
Comprehensive Analysis of the Function and Prognostic Value of TAS2Rs Family-Related Genes in Colon Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Differential Expression and Prognostic Value Analysis of TAS2Rs Gene Family in Colon Cancer
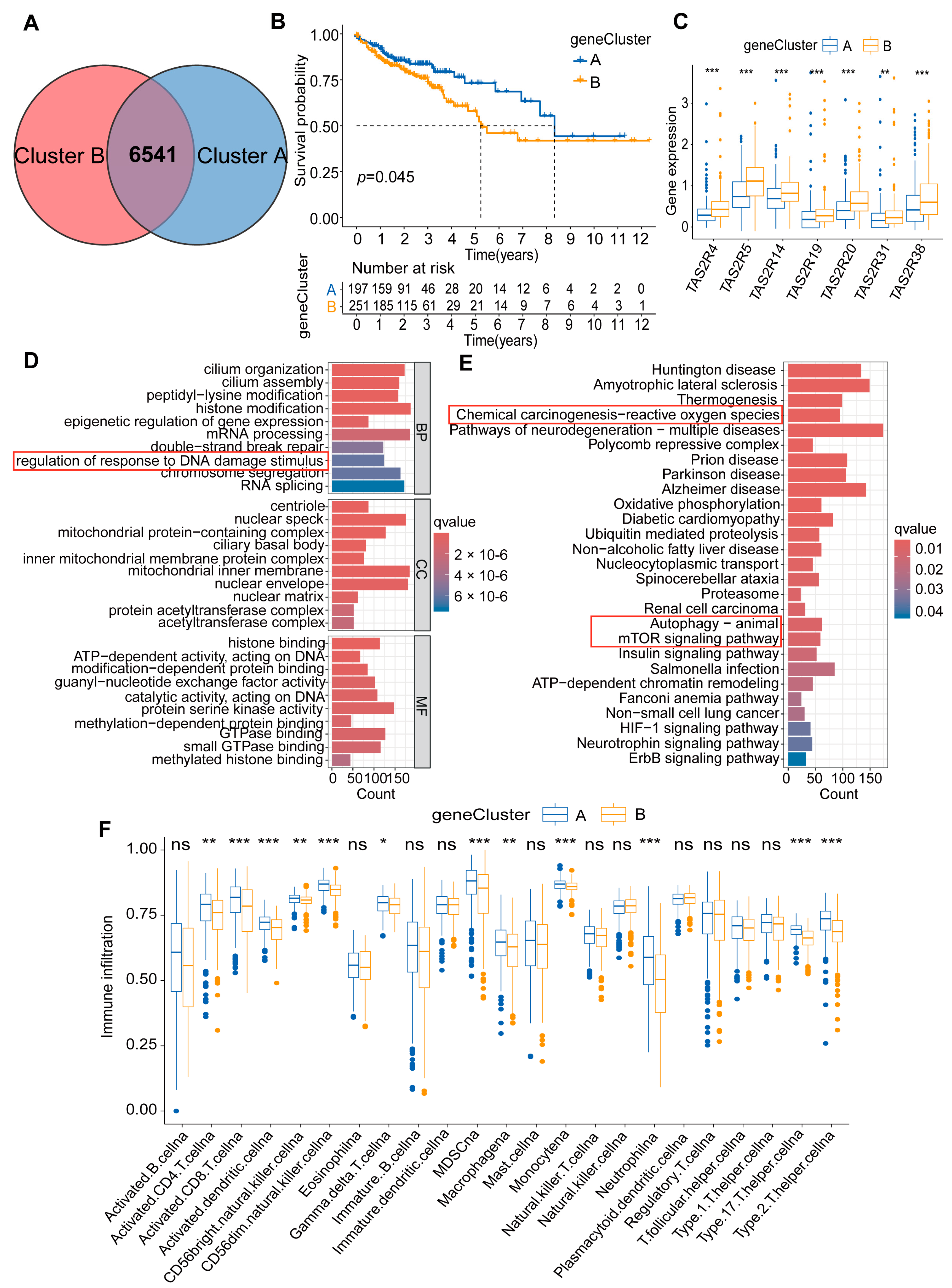
2.2. Constructing Colon Cancer Molecular Subtypes Based on Seven TAS2Rs Prognostic Genes
2.3. Immune Characteristics of Two Molecular Subtypes of Colon Cancer
2.4. Generation and Function of DEGs in Two Molecular Subtypes of Colon Cancer
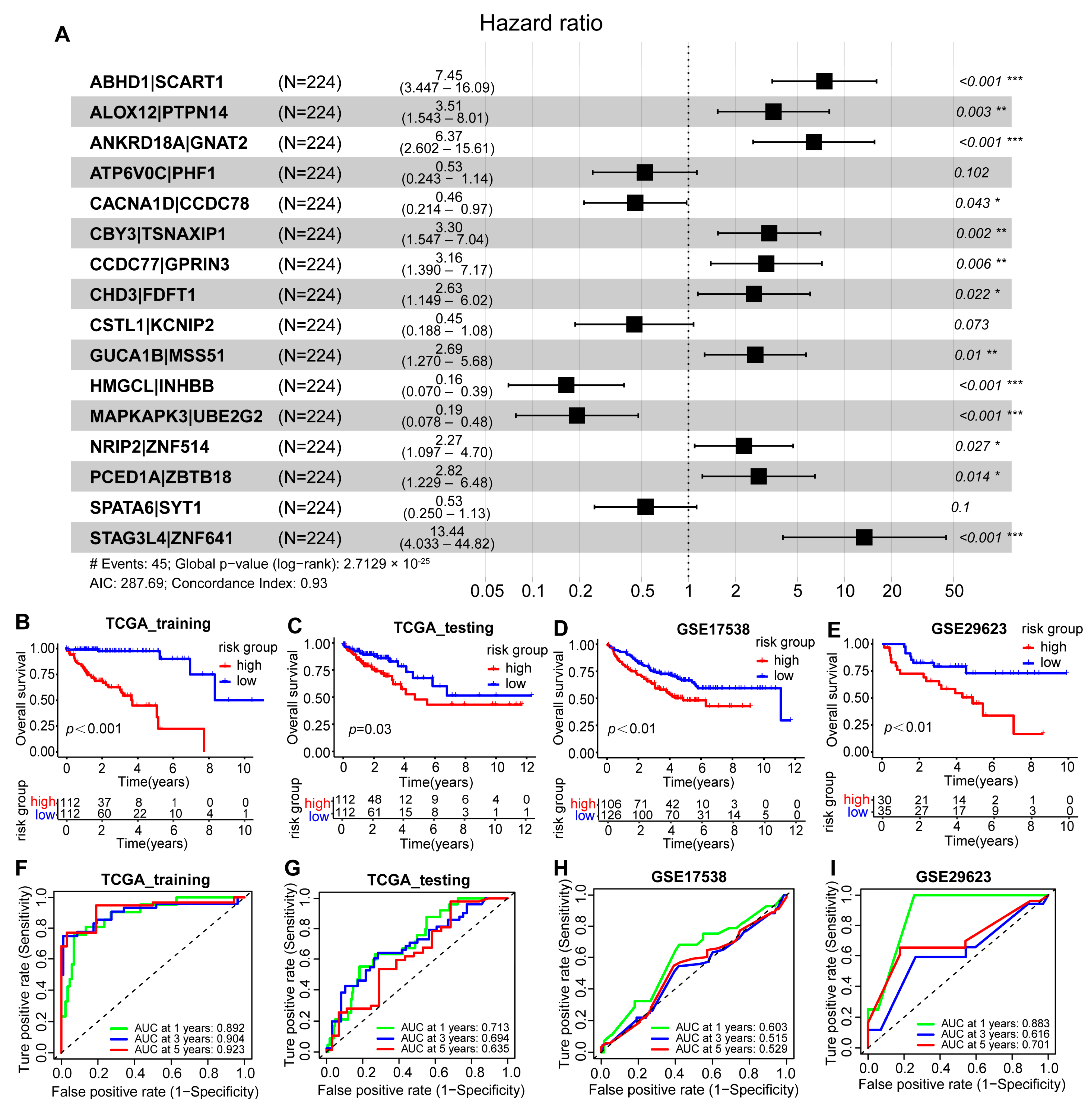
2.5. Construction and Validation of the Scoring Model Based on Two Molecular Subtypes of Colon Cancer
ANKRD18A|GNAT2 + 0.526 × ATP6V0C|PHF1 + 0.457 × CACNA1D|CCDC78 +
3.300 × CBY3|TSNAXIP1 + 3.158 × CCDC77|GPRIN3 + 2.630 × CHD3|FDFT1 +
0.450 × CSTL1|KCNIP2 + 2.686 × GUCA1B|MSS51 + 0.165 × HMGCL|IHBB +
0.193 × MAKAPK3|UBE2G2 + 2.270 × NRIP2|ZNF514 + 2.822 × PCED1A|ZBTB18 +
0.531 × SPATA6|SYT1 + 13.444 × STAG3L4|ZNF641
2.6. Construction and Verification of the GBM Scoring Model Nomogram
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Human Colon Cancer Tissue Samples
4.3. Real-Time PCR (qPCR)
4.4. Source and Pretreatment of Colon Cancer Datasets
4.5. Unsupervised Clustering of Seven TAS2Rs Prognostic Genes
4.6. PD-1/CTLA4 Treatment Response
4.7. Single Sample Gene-Set Enrichment Analysis (ssGSEA)
4.8. Identification of DEGs in the Two Subtypes of Colon Cancer
4.9. Functional Enrichment Analysis of DEGs
4.10. Gene-Set Enrichment Analysis (GSVA)
4.11. Construction of Prognostic Models
- Characteristic: “Gene A|Gene B”
- = {1, Expression(A) > Expression(B);
- 0, Expression(A) ≤ Expression(B)}
4.12. Validation of Prognostic Models
4.13. Construction and Validation of Nomograms
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Global cancer burden growing, amidst mounting need for services. Saudi Med. J. 2024, 45, 326–327.
- Cheng, E.; Ou, F.S.; Ma, C.; Spiegelman, D.; Zhang, S.; Zhou, X.; Bainter, T.M.; Saltz, L.B.; Niedzwiecki, D.; Mayer, R.J.; et al. Diet- and Lifestyle-Based Prediction Models to Estimate Cancer Recurrence and Death in Patients with Stage III Colon Cancer (CALGB 89803/Alliance). J. Clin. Oncol. 2022, 40, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Ciardiello, D.; Martini, G.; Napolitano, S.; Tabernero, J.; Cervantes, A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J. Clin. 2022, 72, 372–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rajput, A.; Jin, N.; Wang, J. Mechanisms of Immunosuppression in Colorectal Cancer. Cancers 2020, 12, 3850. [Google Scholar] [CrossRef] [PubMed]
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Lao, Y.; Yu, W.; Zhang, X.; Jiang, M.; Zhu, C. Progress in the Application of Immune Checkpoint Inhibitor-Based Immunotherapy for Targeting Different Types of Colorectal Cancer. Front. Oncol. 2021, 11, 764618. [Google Scholar] [CrossRef] [PubMed]
- Bortolomeazzi, M.; Keddar, M.R.; Montorsi, L.; Acha-Sagredo, A.; Benedetti, L.; Temelkovski, D.; Choi, S.; Petrov, N.; Todd, K.; Wai, P.; et al. Immunogenomics of Colorectal Cancer Response to Checkpoint Blockade: Analysis of the KEYNOTE 177 Trial and Validation Cohorts. Gastroenterology 2021, 161, 1179–1193. [Google Scholar] [CrossRef] [PubMed]
- Kanikarla Marie, P.; Haymaker, C.; Parra, E.R.; Kim, Y.U.; Lazcano, R.; Gite, S.; Lorenzini, D.; Wistuba, I., II; Tidwell, R.S.S.; Song, X.; et al. Pilot Clinical Trial of Perioperative Durvalumab and Tremelimumab in the Treatment of Resectable Colorectal Cancer Liver Metastases. Clin. Cancer Res. 2021, 27, 3039–3049. [Google Scholar] [CrossRef]
- de Melo Gagliato, D.; Cortes, J.; Curigliano, G.; Loi, S.; Denkert, C.; Perez-Garcia, J.; Holgado, E. Tumor-infiltrating lymphocytes in Breast Cancer and implications for clinical practice. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 527–537. [Google Scholar] [CrossRef]
- Global, regional, and national burden of colorectal cancer and its risk factors, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 627–647. [CrossRef]
- Lambert, J.D.; VanDusen, S.R.; Cockroft, J.E.; Smith, E.C.; Greenwood, D.C.; Cade, J.E. Bitter taste sensitivity, food intake, and risk of malignant cancer in the UK Women’s Cohort Study. Eur. J. Nutr. 2019, 58, 2111–2121. [Google Scholar] [CrossRef]
- Xu, W.; Wu, L.; Liu, S.; Liu, X.; Cao, X.; Zhou, C.; Zhang, J.; Fu, Y.; Guo, Y.; Wu, Y.; et al. Structural basis for strychnine activation of human bitter taste receptor TAS2R46. Science 2022, 377, 1298–1304. [Google Scholar] [CrossRef]
- Yamaki, M.; Saito, H.; Isono, K.; Goto, T.; Shirakawa, H.; Shoji, N.; Satoh-Kuriwada, S.; Sasano, T.; Okada, R.; Kudoh, K.; et al. Genotyping Analysis of Bitter-Taste Receptor Genes TAS2R38 and TAS2R46 in Japanese Patients with Gastrointestinal Cancers. J. Nutr. Sci. Vitaminol. 2017, 63, 148–154. [Google Scholar] [CrossRef]
- Singh, N.; Chakraborty, R.; Bhullar, R.P.; Chelikani, P. Differential expression of bitter taste receptors in non-cancerous breast epithelial and breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 499–503. [Google Scholar] [CrossRef]
- Kwatra, D.; Venugopal, A.; Standing, D.; Ponnurangam, S.; Dhar, A.; Mitra, A.; Anant, S. Bitter melon extracts enhance the activity of chemotherapeutic agents through the modulation of multiple drug resistance. J. Pharm. Sci. 2013, 102, 4444–4454. [Google Scholar] [CrossRef]
- Lamy, V.; Roussi, S.; Chaabi, M.; Gossé, F.; Schall, N.; Lobstein, A.; Raul, F. Chemopreventive effects of lupulone, a hop {beta}-acid, on human colon cancer-derived metastatic SW620 cells and in a rat model of colon carcinogenesis. Carcinogenesis 2007, 28, 1575–1581. [Google Scholar] [CrossRef]
- Jeruzal-Świątecka, J.; Fendler, W.; Pietruszewska, W. Clinical Role of Extraoral Bitter Taste Receptors. Int. J. Mol. Sci. 2020, 21, 5156. [Google Scholar] [CrossRef]
- Behrens, M.; Meyerhof, W. Gustatory and extragustatory functions of mammalian taste receptors. Physiol. Behav. 2011, 105, 4–13. [Google Scholar] [CrossRef]
- Wu, S.V.; Rozengurt, N.; Yang, M.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Expression of bitter taste receptors of the T2R family in the gastrointestinal tract and enteroendocrine STC-1 cells. Proc. Natl. Acad. Sci. USA 2002, 99, 2392–2397. [Google Scholar] [CrossRef] [PubMed]
- Talmon, M.; Pollastro, F.; Fresu, L.G. The Complex Journey of the Calcium Regulation Downstream of TAS2R Activation. Cells 2022, 11, 3638. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.T.; Herz, C.; Ruf, P.; Stetter, R.; Lamy, E. Human T2R38 Bitter Taste Receptor Expression in Resting and Activated Lymphocytes. Front. Immunol. 2018, 9, 2949. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Xu, Y.; Cao, Z. Absinthin attenuates LPS-induced ALI through MIP-1α-mediated inflammatory cell infiltration. Exp. Lung Res. 2015, 41, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Panebra, A.; Pera, T.; Tiegs, B.C.; Hershfeld, A.; Kenyon, L.C.; Deshpande, D.A. Antimitogenic effect of bitter taste receptor agonists on airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L365–L376. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lee, J.; Choi, I.J.; Kim, Y.W.; Ryu, K.W.; Kim, J. Genetic Variation in the TAS2R38 Bitter Taste Receptor and Gastric Cancer Risk in Koreans. Sci. Rep. 2016, 6, 26904. [Google Scholar] [CrossRef]
- Barontini, J.; Antinucci, M.; Tofanelli, S.; Cammalleri, M.; Dal Monte, M.; Gemignani, F.; Vodicka, P.; Marangoni, R.; Vodickova, L.; Kupcinskas, J.; et al. Association between polymorphisms of TAS2R16 and susceptibility to colorectal cancer. BMC Gastroenterol. 2017, 17, 104. [Google Scholar] [CrossRef]
- Wang, P.; Li, Y.; Reddy, C.K. Machine Learning for Survival Analysis: A Survey. ACM Comput. Surv. 2019, 51, 110. [Google Scholar] [CrossRef]
- Abbasi, B.; Goldenholz, D.M. Machine learning applications in epilepsy. Epilepsia 2019, 60, 2037–2047. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef]
- Shu, J.; Li, Y.; Wang, S.; Xi, B.; Ma, J. Disease gene prediction with privileged information and heteroscedastic dropout. Bioinformatics 2021, 37, i410–i417. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.W.; Sun, J.; Wang, Y.; Chen, C.P.; Tao, T.; Ma, M.; Chen, X.; Zhang, X.N.; Yang, L.Y.; Zhang, Z.L.; et al. Tas2R activation relaxes airway smooth muscle by release of Gα(t) targeting on AChR signaling. Proc. Natl. Acad. Sci. USA 2022, 119, e2121513119. [Google Scholar] [CrossRef] [PubMed]
- Delon, C.; Brown, K.F.; Payne, N.W.S.; Kotrotsios, Y.; Vernon, S.; Shelton, J. Differences in cancer incidence by broad ethnic group in England, 2013-2017. Br. J. Cancer 2022, 126, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Fwelo, P.; Afolayan, O.; Nwosu, K.O.S.; Ojaruega, A.A.; Ahaiwe, O.; Olateju, O.A.; Ezeigwe, O.J.; Adekunle, T.E.; Bangolo, A. Racial and ethnic differences in colon cancer surgery type performed and delayed treatment among people 45 years old and older in the USA between 2007 and 2017: Mediating effect on survival. Surg. Oncol. 2023, 50, 101983. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tu, Y.X.; Chen, L.; Zhang, Y.; Pan, X.L.; Yang, S.Q.; Zhang, S.J.; Li, S.H.; Yu, K.C.; Song, S.; et al. Male-Biased Gut Microbiome and Metabolites Aggravate Colorectal Cancer Development. Adv. Sci. 2023, 10, e2206238. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Jyotaki, M.; Quinlan, J.; Feng, S.; Zhou, M.; Jiang, P.; Matsumoto, I.; Huang, L.; Ninomiya, Y.; Margolskee, R.F.; et al. Lipopolysaccharide increases bitter taste sensitivity via epigenetic changes in Tas2r gene clusters. iScience 2023, 26, 106920. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhu, J.; Kong, W.; Li, P.; Zhu, S. Expression and Prognostic Characteristics of m6A RNA Methylation Regulators in Colon Cancer. Int. J. Mol. Sci. 2021, 22, 2134. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Liu, X.; Li, M.; Zhao, Y.; Li, J.; Wen, Z.; Liu, M.; Yang, M.; Fu, B.; Wei, M. Analysis of 5-Methylcytosine Regulators and DNA Methylation-Driven Genes in Colon Cancer. Front. Cell Dev. Biol. 2021, 9, 657092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Gerard, C.L.; Delyon, J.; Wicky, A.; Homicsko, K.; Cuendet, M.A.; Michielin, O. Turning tumors from cold to inflamed to improve immunotherapy response. Cancer Treat. Rev. 2021, 101, 102227. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Brea, E.J.; Oh, C.Y.; Manchado, E.; Budhu, S.; Gejman, R.S.; Mo, G.; Mondello, P.; Han, J.E.; Jarvis, C.A.; Ulmert, D.; et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol. Res. 2016, 4, 936–947. [Google Scholar] [CrossRef]
- Kruse, B.; Buzzai, A.C.; Shridhar, N.; Braun, A.D.; Gellert, S.; Knauth, K.; Pozniak, J.; Peters, J.; Dittmann, P.; Mengoni, M.; et al. CD4(+) T cell-induced inflammatory cell death controls immune-evasive tumours. Nature 2023, 618, 1033–1040. [Google Scholar] [CrossRef]
- Demler, O.V.; Pencina, M.J.; D’Agostino, R.B., Sr. Misuse of DeLong test to compare AUCs for nested models. Stat. Med. 2012, 31, 2577–2587. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Gupta, P.; Chiang, S.F.; Sahoo, P.K.; Mohapatra, S.K.; You, J.F.; Onthoni, D.D.; Hung, H.Y.; Chiang, J.M.; Huang, Y.; Tsai, W.S. Prediction of Colon Cancer Stages and Survival Period with Machine Learning Approach. Cancers 2019, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Ben Hamida, A.; Devanne, M.; Weber, J.; Truntzer, C.; Derangère, V.; Ghiringhelli, F.; Forestier, G.; Wemmert, C. Deep learning for colon cancer histopathological images analysis. Comput. Biol. Med. 2021, 136, 104730. [Google Scholar] [CrossRef]
- Taieb, J.; Le Malicot, K.; Shi, Q.; Penault-Llorca, F.; Bouché, O.; Tabernero, J.; Mini, E.; Goldberg, R.M.; Folprecht, G.; Luc Van Laethem, J.; et al. Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. J. Natl. Cancer Inst. 2017, 109, djw272. [Google Scholar] [CrossRef] [PubMed]
- Westcott, P.M.K.; Muyas, F.; Hauck, H.; Smith, O.C.; Sacks, N.J.; Ely, Z.A.; Jaeger, A.M.; Rideout, W.M., 3rd; Zhang, D.; Bhutkar, A.; et al. Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity. Nat. Genet. 2023, 55, 1686–1695. [Google Scholar] [CrossRef]
- Carrai, M.; Steinke, V.; Vodicka, P.; Pardini, B.; Rahner, N.; Holinski-Feder, E.; Morak, M.; Schackert, H.K.; Görgens, H.; Stemmler, S.; et al. Association between TAS2R38 gene polymorphisms and colorectal cancer risk: A case-control study in two independent populations of Caucasian origin. PLoS ONE 2011, 6, e20464. [Google Scholar] [CrossRef]
- Singh, N.; Shaik, F.A.; Myal, Y.; Chelikani, P. Chemosensory bitter taste receptors T2R4 and T2R14 activation attenuates proliferation and migration of breast cancer cells. Mol. Cell Biochem. 2020, 465, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Vobugari, N.; Raja, V.; Sethi, U.; Gandhi, K.; Raja, K.; Surani, S.R. Advancements in Oncology with Artificial Intelligence—A Review Article. Cancers 2022, 14, 1349. [Google Scholar] [CrossRef] [PubMed]
- Mitry, E.; Guiu, B.; Cosconea, S.; Jooste, V.; Faivre, J.; Bouvier, A.M. Epidemiology, management and prognosis of colorectal cancer with lung metastases: A 30-year population-based study. Gut 2010, 59, 1383–1388. [Google Scholar] [CrossRef]
- Wei, R.; Wang, J.; Wang, X.; Xie, G.; Wang, Y.; Zhang, H.; Peng, C.Y.; Rajani, C.; Kwee, S.; Liu, P.; et al. Clinical prediction of HBV and HCV related hepatic fibrosis using machine learning. EBioMedicine 2018, 35, 124–132. [Google Scholar] [CrossRef]
- Li, X.; Yang, L.; Yuan, Z.; Lou, J.; Fan, Y.; Shi, A.; Huang, J.; Zhao, M.; Wu, Y. Multi-institutional development and external validation of machine learning-based models to predict relapse risk of pancreatic ductal adenocarcinoma after radical resection. J. Transl. Med. 2021, 19, 281. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m (6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006, 34, D322–D326. [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Bao, J.; Zhan, M.; Wang, J.; Li, X.; Gu, X.; Song, S.; Yang, Q.; Liu, Y.; Wang, Z.; et al. Comprehensive Analysis of m5C Methylation Regulatory Genes and Tumor Microenvironment in Prostate Cancer. Front. Immunol. 2022, 13, 914577. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Kong, W.; Zhu, J.; Huang, L.; Wang, S.; Bi, S.; Xie, Z. The genetic algorithm-aided three-stage ensemble learning method identified a robust survival risk score in patients with glioma. Brief. Bioinform. 2022, 23, bbac344. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, S.; Zhu, J.; Huang, L.; Feng, W.; Peng, L.; Leng, L.; Wang, Y.; Shan, P.; Kong, W.; Zhu, S. Comprehensive Analysis of the Function and Prognostic Value of TAS2Rs Family-Related Genes in Colon Cancer. Int. J. Mol. Sci. 2024, 25, 6849. https://doi.org/10.3390/ijms25136849
Bi S, Zhu J, Huang L, Feng W, Peng L, Leng L, Wang Y, Shan P, Kong W, Zhu S. Comprehensive Analysis of the Function and Prognostic Value of TAS2Rs Family-Related Genes in Colon Cancer. International Journal of Molecular Sciences. 2024; 25(13):6849. https://doi.org/10.3390/ijms25136849
Chicago/Turabian StyleBi, Suzhen, Jie Zhu, Liting Huang, Wanting Feng, Lulu Peng, Liangqi Leng, Yin Wang, Peipei Shan, Weikaixin Kong, and Sujie Zhu. 2024. "Comprehensive Analysis of the Function and Prognostic Value of TAS2Rs Family-Related Genes in Colon Cancer" International Journal of Molecular Sciences 25, no. 13: 6849. https://doi.org/10.3390/ijms25136849