
Identifying Core Genes Related to Low-Temperature Stress Resistance in Quinoa Seedlings Based on WGCNA

Abstract
1. Introduction
2. Results
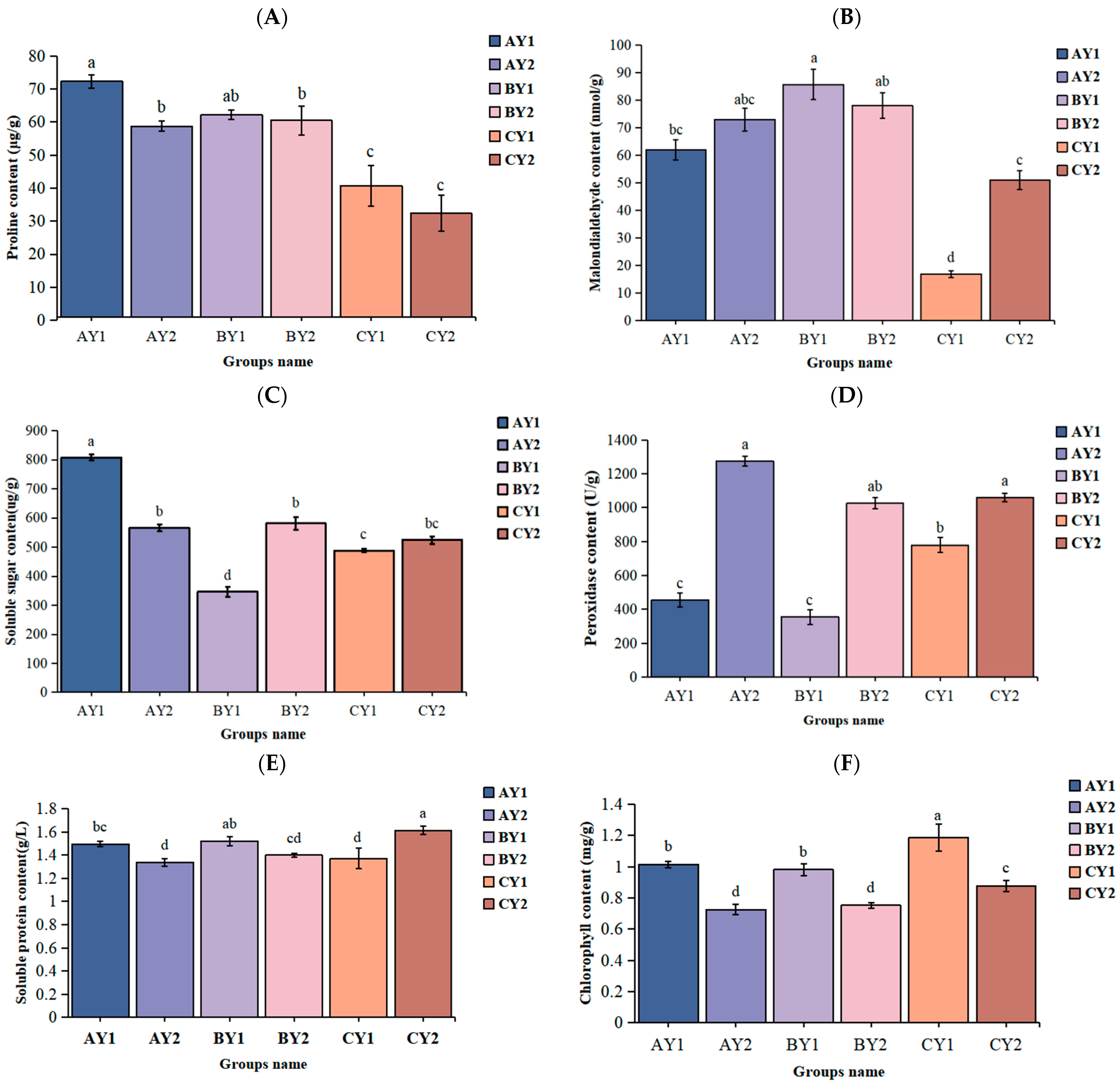
2.1. Physiological Changes in Quinoa Seedlings under Low-Temperature Stress
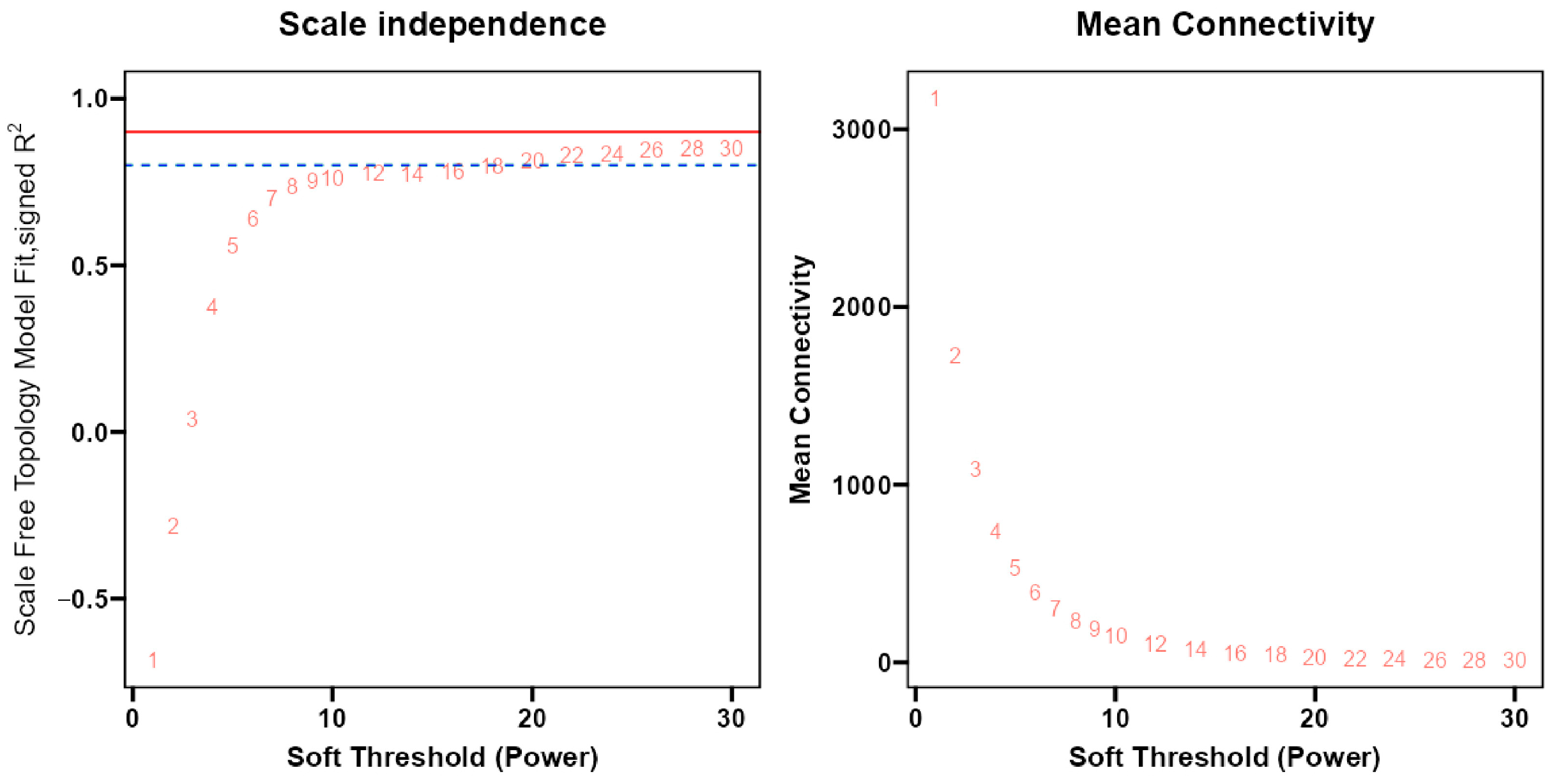
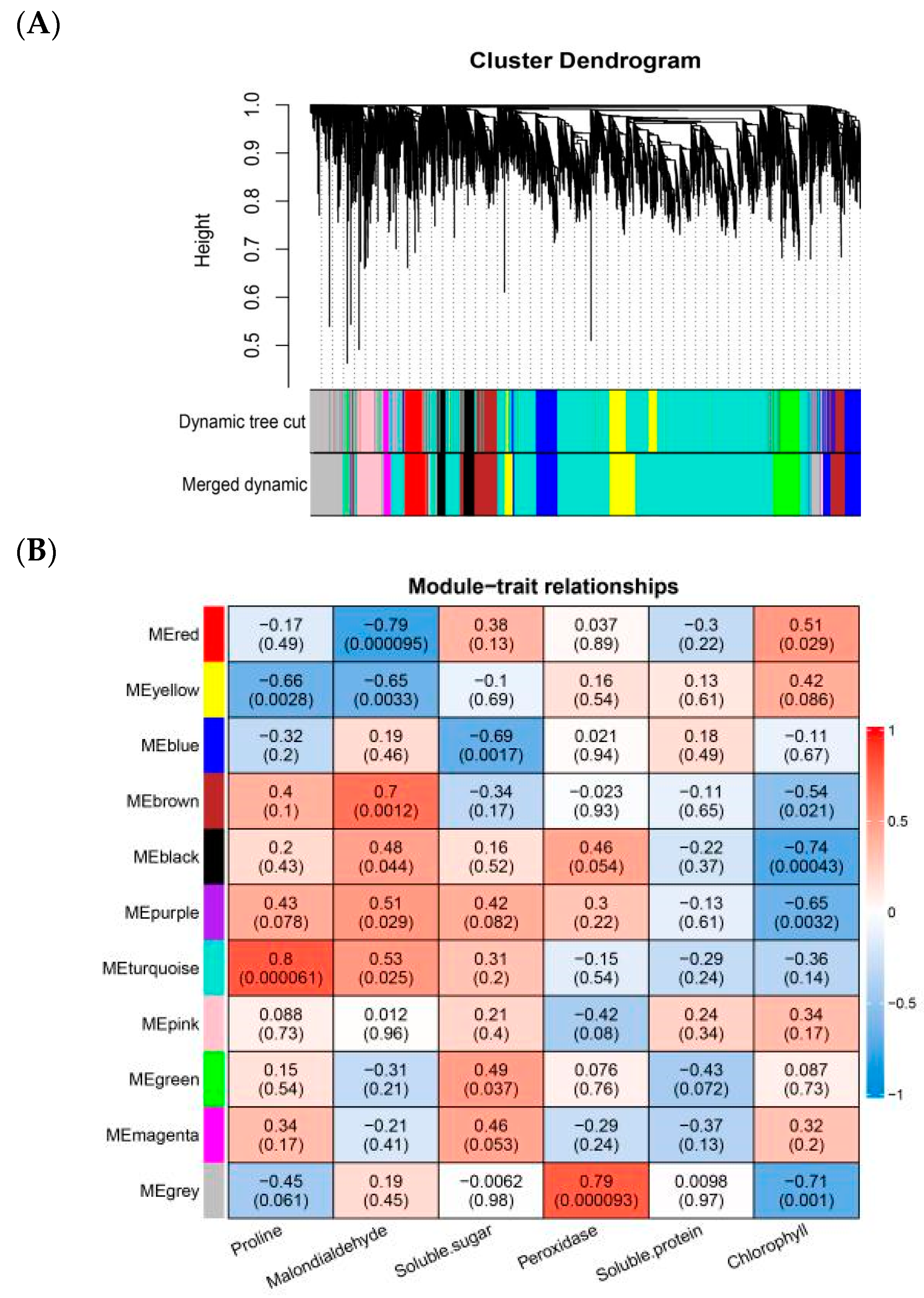
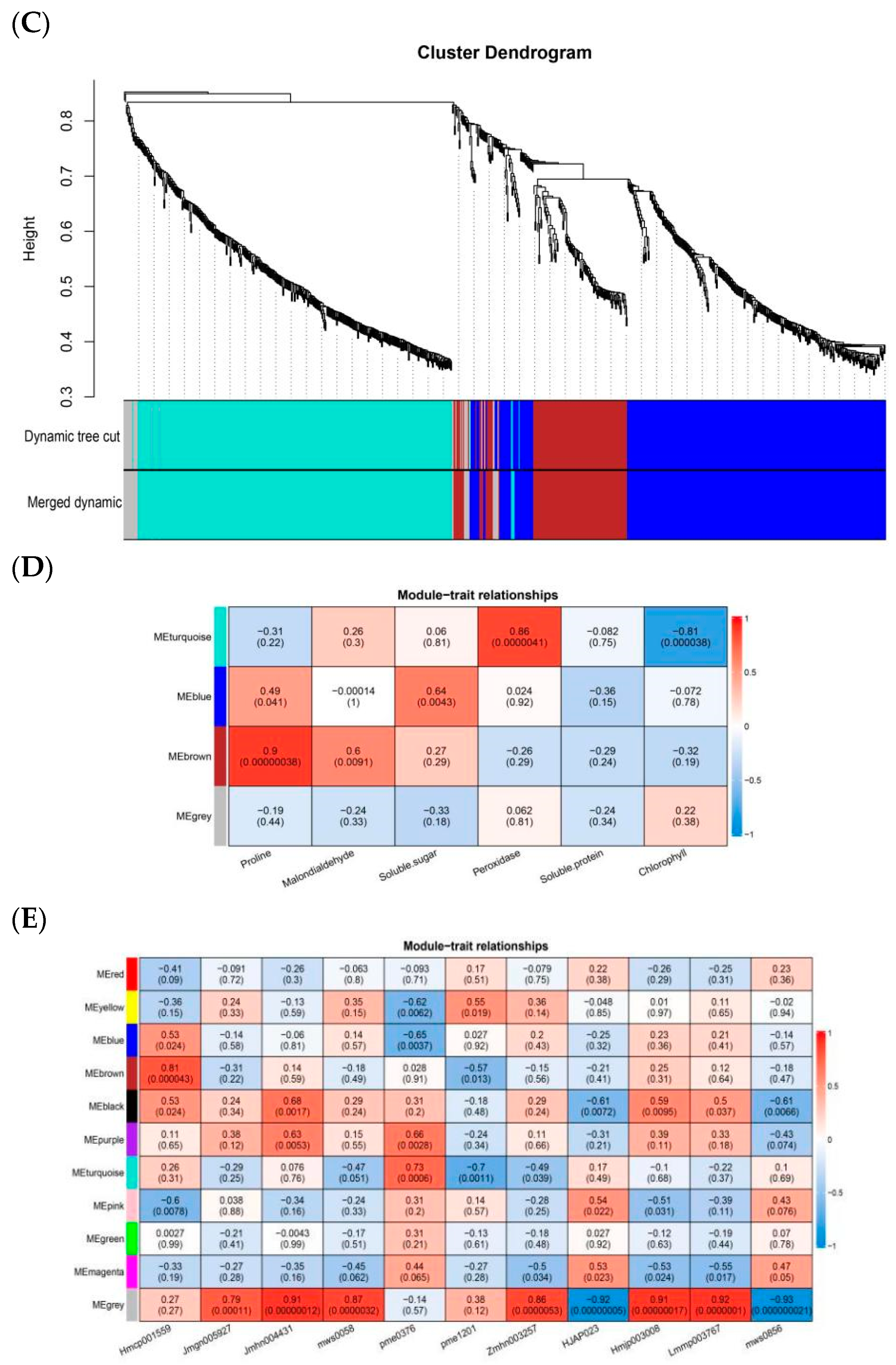
2.2. Construction of Gene Co-Expression Network
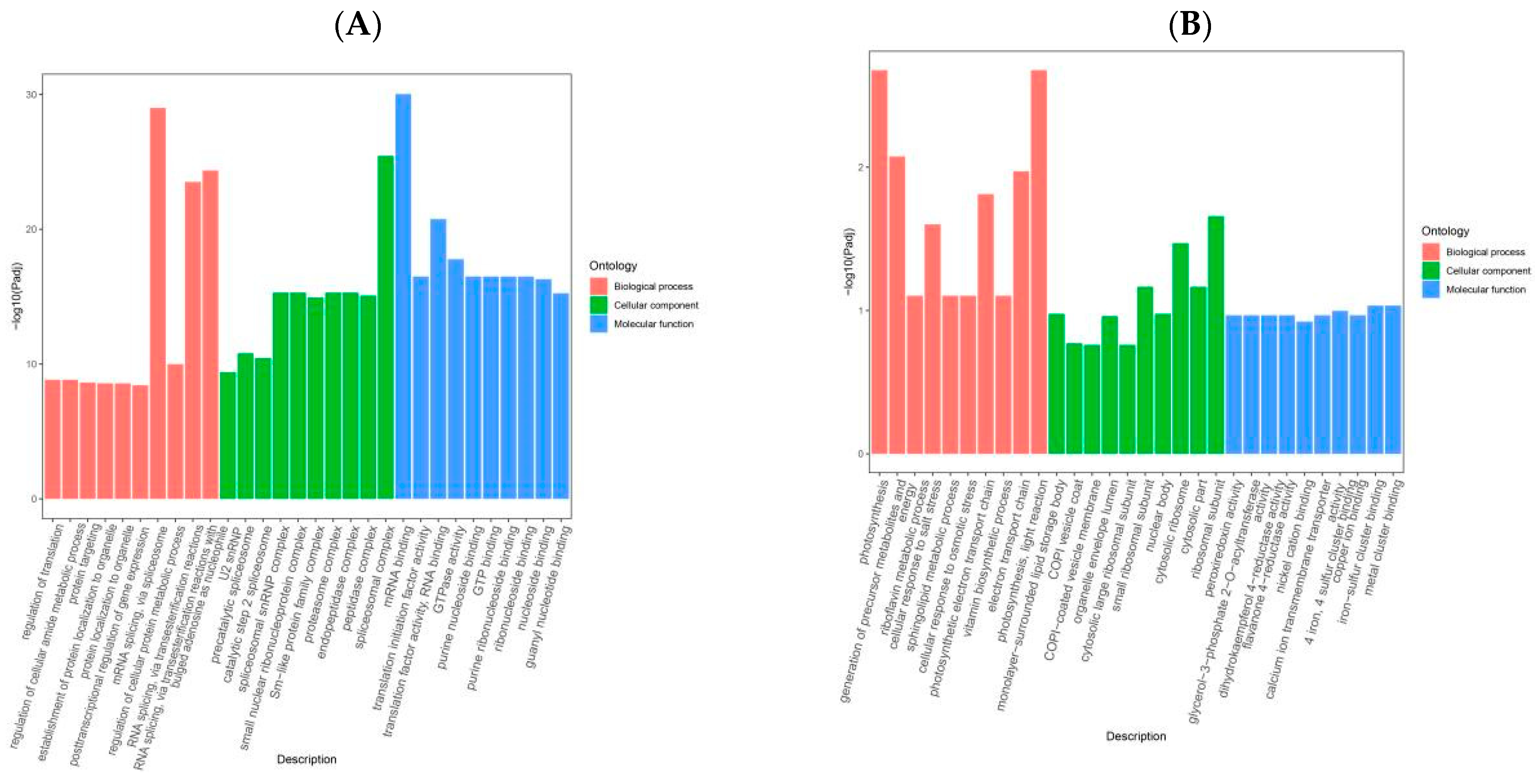
2.3. GO and KEGG Enrichment Analysis of Relevant Specificity Modules
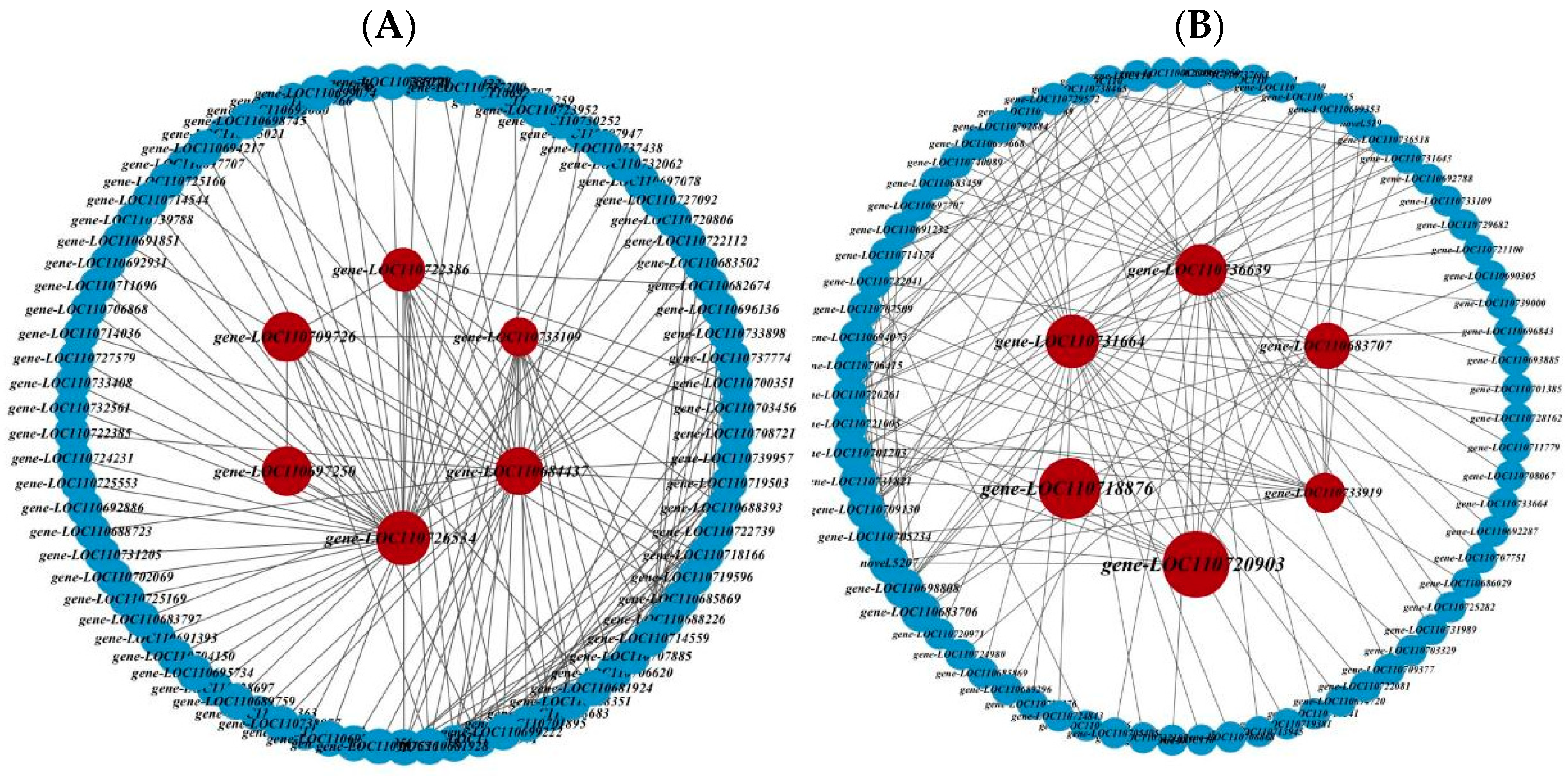
2.4. Identification of Core Genes and Construction of Interaction Network
2.5. Transcription Factors under Low-Temperature Stress
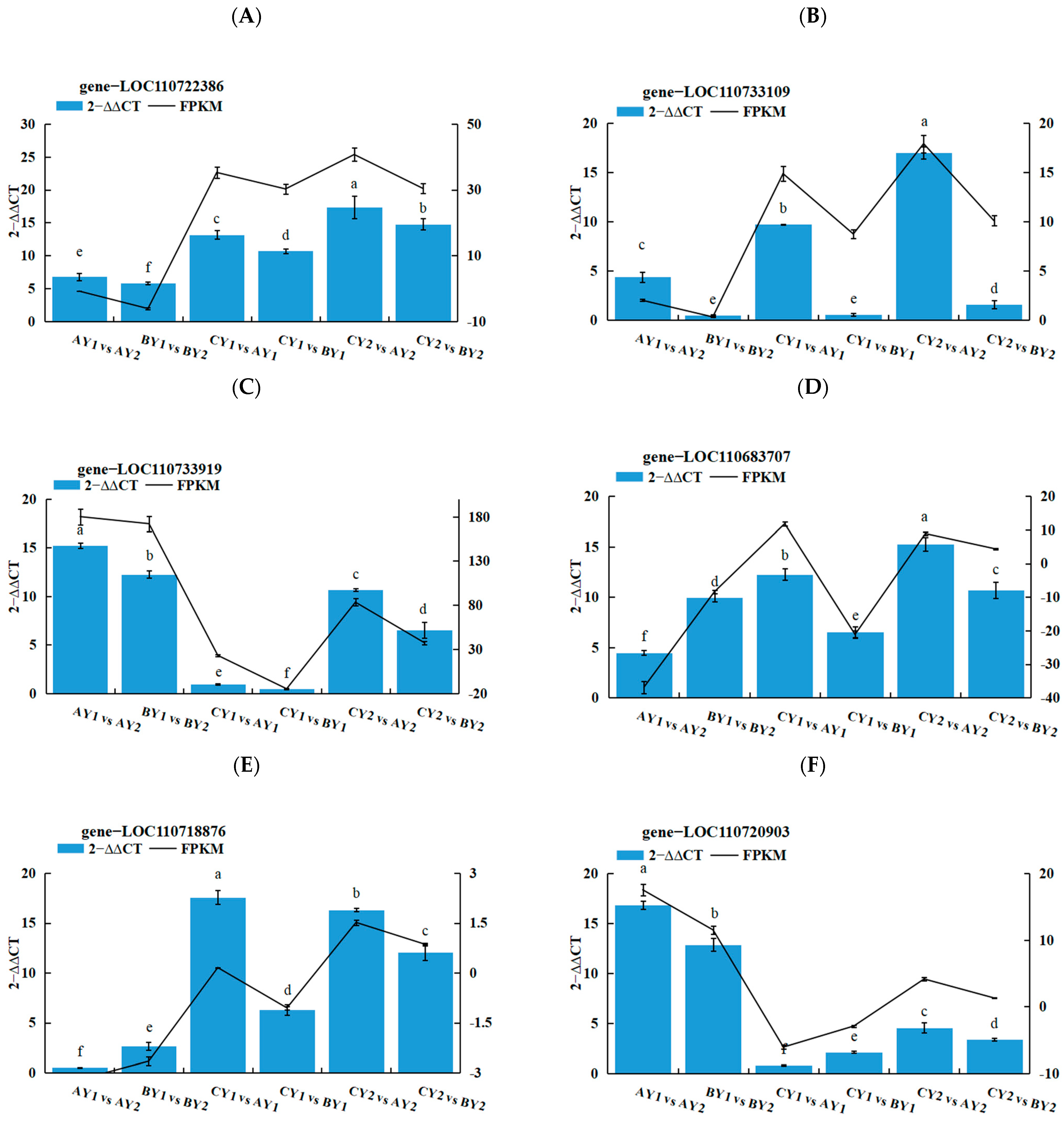
2.6. Real-Time Fluorescence Quantitative PCR Validation
3. Discussion
4. Materials and Methods
4.1. Quinoa Planting and Growing Conditions
4.2. Data Acquisition
4.3. Physiological Indicators
4.4. Construction of Weighted Gene Co-Expression Network Analysis
4.5. Identification of Specific Modules and GO and KEGG Functional Enrichment Analysis
4.6. Identification of Core Genes of Specific Modules and Construction of Gene Interaction Network
4.7. Identification of Transcription Factors
4.8. Real-Time Fluorescence Quantitative PCR Validation
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DaMatta, F.M.; Ramalho, J.D.C. Impacts of drought and temperature stress on coffee physiology and production: A review. Brazil. J. Plant Physiol. 2006, 18, 55–81. [Google Scholar] [CrossRef]
- Zhang, N.; Huo, W.; Zhang, L.; Chen, F.; Cui, D. Identification of winter-responsive proteins in bread wheat using proteomics analysis and virus-induced gene silencing (VIGS). Mol. Cell. Proteom. 2016, 15, 2954–2969. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Qi, X.; Wang, R.; Lin, M.; Fang, J. Evaluation of freezing tolerance in Actinidia germplasm based on relative electrolyte leakage. Hortic. Environ. Biote. 2020, 61, 755–765. [Google Scholar] [CrossRef]
- Chen, L.; Hu, W.; Mishra, N.; Wei, J.; Lu, H.; Hou, Y.; Shen, G. AKR2A interacts with KCS1 to improve VLCFAs contents and chilling tolerance of Arabidopsis thaliana. Plant J. 2020, 103, 1575–1589. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, L.; Baral, R.; Paudel, D.; Min, D.; Makaju, S.O.; Poudel, H.P.; Missaoui, A.M. Cold stress in plants: Strategies to improve cold tolerance in forage species. Plant Stress 2022, 4, 100081. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, L.; Song, A.; Wang, H.; Chen, S.; Jiang, J.; Chen, F. Chrysanthemum (Chrysanthemum morifolium) CmICE2 conferred freezing tolerance in Arabidopsis. Plant Physiol. Biochem. 2020, 146, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.; Clément, C.; Barka, E.A. Physiological and molecular changes in plants grown at low temperatures. Planta 2012, 235, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Sanghera, G.S.; Wani, S.H.; Hussain, W.; Singh, N.B. Engineering cold stress tolerance in crop plants. Curr. Genom. 2011, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Askari-Khorasgani, O.; Hatterman-Valenti, H.; Flores, F.B.; Pessarakli, M. Managing plant-environment-symbiont interactions to promote plant performance under low temperature stress. J. Plant Nutr. 2019, 42, 2010–2027. [Google Scholar] [CrossRef]
- Guo, X.; Liu, D.; Chong, K. Cold signaling in plants: Insights into mechanisms and regulation. J. Integr. Plant Biol. 2018, 60, 745–756. [Google Scholar] [CrossRef]
- Beck, E.H.; Fettig, S.; Knake, C.; Hartig, K.; Bhattarai, T. Specific and unspecific responses of plants to cold and drought stress. J. Biosci. 2007, 32, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Maleki, M.; Ghorbanpour, M. Cold tolerance in plants: Molecular machinery deciphered. In Biochemical, Physiological and Molecular Avenues for Combating Abiotic Stress Tolerance in Plants; Shabir, H.W., Ed.; Academic Press: London, UK, 2018; pp. 57–71. [Google Scholar]
- Ritonga, F.N.; Ngatia, J.N.; Wang, Y.; Khoso, M.A.; Farooq, U.; Chen, S. AP2/ERF, an important cold stress–related transcription factor family in plants: A review. Physiol. Mol. Biol. Plants 2021, 27, 1953–1968. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, S.; Verma, S.; Kumar, S.; Kumari, S.; Mishra, B.N. Transcriptional regulation and signalling of cold stress response in plants: An overview of current understanding. Environ. Exp. Bot. 2020, 180, 104243. [Google Scholar] [CrossRef]
- Nakashima, K.; Yamaguchi-Shinozaki, K. Regulons involved in osmotic stress-responsive and cold stress-responsive gene expression in plants. Physiol. Plant. 2006, 126, 62–71. [Google Scholar] [CrossRef]
- Yu, T.; Zhou, H.; Liu, Z.; Zhai, H.; Liu, Q. The sweet potato transcription factor IbbHLH33 enhances chilling tolerance in transgenic tobacco. Czech J. Genet. Plant Breed. 2022, 58, 210–222. [Google Scholar] [CrossRef]
- Wang, Y.X.; Ya, H.U.; Chen, B.H.; Zhu, Y.F.; Dawuda, M.M.; Svetla, S. Physiological mechanisms of resistance to cold stress associated with 10 elite apple rootstocks. J. Integr. Agric. 2018, 17, 857–866. [Google Scholar] [CrossRef]
- Bhargava, A.; Shukla, S.; Ohri, D. Chenopodium quinoa—An Indian perspectiveJ. Ind. Crops Prod. 2006, 23, 73–87. [Google Scholar] [CrossRef]
- James, L.E.A. Quinoa (Chenopodium quinoa Willd.): Composition, chemistry, nutritional, and functional properties. Adv. Food Nutr. Res. 2009, 58, 1–31. [Google Scholar]
- Scanlin, L.; Lewis, K.A. Quinoa as a Sustainable Protein Source: Production, Nutrition, and Processing. In Sustainable Protein Sources, 1st ed.; Nadathur, S.R., Wanasundara, J.P.D., Eds.; Academic Press: New York, NY, USA, 2017; pp. 223–238. [Google Scholar]
- Chaudhary, N.; Walia, S.; Kumar, R. Functional composition, physiological effect and agronomy of future food quinoa (Chenopodium quinoa Willd.): A review. J. Food Compos. Anal. 2023, 118, 105192. [Google Scholar] [CrossRef]
- Vilcacundo, R.; Hernández-Ledesma, B. Nutritional and biological value of quinoa (Chenopodium quinoa Willd.). Curr. Opin. Food Sci. 2017, 14, 1–6. [Google Scholar] [CrossRef]
- Murphy, K.; Matanguihan, J. Quinoa: Improvement and Sustainable Production (Murphy/Quinoa: Improvement and Sustainable Production). In Nutritional Properties of Quinoa; Murphy, K., Matanguihan, J., Eds.; Academic Press: Hoboken, NJ, USA, 2015; pp. 1–18. [Google Scholar]
- Jacobsen, S.E.; Monteros, C.; Christiansen, J.L.; Bravo, L.A.; Corcuera, L.J.; Mujica, A. Plant responses of quinoa (Chenopodium quinoa Willd.) to frost at various phenological stages. Eur. J. Agron. 2005, 22, 131–139. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinforma 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wu, X.; Tao, Y.; Yan, G.; Wang, X.; Cao, S.; He, W. Mining Candidate Genes Related to Heavy Metals in Mature Melon (Cucumis melo L.) Peel and Pulp Using WGCNA. Genes 2022, 13, 1767. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cao, K.; Li, Y.; Wu, J.; Fan, J.; Ding, T.; Wang, L. Identification of co–expressed networks and key genes associated with organic acid in peach fruit. Sci. Hortic. 2023, 307, 111496. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, R.; Xing, X.; Hou, W.; Mu, X.; Zhang, J.; Wang, P. Transcriptome and weighted gene co–expression network analysis reveal key genes involved in the proanthocyanidin biosynthesis in Cerasus humilis. Sci. Hortic. 2024, 325, 112717. [Google Scholar] [CrossRef]
- Wang, W.; Pu, Y.; Wen, H.; Lu, D.; Yan, M.; Liu, M.; Wu, C. Transcriptome and weighted gene co–expression network analysis of jujube (Ziziphus jujuba Mill.) fruit reveal putative genes involved in proanthocyanin biosynthesis and regulation. Food Sci. Hum. Well. 2023, 12, 1557–1570. [Google Scholar] [CrossRef]
- Xie, H.; Wang, Q.; Zhang, P.; Zhang, X.; Huang, T.; Guo, Y.; Qin, P. Transcriptomic and metabolomic analysis of the response of quinoa seedlings to low temperatures. Biomol. Ther. 2022, 12, 977. [Google Scholar] [CrossRef]
- Sierra-Almeida, A.; Cavieres, L.A.; Bravo, L.A. Freezing resistance varies within the growing season and with elevation in high-Andean species of central Chile. New Phytol. 2009, 182, 461–469. [Google Scholar] [CrossRef]
- Gamboa, C.; Bojacá, C.R.; Schrevens, E.; Maertens, M. Sustainability of smallholder quinoa production in the Peruvian Andes. J. Clean. Prod. 2020, 264, 121657. [Google Scholar] [CrossRef]
- Yadav, S.K. Cold stress tolerance mechanisms in plants. A review. Agron. Sustain. Dev. 2010, 30, 515–527. [Google Scholar] [CrossRef]
- Yan, C.; Zhang, N.; Wang, Q.; Fu, Y.; Wang, F.; Su, Y.; Liao, H. The effect of low temperature stress on the leaves and microRNA expression of potato seedlings. Front. Ecol. Evol. 2021, 9, 727081. [Google Scholar] [CrossRef]
- Kaur, A.; Gupta, N.; Sharma, S.; Singh, P.; Singh, S. Physiological and biochemical characterization of chickpea genotypes for cold tolerance at reproductive stage. S. Afr. J. Bot. 2022, 150, 488–499. [Google Scholar] [CrossRef]
- Uzal, O. Effects of proline treatments on plant growth, lipid peroxidation and antioxidant enzyme activities of tomato (Solanum lycopersicum L.) seedlings under chilling stress. Gesunde Pflanz. 2022, 74, 729–736. [Google Scholar] [CrossRef]
- Hossain, M.A.; Fujita, M. Evidence for a role of exogenous glycinebetaine and proline in antioxidant defense and methylglyoxal detoxification systems in mung bean seedlings under salt stress. Physiol. Mol. Biol. Plants 2010, 16, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Galleano, M.; Verstraeten, S.V.; Oteiza, P.I.; Fraga, C.G. Antioxidant actions of flavonoids: Thermodynamic and kinetic analysis. Arch. Biochem. Biophys. 2010, 501, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Bhowmik, P.C.; Shetty, K. Cold acclimation responses of three cool–season turfgrasses and the role of proline–associated pentose phosphate pathway. J. Am. Soc. Horticult. Sci. 2009, 134, 210–220. [Google Scholar] [CrossRef]
- Xu, Y.; Zeng, X.; Wu, J.; Zhang, F.; Li, C.; Jiang, J.; Sun, W. iTRAQ–based quantitative proteome revealed metabolic changes in winter turnip rape (Brassica rapa L.) under cold stress. Int. J. Mol. Sci. 2018, 19, 3346. [Google Scholar] [CrossRef]
- Yao, C.; Li, W.; Liang, X.; Ren, C.; Liu, W.; Yang, G.; Han, D. Molecular cloning and characterization of MbMYB108, a Malus baccata MYB transcription factor gene, with functions in tolerance to cold and drought stress in transgenic Arabidopsis thaliana. Int. J. Mol. Sci. 2022, 23, 4846. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Li, X.; Li, Y.; Yang, G.; Liu, W.; Shao, B.; Han, D. Overexpression of a Malus baccata MYB transcription factor gene MbMYB4 increases cold and drought tolerance in Arabidopsis thaliana. Int. J. Mol. Sci. 2022, 23, 1794. [Google Scholar] [CrossRef]
- Hou, X.M.; Zhang, H.F.; Liu, S.Y.; Wang, X.K.; Zhang, Y.M.; Meng, Y.C.; Chen, R.G. The NAC transcription factor CaNAC064 is a regulator of cold stress tolerance in peppers. Plant sci. 2020, 291, 110346. [Google Scholar] [CrossRef]
- Pang, X.; Xue, M.; Ren, M.; Nan, D.; Wu, Y.; Guo, H. Ammopiptanthus mongolicus stress–responsive NAC gene enhances the tolerance of transgenic Arabidopsis thaliana to drought and cold stresses. Genet. Mol. Biol. 2019, 42, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, X.; Wang, X.; Xi, Z. An AP2/ERF transcription factor VvERF63 positively regulates cold tolerance in Arabidopsis and grape leaves. Environ. Exp. Bot. 2023, 205, 105124. [Google Scholar] [CrossRef]
- Bolt, S.; Zuther, E.; Zintl, S.; Hincha, D.K.; Schmülling, T. ERF105 is a transcription factor gene of Arabidopsis thaliana required for freezing tolerance and cold acclimation. Plant Cell Environ. 2017, 40, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, Z.; Zhang, H.; Wang, X.C.; Huang, R. Transcriptional modulation of ethylene response factor protein JERF3 in the oxidative stress response enhances tolerance of tobacco seedlings to salt, drought, and freezing. Plant physiol. 2008, 148, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zheng, Y.; Teixeira, J.A.; Yu, X. Comparative transcriptomics and WGCNA reveal candidate genes involved in petaloid stamens in Paeonia lactiflora. J. Hortic. Sci. Biotechnol. 2021, 96, 588–603. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Huang, T.; Zhang, X.; Zhang, P.; Xie, H.; Liu, J.; Li, L.; Kong, Z.; Qin, P. Transcriptome and metabolome analyses revealed the response mechanism of quinoa seedlings to different phosphorus stresses. Int. J. Mol. Sci. 2022, 23, 4704. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Zhang, X.; Wang, Q.; Guo, Y.; Xie, H.; Li, L.; Zhang, P.; Liu, J.; Qin, P. Metabolome and transcriptome profiles in quinoa seedlings in response to potassium supply. BMC Plant Biol. 2022, 22, 604. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, P.; Liu, J.; Wang, H.; Liu, J.; Li, H.; Qin, P. Integrated Metabolomic and Transcriptomic Analysis of the Quinoa Seedling Response to High Relative Humidity Stress. Biomolecules 2023, 13, 1352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Feng, S.; Yu, X.; Zhou, A. Morphological and physiological responses of Dianthus spiculifolius high wax mutant to low–temperature stress. J. Plant Physiol. 2022, 275, 153762. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, X.; Li, X.; Ren, M.; Tong, Y.; Wu, X.; Chen, S. Comprehensive transcriptome and WGCNA analysis reveals the potential function of anthocyanins in low–temperature resistance of a red flower mutant tobacco. Genomics 2023, 115, 110728. [Google Scholar] [CrossRef]
- Chen, X.; Mei, Q.; Liang, W.; Sun, J.; Wang, X.; Zhou, J.; Chen, J. Gene mapping, genome–wide transcriptome analysis, and WGCNA reveals the molecular mechanism for triggering programmed cell death in rice mutant pir1. Plants 2020, 9, 1607. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinf. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Li, M.Z.; Wang, S.M.; Yin, H.J. Revisiting the role of plant transcription factors in the battle against abiotic stress. Int. J. Mol. Sci. 2018, 19, 1634. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Mejia-Guerra, M.K.; Kurz, K.; Liang, X.; Welch, L.; Grotewold, E. AGRIS: The Arabidopsis gene regulatory information server, an update. Nucleic Acids Res. 2010, 39, D1118–D1122. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.H.; Kuznetsov, A.; Ananiev, V.; Asztalos, A.; Borodin, E.; Evgeniev, V.; Schneider, V.A. Accessing NCBI data using the NCBI sequence viewer and genome data viewer (GDV). Genome Res. 2021, 31, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Tian, C.; Jiang, Q.; Wang, F.; Wang, G.L.; Xu, Z.S.; Xiong, A.S. Selection of suitable reference genes for qPCR normalization under abiotic stresses and hormone stimuli in carrot leaves. PLoS ONE 2015, 10, e0117569. [Google Scholar] [CrossRef]
- Chen, C.; Wu, J.; Hua, Q.; Tel-Zur, N.; Xie, F.; Zhang, Z.; Qin, Y. Identification of reliable reference genes for quantitative real–time PCR normalization in pitaya. Plant Methods 2019, 15, 1–12. [Google Scholar] [CrossRef]
- Taniguchi, K.; Kajiyama, T.; Kambara, H. Quantitative analysis of gene expression in a single cell by qPCR. Nat. Methods 2009, 6, 503–506. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proline | Malondialdehyde | Soluble Sugar | Peroxidase | Soluble Protein | Total Chlorophyll | |
|---|---|---|---|---|---|---|
| Proline | 1 | |||||
| Malondialdehyde | −0.519 * | 1 | ||||
| Soluble sugar | 0.414 | −0.059 | 1 | |||
| Peroxidase | −0.395 | −0.022 | 0.037 | 1 | ||
| Soluble protein | −0.186 | 0.168 | −0.095 | −0.397 | 1 | |
| Total chlorophyll | −0.199 | −0.692 ** | −0.088 | −0.599 ** | 0.112 | 1 |
| Module | Module Candidate Hub Genes | TFs Family | Gene Function |
|---|---|---|---|
| Turquoise | gene-LOC110684437 | AP2/ERF | Ethylene-responsive transcription factor RAP2-12-like |
| gene-LOC110697250 | - | Pre-mRNA-processing factor 19-like | |
| gene-LOC110709726 | - | 14-3-3 protein 10-like | |
| gene-LOC110726534 | - | Eukaryotic peptide chain release factor subunit 1-3 | |
| gene-LOC110722386 | - | Zinc finger CCCH domain-containing protein 40-like | |
| gene-LOC110733109 | - | E3 ubiquitin-protein ligase Hakai-like | |
| Gray | gene-LOC110731664 | MYB-related | Protein LHY-like |
| gene-LOC110736639 | NAC | NAC domain-containing protein 2-like | |
| gene-LOC110733919 | C2C2-CO-like | Zinc finger protein CONSTANS-LIKE 6-like | |
| gene-LOC110683707 | - | 40S ribosomal protein S23 | |
| gene-LOC110718876 | - | Uncharacterized LOC110718876 | |
| gene-LOC110720903 | AP2/ERF | Ethylene-responsive transcription factor 3-like |
| Quantity | Gene-ID | NCBI-Gene ID | Primer | 5′ to 3′ |
|---|---|---|---|---|
| 1 | gene-LOC110722386 | 110722386 | Forward primer | AAGAGTGATGTCAATAGG |
| Reverse primer | CCGTAAGAGGATTCATAA | |||
| 2 | gene-LOC110733109 | 110733109 | Forward primer | GACTCAATTTGCTATCTATG |
| Reverse primer | TCACTCATCTCTTCTCTT | |||
| 3 | gene-LOC110733919 | 110733919 | Forward primer | AAGCAAGAGTATCAAGAT |
| Reverse primer | TCAACTTCCTAACTTCATA | |||
| 4 | gene-LOC110683707 | 110683707 | Forward primer | CTTACAAGAAGTCACATCT |
| Reverse primer | AGTTCAAACAACCATCAT | |||
| 5 | gene-LOC110718876 | 110718876 | Forward primer | GGATTGGATGAGAATAACT |
| Reverse primer | GCTCTTAACCTCTGTAAC | |||
| 6 | gene-LOC110720903 | 110720903 | Forward primer | TCTAATCACCACCATCAA |
| Reverse primer | CTCAACCGTACTACTCAT | |||
| Internal reference gene | ACT-1 | 818339 | Forward primer | GTCCACAGAAAGTGCTTCTAAG |
| Reverse primer | AACAACTCCTCACCTTCTCATG |
| Component | Volume (µL) |
|---|---|
| 2× PerfectStartTM SYBR qPCR Supermix | 10 |
| Passive Reference Dye (50×) (optional) | 0.4 |
| Nuclease-free Water | 6.8 |
| Forward Primer (10 μM) | 0.4 |
| Reverse Primer (10 μM) | 0.4 |
| cDNA (200 µg/µL) | 2 |
| Total volume | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Jiang, G.; Wang, X.; Bai, Y.; Zhang, P.; Liu, J.; Li, L.; Huang, L.; Qin, P. Identifying Core Genes Related to Low-Temperature Stress Resistance in Quinoa Seedlings Based on WGCNA. Int. J. Mol. Sci. 2024, 25, 6885. https://doi.org/10.3390/ijms25136885
Zhang L, Jiang G, Wang X, Bai Y, Zhang P, Liu J, Li L, Huang L, Qin P. Identifying Core Genes Related to Low-Temperature Stress Resistance in Quinoa Seedlings Based on WGCNA. International Journal of Molecular Sciences. 2024; 25(13):6885. https://doi.org/10.3390/ijms25136885
Chicago/Turabian StyleZhang, Lingyuan, Guofei Jiang, Xuqin Wang, Yutao Bai, Ping Zhang, Junna Liu, Li Li, Liubin Huang, and Peng Qin. 2024. "Identifying Core Genes Related to Low-Temperature Stress Resistance in Quinoa Seedlings Based on WGCNA" International Journal of Molecular Sciences 25, no. 13: 6885. https://doi.org/10.3390/ijms25136885
APA StyleZhang, L., Jiang, G., Wang, X., Bai, Y., Zhang, P., Liu, J., Li, L., Huang, L., & Qin, P. (2024). Identifying Core Genes Related to Low-Temperature Stress Resistance in Quinoa Seedlings Based on WGCNA. International Journal of Molecular Sciences, 25(13), 6885. https://doi.org/10.3390/ijms25136885