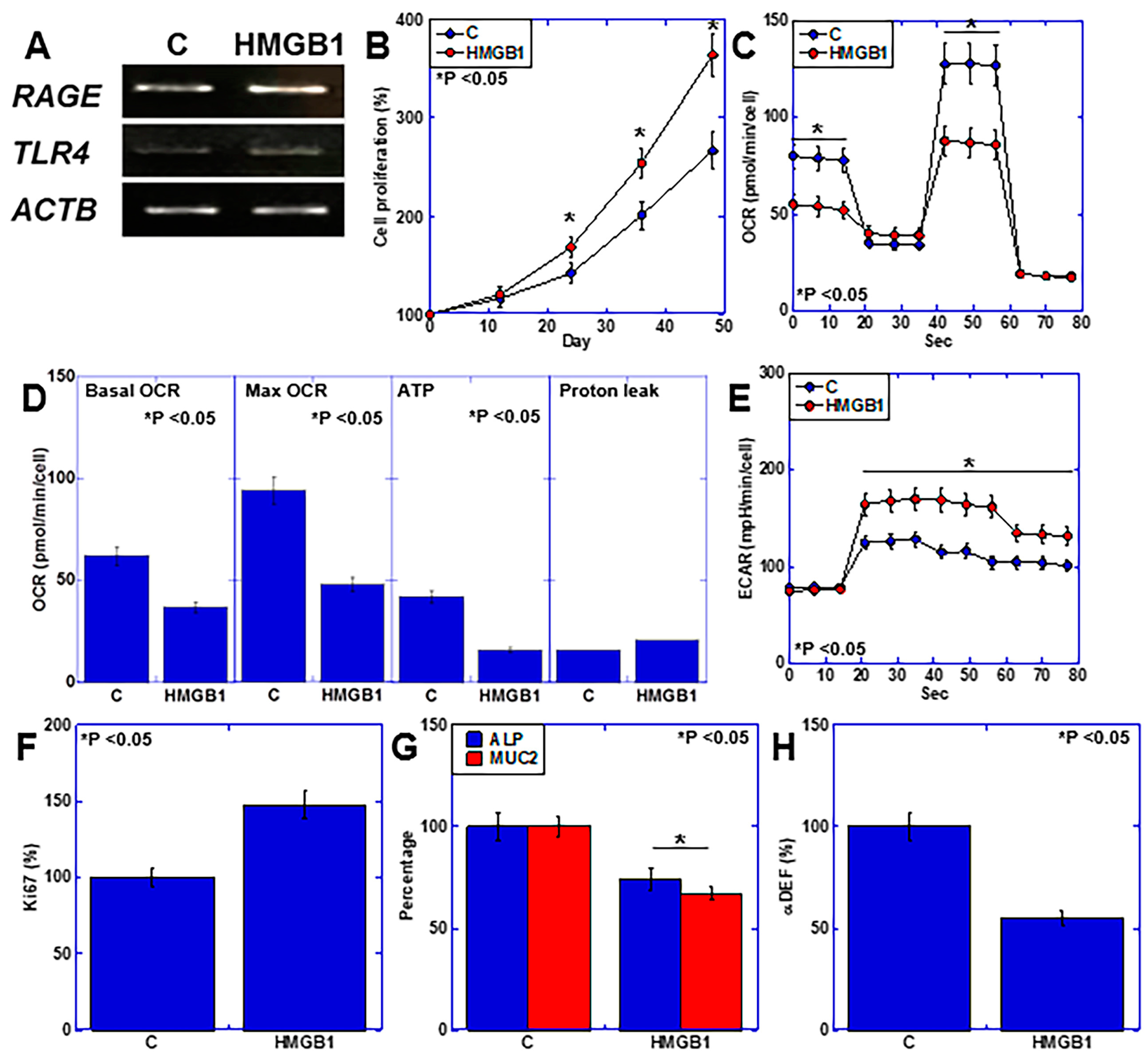
2.4. Effects of HMGB1 on Intestinal Epithelial Cells
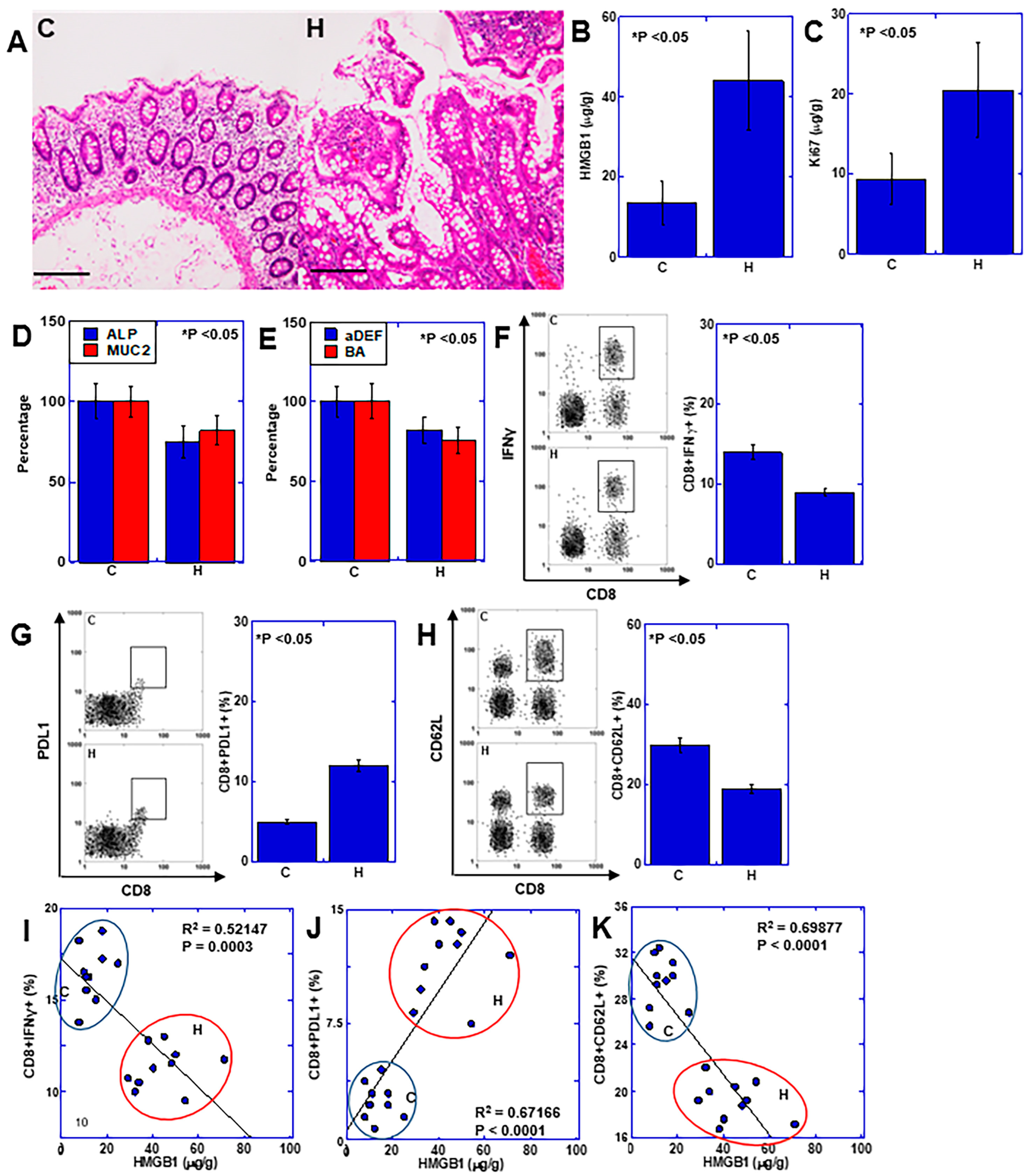
2.5. Role of HMGB1 in Human UC-Associated Carcinogenesis
2.6. Effects of HMGB1 on Hyperplastic Mucosa Surrounding CRC
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Mouse AOM-Induced Colon Carcinogenesis Model
4.3. Mouse DSS-Related Colitis Model
4.4. Colon Specimen Preparation
4.5. Protein Extraction
4.6. Enzyme-Linked Immunosorbent Assay
4.7. Cell Line
4.8. Flow Cytometry
4.9. Spleen Cell Isolation
4.10. Reverse Transcription–Polymerase Chain Reaction (RT-PCR)
4.11. In Vitro Activation of CD8+ Cells
4.12. Patient Information
4.13. Patients with Sporadic CRC
4.14. Patients with UC
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lukas, M. Inflammatory bowel disease as a risk factor for colorectal cancer. Dig. Dis. 2010, 28, 619–624. [Google Scholar] [CrossRef]
- Breynaert, C.; Vermeire, S.; Rutgeerts, P.; Van Assche, G. Dysplasia and colorectal cancer in inflammatory bowel disease: A result of inflammation or an intrinsic risk? Acta Gastroenterol. Belg. 2008, 71, 367–372. [Google Scholar]
- Gyde, S.N.; Prior, P.; Thompson, H.; Waterhouse, J.A.; Allan, R.N. Survival of patients with colorectal cancer complicating ulcerative colitis. Gut 1984, 25, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Altobelli, E.; Latella, G.; Morroni, M.; Licini, C.; Tossetta, G.; Mazzucchelli, R.; Profeta, V.F.; Coletti, G.; Leocata, P.; Castellucci, M.; et al. Low HtrA1 expression in patients with long-standing ulcerative colitis and colorectal cancer. Oncol. Rep. 2017, 38, 418–426. [Google Scholar] [CrossRef]
- Casalegno Garduño, R.; Däbritz, J. New Insights on CD8(+) T Cells in Inflammatory Bowel Disease and Therapeutic Approaches. Front. Immunol. 2021, 12, 738762. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, T. Transcription Factors That Regulate the Pathogenesis of Ulcerative Colitis. Biomed. Res. Int. 2020, 2020, 7402657. [Google Scholar] [CrossRef] [PubMed]
- Corridoni, D.; Antanaviciute, A.; Gupta, T.; Fawkner-Corbett, D.; Aulicino, A.; Jagielowicz, M.; Parikh, K.; Repapi, E.; Taylor, S.; Ishikawa, D.; et al. Single-cell atlas of colonic CD8(+) T cells in ulcerative colitis. Nat. Med. 2020, 26, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Chihara, Y.; Takahashi, T. Co-expression of receptor for advanced glycation end products and the ligand amphoterin associates closely with metastasis of colorectal cancer. Oncol. Rep. 2003, 10, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, H.; Luo, Y.; Kuniyasu, H. Non-histone nuclear factor HMGB1 as a therapeutic target in colorectal cancer. Expert Opin. Ther. Targets 2011, 15, 183–193. [Google Scholar] [CrossRef]
- Luo, Y.; Yoneda, J.; Ohmori, H.; Sasaki, T.; Shimbo, K.; Eto, S.; Kato, Y.; Miyano, H.; Kobayashi, T.; Sasahira, T.; et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res. 2014, 74, 330–340. [Google Scholar] [CrossRef]
- Shimomoto, T.; Luo, Y.; Ohmori, H.; Chihara, Y.; Fujii, K.; Sasahira, T.; Denda, A.; Kuniyasu, H. Advanced glycation end products (AGE) induce the receptor for AGE in the colonic mucosa of azoxymethane-injected Fischer 344 rats fed with a high-linoleic acid and high-glucose diet. J. Gastroenterol. 2012, 47, 1073–1083. [Google Scholar] [CrossRef]
- Muire, P.J.; Schwacha, M.G.; Wenke, J.C. Systemic T Cell Exhaustion Dynamics Is Linked to Early High Mobility Group Box Protein 1 (HMGB1) Driven Hyper-Inflammation in a Polytrauma Rat Model. Cells 2021, 10, 1646. [Google Scholar] [CrossRef]
- Liu, Z.; Falo, L.D., Jr.; You, Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J. Immunol. 2011, 187, 118–125. [Google Scholar] [CrossRef]
- Davé, S.H.; Tilstra, J.S.; Matsuoka, K.; Li, F.; DeMarco, R.A.; Beer-Stolz, D.; Sepulveda, A.R.; Fink, M.P.; Lotze, M.T.; Plevy, S.E. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J. Leukoc. Biol. 2009, 86, 633–643. [Google Scholar] [CrossRef]
- Shen, Z.H.; Zhu, C.X.; Quan, Y.S.; Yang, Z.Y.; Wu, S.; Luo, W.W.; Tan, B.; Wang, X.Y. Relationship between intestinal microbiota and ulcerative colitis: Mechanisms and clinical application of probiotics and fecal microbiota transplantation. World J. Gastroenterol. 2018, 24, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I. Development of ulcerative colitis and its associated colorectal neoplasia as a model of the organ-specific chronic inflammation-carcinoma sequence. Pathol. Int. 2012, 62, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Biscetti, F.; Flex, A.; Alivernini, S.; Tolusso, B.; Gremese, E.; Ferraccioli, G. The Role of High-Mobility Group Box-1 and Its Crosstalk with Microbiome in Rheumatoid Arthritis. Mediators Inflamm. 2017, 2017, 5230374. [Google Scholar] [CrossRef]
- Yao, D.; Dai, W.; Dong, M.; Dai, C.; Wu, S. MUC2 and related bacterial factors: Therapeutic targets for ulcerative colitis. EBioMedicine 2021, 74, 103751. [Google Scholar] [CrossRef]
- Shukla, P.K.; Meena, A.S.; Rao, V.; Rao, R.G.; Balazs, L.; Rao, R. Human Defensin-5 Blocks Ethanol and Colitis-Induced Dysbiosis, Tight Junction Disruption and Inflammation in Mouse Intestine. Sci. Rep. 2018, 8, 16241. [Google Scholar] [CrossRef] [PubMed]
- Gospodarowicz, M.; Wittekind, C.; Sobin, L.H. UICC TNM Classification of Malignant Tumours, 7th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2009. [Google Scholar]
- Japanese Society for Cancer of the Colon and Rectum. Japanese Classification of Colorectal, Appendiceal, and Anal Carcinoma, 3rd ed.; Kanehara & Co. Ltd.: Kyoto, Japan, 2019. [Google Scholar]
- Kuniyasu, H.; Yasui, W.; Shinohara, H.; Yano, S.; Ellis, L.M.; Wilson, M.R.; Bucana, C.D.; Rikita, T.; Tahara, E.; Fidler, I.J. Induction of angiogenesis by hyperplastic colonic mucosa adjacent to colon cancer. Am. J. Pathol. 2000, 157, 1523–1535. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Oue, N.; Shigeishi, H.; Ito, R.; Kato, Y.; Yokozaki, H.; Yasui, W. Prospective study of Ki-67 labeling index in the mucosa adjacent to cancer as a marker for colorectal cancer metastasis. J. Exp. Clin. Cancer Res. 2001, 20, 543–548. [Google Scholar] [PubMed]
- Yu, A.I.; Zhao, L.; Eaton, K.A.; Ho, S.; Chen, J.; Poe, S.; Becker, J.; Gonzalez, A.; McKinstry, D.; Hasso, M.; et al. Gut Microbiota Modulate CD8 T Cell Responses to Influence Colitis-Associated Tumorigenesis. Cell Rep. 2020, 31, 107471. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, V.V.; Shmagel, K.V.T. Lymphocyte Metabolic Features and Techniques to Modulate Them. Biochemistry 2023, 88, 1857–1873. [Google Scholar] [PubMed]
- Chen, H.; Yang, T.; Zhu, L.; Zhao, Y. Cellular metabolism on T-cell development and function. Int. Rev. Immunol. 2015, 34, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, H.; Zhang, D.; Ma, Y.; Zhu, B. Metabolic plasticity and regulation of T cell exhaustion. Immunology 2022, 167, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- de Lima, M.H.F.; Machado, C.C.; Nascimento, D.C.; Silva, C.M.S.; Toller-Kawahisa, J.E.; Rodrigues, T.S.; Veras, F.P.; Pontelli, M.C.; Castro, I.A.; Zamboni, D.S.; et al. The TIGIT(+) T regulatory cells subset associates with nosocomial infection and fatal outcome in COVID-19 patients under mechanical ventilation. Sci. Rep. 2023, 13, 13599. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.J.; Murray, G.I.; Hapca, S.; Hay, A.; Craig, S.G.; Humphries, M.P.; James, J.A.; Salto-Tellez, M.; Brice, D.P.; Berry, S.H.; et al. Subcellular Epithelial HMGB1 Expression Is Associated with Colorectal Neoplastic Progression, Male Sex, Mismatch Repair Protein Expression, Lymph Node Positivity, and an ‘Immune Cold’ Phenotype Associated with Poor Survival. Cancers 2023, 15, 1865. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, T.; An, N.; Sun, Y.; Xu, P.; Han, P.; Zhao, Y.; Wang, L.; Ni, X.; Li, Y.; et al. Enhancing regulatory T-cell function via inhibition of high mobility group box 1 protein signaling in immune thrombocytopenia. Haematologica 2023, 108, 843–858. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, P.; Liu, X.; Liu, H.; Wang, J.; Wang, J.; Xiao, J.; Nie, D.; Ma, L. HMGB1 is increased in patients with immune thrombocytopenia and negatively associates with Tregs. Thromb. Res. 2022, 213, 128–136. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Jin, H.; Xu, Y. T Cell Metabolism: A New Perspective on Th17/Treg Cell Imbalance in Systemic Lupus Erythematosus. Front. Immunol. 2020, 11, 1027. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Li, N.; Gao, G.; He, Q.; Kwok, L.Y.; Zhang, H. Dynamic Changes of the Gut Microbiota and Its Functional Metagenomic Potential during the Development of Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2024, 25, 3768. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Hesse, L.; Wu, X.; Allam, V.; van Oldeniel, D.; Bhiekharie, L.J.; Phipps, S.; Oliver, B.G.; Gosens, R.; Sukkar, M.B.; et al. LL-37 and HMGB1 induce alveolar damage and reduce lung tissue regeneration via RAGE. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L641–L652. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.R.; Song, F.; Young, M.A.; Hassan, N.; Antoine, D.J.; Gemici, N.B.; Clarke, A.R.; Jenkins, J.R. Secreted HMGB1 from Wnt activated intestinal cells is required to maintain a crypt progenitor phenotype. Oncotarget 2016, 7, 51665–51673. [Google Scholar] [CrossRef] [PubMed]
- Eymael, J.; van den Broek, M.; Miesen, L.; Monge, V.V.; van den Berge, B.T.; Mooren, F.; Velez, V.L.; Dijkstra, J.; Hermsen, M.; Bándi, P.; et al. Human scattered tubular cells represent a heterogeneous population of glycolytic dedifferentiated proximal tubule cells. J. Pathol. 2023, 259, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Gómez, H. Reprogramming Metabolism to Enhance Kidney Tolerance during Sepsis: The Role of Fatty Acid Oxidation, Aerobic Glycolysis, and Epithelial De-Differentiation. Nephron 2023, 147, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Li, J.Y.; Xu, J.H.; Xia, Z.S.; Cheng, D.; Zhong, W.; Lai, Y.; Yu, T.; Chen, Q.K. Butyrate suppresses abnormal proliferation in colonic epithelial cells under diabetic state by targeting HMGB1. J. Pharmacol. Sci. 2019, 139, 266–274. [Google Scholar] [CrossRef]
- Jackson, D.N.; Theiss, A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 2020, 11, 285–304. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, D.; Sun, L. Clinical Significance of High-Mobility Group Box 1 Protein (HMGB1) and Nod-Like Receptor Protein 3 (NLRP3) in Patients with Ulcerative Colitis. Med. Sci. Monit. 2020, 26, e919530. [Google Scholar] [CrossRef]
- Nakov, R. New markers in ulcerative colitis. Clin. Chim. Acta 2019, 497, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Palone, F.; Vitali, R.; Cucchiara, S.; Pierdomenico, M.; Negroni, A.; Aloi, M.; Nuti, F.; Felice, C.; Armuzzi, A.; Stronati, L. Role of HMGB1 as a suitable biomarker of subclinical intestinal inflammation and mucosal healing in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Vitali, R.; Palone, F.; Cucchiara, S.; Negroni, A.; Cavone, L.; Costanzo, M.; Aloi, M.; Dilillo, A.; Stronati, L. Dipotassium Glycyrrhizate Inhibits HMGB1-Dependent Inflammation and Ameliorates Colitis in Mice. PLoS ONE 2013, 8, e66527. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case# | Sex | Age | Disease | Subtype | Cancer | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Duration (1) | Site (2) | Histology (3) | pT (2) | pN (2) | M (2) | pStage (2) | ||||
| 1 | F | 38 | 16 | Pancolitis | S | tub1 | pT3 | pN0 | M0 | IIA |
| 2 | M | 44 | 24 | Pancolitis | R | tub1 | pT3 | pN0 | M0 | IIA |
| 3 | F | 41 | 20 | Left colon-rectal | S | tub2 | pT3 | pN1 | M0 | IIB |
| 4 | F | 50 | 28 | Pancolitis | S | tub2 | pT2 | pN0 | M0 | I |
| 5 | M | 39 | 18 | Pancolitis | R | tub2 | pT3 | pN0 | M0 | IIA |
| Case# | Sex | Age | Site (1) | Histology (2) | pT (1) | pN (1) | M (1) | pStage (1) |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 94 | A | por | pT3 | pN1 | M0 | IIIB |
| 2 | M | 66 | S | tub2 | pT4b | pN2 | M0 | IIIC |
| 3 | M | 71 | S | tub2 | pT4b | pN1 | M0 | IIIC |
| 4 | F | 83 | A | tub1 | pT4a | pN1 | M0 | IIIB |
| 5 | F | 61 | C | tub2 | pT4a | pN1 | M0 | IIIB |
| 6 | M | 67 | R | muc | pT4a | pN1 | M0 | IIIB |
| 7 | F | 62 | S | tub2 | pT3 | pN1 | M0 | IIIB |
| 8 | F | 86 | T | tub1 | pT3 | pN1 | M0 | IIIB |
| 9 | M | 59 | S | tub2 | pT3 | pN1 | M0 | IIIB |
| 10 | F | 88 | S | tub1 | pT3 | pN1 | M0 | IIIB |
| Target | Species | Cat# | Company |
|---|---|---|---|
| Ki67 | mouse | FY-EM8749 | Axel, Osaka, Japan |
| Ki67 | human | ab253221 | Abcam, Waltham, MA, USA |
| ALP | mouse | ab285274 | Abcam, Waltham, MA, USA |
| ALP | human | ab285149 | Abcam, Waltham, MA, USA |
| MUC2 | mouse | M0EB0548 | AssayGenie, Dublin, Ireland |
| MUC2 | human | ab282871 | Abcam, Waltham, MA, USA |
| αDefensin | mouse | EK11275 | Signalway Antibody, Greenbelt, MA, USA |
| αDefensin | human | EL006657HU | Cusabio, Houston, TX, USA |
| Butyric acid | - | abx258338 | Abbexa Ltd. Cambridge, UK |
| Antibody | Species | Cat# | Company |
|---|---|---|---|
| CD8 | Mouse | Cat #11-0081-82 | Thermo Fisher, Tokyo, Japan |
| CD8 | Human | Cat #12-0088-42 | Thermo Fisher, Tokyo, Japan |
| FOXP3 | Mouse | #700914 | Thermo Fisher, Tokyo, Japan |
| FOXP3 | Human | #700914 | Thermo Fisher, Tokyo, Japan |
| PDL1 | Mouse | Cat #14-5982-82 | Thermo Fisher, Tokyo, Japan |
| PDL1 | Human | #14-5983-82 | Thermo Fisher, Tokyo, Japan |
| IFNγ | Mouse | #47-7311-82 | Thermo Fisher, Tokyo, Japan |
| IFNγ | Human | #53-7319-42 | Thermo Fisher, Tokyo, Japan |
| CD62L | Mouse | #12-0621-82 | Thermo Fisher, Tokyo, Japan |
| CD62L | Human | ab317478 | Abcam, Waltham, MA, USA |
| Gene | Species | ID | Upper | Lower |
|---|---|---|---|---|
| ACTB | Mouse | NM_007393.5 | agccatgtacgtagccatcc | ctctcagctgtggtggtgaa |
| RAGE | Mouse | L33412.1 | aattgtggatcctgcctctg | aaggtaggatgggtggttcc |
| TLR4 | Mouse | NM_021297.3 | gcagcacctggattttcagc | ccttgacccactgcaggaat |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasaki, T.; Fujiwara-Tani, R.; Luo, Y.; Ogata, R.; Sasaki, R.; Ikemoto, A.; Nishiguchi, Y.; Nakashima, C.; Kishi, S.; Fujii, K.; et al. Effects of High-Mobility Group Box-1 on Mucosal Immunity and Epithelial Differentiation in Colitic Carcinoma. Int. J. Mol. Sci. 2024, 25, 6846. https://doi.org/10.3390/ijms25136846
Sasaki T, Fujiwara-Tani R, Luo Y, Ogata R, Sasaki R, Ikemoto A, Nishiguchi Y, Nakashima C, Kishi S, Fujii K, et al. Effects of High-Mobility Group Box-1 on Mucosal Immunity and Epithelial Differentiation in Colitic Carcinoma. International Journal of Molecular Sciences. 2024; 25(13):6846. https://doi.org/10.3390/ijms25136846
Chicago/Turabian StyleSasaki, Takamitsu, Rina Fujiwara-Tani, Yi Luo, Ruiko Ogata, Rika Sasaki, Ayaka Ikemoto, Yukiko Nishiguchi, Chie Nakashima, Shingo Kishi, Kiyomu Fujii, and et al. 2024. "Effects of High-Mobility Group Box-1 on Mucosal Immunity and Epithelial Differentiation in Colitic Carcinoma" International Journal of Molecular Sciences 25, no. 13: 6846. https://doi.org/10.3390/ijms25136846