
First Examples of s-Metal Complexes with Subporphyrazine and Its Phenylene-Annulated Derivatives: DFT Calculations




Abstract
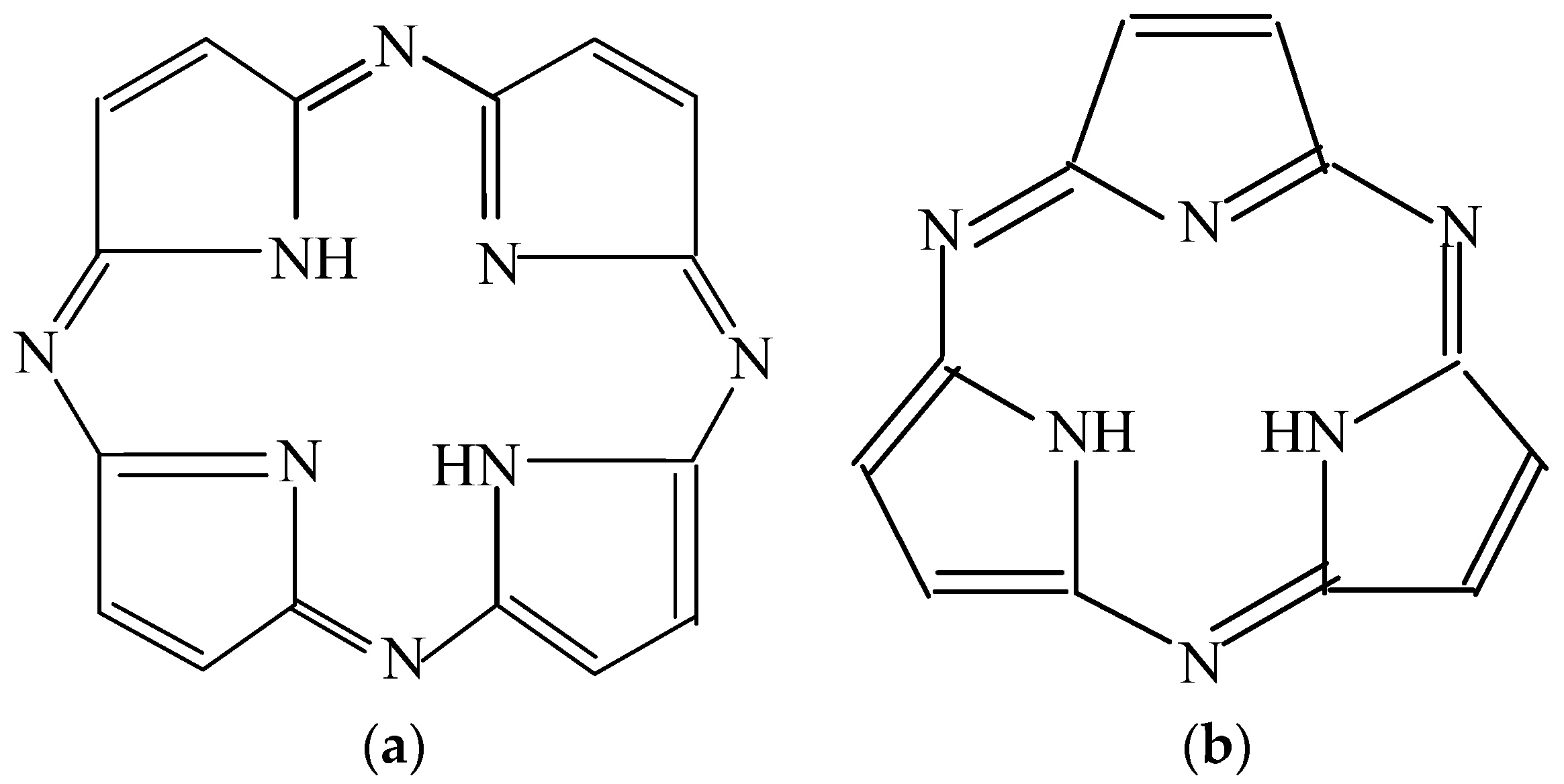
1. Introduction
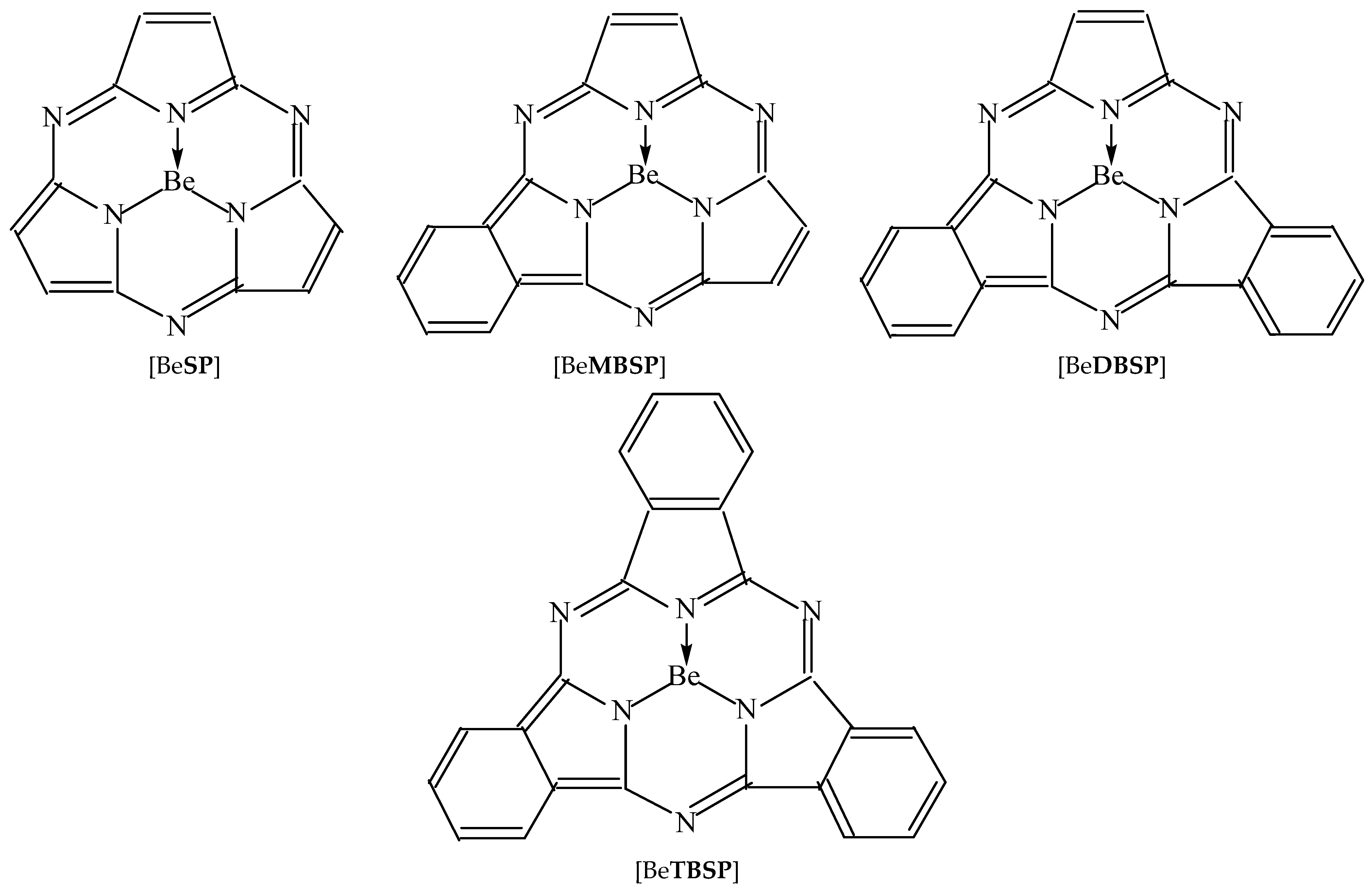
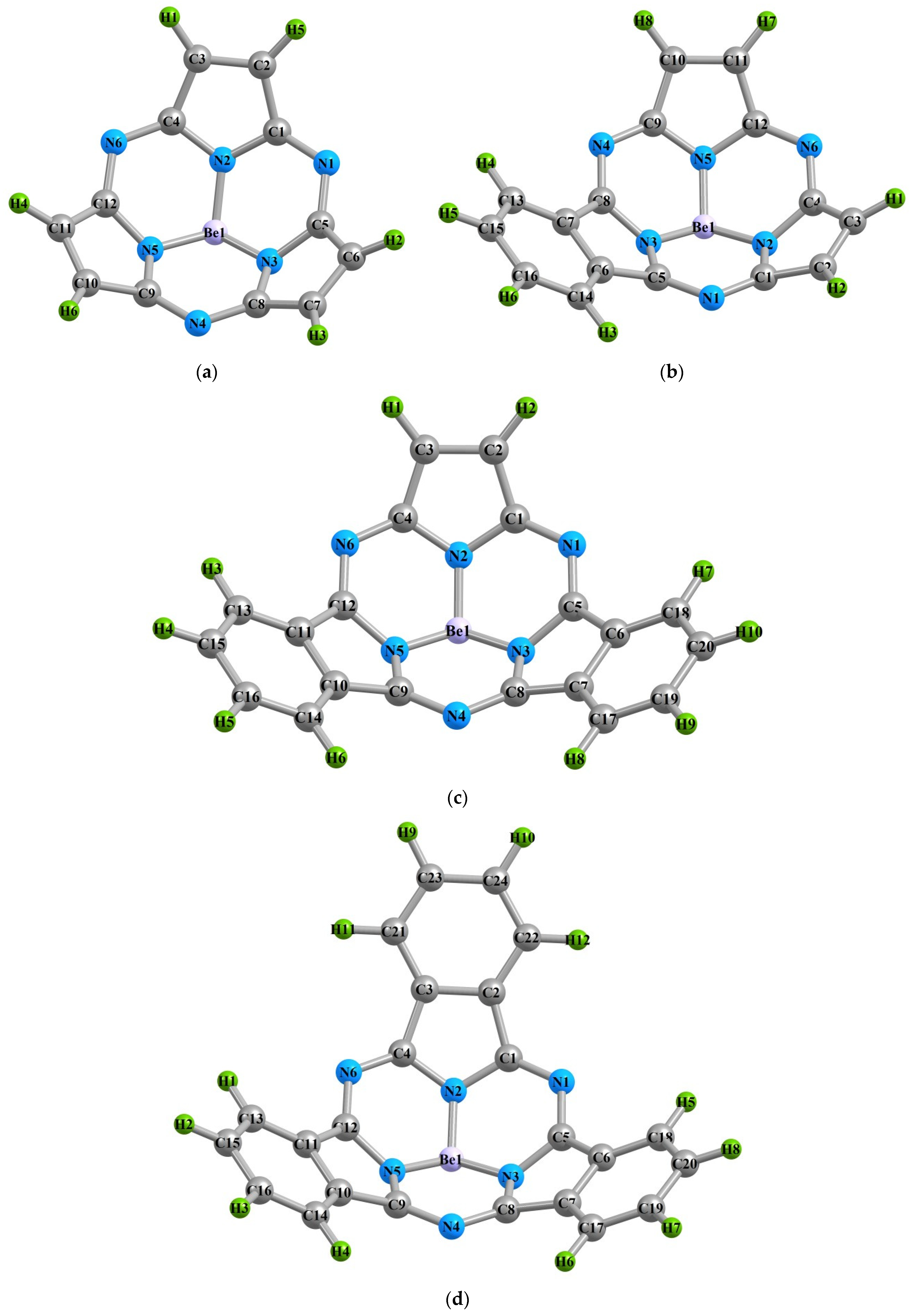
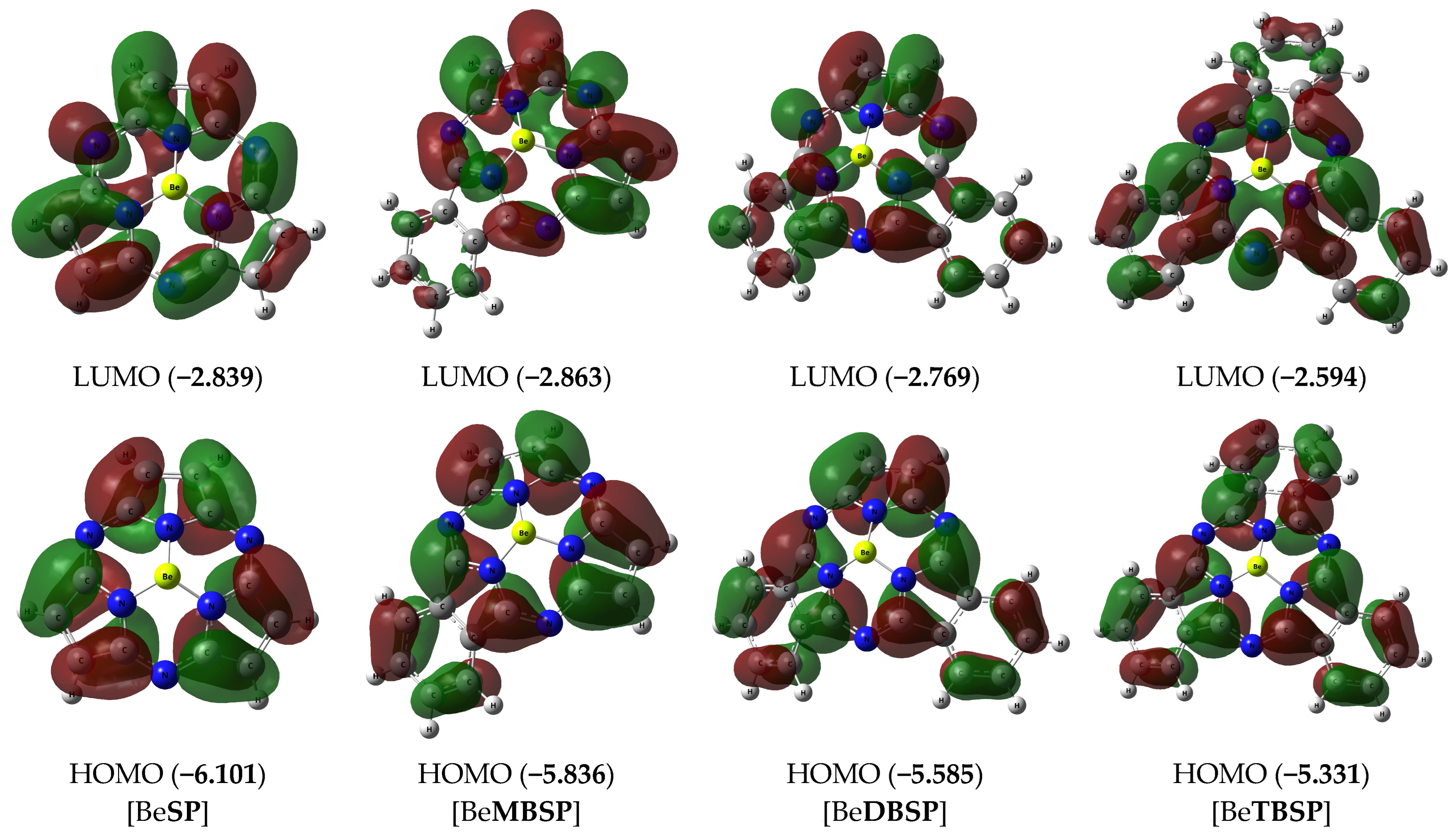
2. Results and Discussion
3. Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kasuda, K.; Tsutsui, M. Some new developments in the chemistry of metallophthalocyanines. Coord. Chem. Rev. 1980, 32, 67–95. [Google Scholar] [CrossRef]
- Sliva, W.; Mianovska, B. Metalloporphyrin arrays. Transit. Met. Chem. 2000, 25, 491–504. [Google Scholar] [CrossRef]
- Mamardashvili, G.M.; Mamardashvili, N.Z.; Koifman, O.I. Self-assembling systems based on porphyrins. Russ. Chem. Rev. 2008, 77, 59–75. [Google Scholar] [CrossRef]
- Khelevina, O.G.; Malyasova, A.S. 40 years with porphyrazines. J. Porphyr. Phthalocyanines 2019, 23, 1261–1264. [Google Scholar] [CrossRef]
- Thomas, A.L. Phthalocyanines. Research & Applications; CRC Press: Boca Raton, FL, USA, 1990; 304p. [Google Scholar]
- Lomova, T.N. Axial Coordinated Metal Porphyrins in Science and Practice; URSS: Moscow, Russia, 2018; 700p. [Google Scholar]
- Claessens, C.G.; González-Rodríguez, D.; Torres, T. Subphthalocyanines: Singular nonplanar aromatic compounds-synthesis, reactivity, and physical properties. Chem. Rev. 2002, 102, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Salomé Rodríguez-Morgade, M.; Esperanza, S.; Torres, T.; Barberá, J. Synthesis, characterization, and properties of subporphyrazines: A new class of nonplanar, aromatic macrocycles with absorption in the green region. Chem.—A Eur. J. 2004, 11, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Gommans, H.; Aernouts, T.; Verreet, B.; Heremans, P.; Medina, A.; Claessens, C.G.; Torres, T. Perfluorinated Subphthalocyanine as a New Acceptor Material in a Small-Molecule Bilayer Organic Solar Cell. Adv. Funct. Mater. 2009, 19, 3435–3439. [Google Scholar] [CrossRef]
- Morse, G.E.; Helander, M.G.; Maka, J.F.; Lu, Z.H.; Bender, T.P. Fluorinated phenoxy boron subphthalocyanines in organic light-emitting diodes. ACS Appl. Mater. Interfaces 2010, 2, 1934–1944. [Google Scholar] [CrossRef]
- Sullivan, P.; Duraud, A.; Hancox, L.; Beaumont, N.; Mirri, G.; Tucker, J.H.R.; Hatton, R.A.; Shipman, M.; Jones, T.S. Halogenated boron subphthalocyanines as light harvesting electron acceptors in organic photovoltaics. Adv. Energy Mater. 2011, 1, 352–355. [Google Scholar] [CrossRef]
- Beaumont, N.; Cho, S.W.; Sullivan, P.; Newby, D.; Smith, K.E.; Jones, T.S. Boron subphthalocyanine chloride as an electron acceptor for high-voltage fullerene-free organic photovoltaics. Adv. Funct. Mater. 2012, 22, 561–566. [Google Scholar] [CrossRef]
- Morse, G.E.; Bender, T.P. Boron subphthalocyanines as organic electronic materials. ACS Appl. Mater. Interfaces 2012, 4, 5055–5068. [Google Scholar] [CrossRef] [PubMed]
- Claessens, C.G.; González-Rodríguez, D.; Rodríguez-Morgade, M.S.; Medina, A.; Torres, T. Subphthalocyanines, Subporphyrazines, and Subporphyrins: Singular Nonplanar Aromatic Systems. Chem. Rev. 2014, 114, 2192–2277. [Google Scholar] [CrossRef] [PubMed]
- Dearden, C.A.; Walker, M.; Beaumont, N.; Hancox, I.; Unsworth, N.K.; Sullivan, P.; McConville, C.F.; Jones, T.S. High voltage hybrid organic photovoltaics using a zinc oxide acceptor and a subphthalocyanine donor. Phys. Chem. Chem. Phys. 2014, 16, 18896–18901. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, N.; Castrucci, J.S.; Sullivan, P.; Morse, G.E.; Paton, A.S.; Lu, Z.H.; Bender, T.P.; Jones, T.S. Acceptor properties of boron subphthalocyanines in fullerene free photovoltaics. J. Phys. Chem. C 2014, 118, 14813–14823. [Google Scholar] [CrossRef]
- Ebenhoch, B.; Prasetya, N.B.A.; Rotello, V.M.; Cooke, G.; Samuel, I.D.W. Solution-processed boron subphthalocyanine derivatives as acceptors for organic bulk-heterojunction solar cells. J. Mater. Chem. A 2015, 3, 7345–7352. [Google Scholar] [CrossRef]
- Castrucci, J.S.; Josey, D.S.; Thibau, E.; Lu, Z.H.; Bender, T.P. Boron Subphthalocyanines as Triplet Harvesting Materials within Organic Photovoltaics. J. Phys. Chem. Lett. 2015, 6, 3121–3125. [Google Scholar] [CrossRef] [PubMed]
- Sfyri, G.; Kumar, C.V.; Sabapathi, G.; Giribabu, L.; Andrikopoulos, K.S.; Stathatos, E.; Lianos, P. Subphthalocyanine as hole transporting material for perovskite solar cells. RSC Adv. 2015, 5, 69813–69818. [Google Scholar] [CrossRef]
- Pakhomov, G.I.; Travkin, V.V.; Hamdoush, M.; Zhabanov, Y.A.; Stuzhin, P.A. Thiadiazole Fused Subporphyrazines as Acceptors in Organic Photovoltaic Cells. Macroheterocycles 2017, 10, 548–551. [Google Scholar] [CrossRef]
- Lee, H.; Ahn, S.W.; Ryu, S.H.; Ryu, B.K.; Lee, M.H.; Cho, S.W.; Smith, K.E.; Jones, T.S. Interfacial electronic structure of Cl6SubPc non-fullerene acceptors in organic photovoltaics using soft X-ray spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 31628–31633. [Google Scholar] [CrossRef] [PubMed]
- Stuzhin, P.A.; Skvortsov, I.A.; Zhabanov, Y.A.; Somov, N.V.; Razgonyaev, O.V.; Nikitin, I.A.; Koifman, O.I. Subphthalocyanine azaanalogues—Boron(III) subporphyrazines with fused pyrazine fragments. Dye. Pigment. 2019, 162, 888–897. [Google Scholar] [CrossRef]
- Hamdoush, M.; Nikitin, K.; Skvortsov, I.; Somov, N.; Zhabanov, Y.A.; Stuzhin, P.A. Influence of heteroatom substitution in benzene rings on structural features and spectral properties of subphthalocyanine dyes. Dye. Pigment. 2019, 170, 107584. [Google Scholar] [CrossRef]
- Mizrahi, A.; Bukuroshi, E.; Vestfrid, J.; Bender, T.P.; Gross, Z. Axial/Peripheral Chloride/Fluoride-Substituted Boron Subphthalocyanines as Electron Acceptors. Inorg. Chem. 2020, 59, 2641–2645. [Google Scholar] [CrossRef] [PubMed]
- Skvortsov, I.A.; Kovkova, U.P.; Zhabanov, Y.A.; Khodov, I.A.; Somov, N.V.; Pakhomov, G.L.; Stuzhin, P.A. Subphthalocyanine-type dye with enhanced electron affinity: Effect of combined azasubstitution and peripheral chlorination. Dye. Pigment. 2021, 185, 108944. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, O.V.; Chachkov, D.V. DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands. Int. J. Mol. Sci. 2020, 21, 9085. [Google Scholar] [CrossRef] [PubMed]
- Chachkov, D.V.; Mikhailov, O.V. Heteroligand Iron(V) Complexes Containing Porphyrazine, trans-Di[benzo]porphyrazine or Tetra[benzo]porphyrazine, Oxo and Fluoro Ligands: DFT Quantum-Chemical Study. Int. J. Mol. Sci. 2023, 24, 6442. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, O.V.; Chachkov, D.V. Molecular Structure of M(N13) Compounds with 12-Membered Nitrogen-Containing Cycle and Axial Nitrogen Atom (M = Mn, Fe): Quantum-Chemical Design by DFT Method. Quantum Rep. 2023, 5, 282–293. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Comparative DFT study of van der Waals complexes: Rare-gas dimers, alkaline-earth dimers, zinc dimer, and zinc-rare-gas dimers. J. Phys. Chem. 2006, 110, 5121–5129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | [BeSP] | [BeMBSP] | [BeDBSP] | [BeTBSP] | ||||
|---|---|---|---|---|---|---|---|---|
| Structural Parameter | B3PW91/ TZVP | M062X/ def2TZVP | B3PW91/ TZVP | M062X/ def2TZVP | B3PW91/ TZVP | M062X/ def2TZVP | B3PW91/ TZVP | M062X/ def2TZVP |
| Be–N bond lengths in chelate node BeN3, pm | ||||||||
| Be1N2 | 153.7 | 153.8 | 153.5 | 153.6 | 153.2 | 153.2 | 153.5 | 153.7 |
| Be1N3 | 153.7 | 153.8 | 154.0 | 154.1 | 153.8 | 154.0 | 153.5 | 153.7 |
| Be1N5 | 153.7 | 153.8 | 153.5 | 153.6 | 153.8 | 154.0 | 153.5 | 153.7 |
| C–N bond lengths in 6-membered chelate rings, pm | ||||||||
| N1C1 | 134.1 | 133.9 | 135.1 | 135.5 | 135.3 | 136.0 | 133.8 | 133.7 |
| C1N2 | 136.6 | 136.2 | 136.1 | 135.4 | 136.7 | 136.3 | 136.6 | 136.2 |
| N2C4 | 136.6 | 136.2 | 137.3 | 137.2 | 136.7 | 136.3 | 136.6 | 136.2 |
| C4N6 | 134.1 | 133.9 | 134.0 | 133.8 | 135.3 | 136.0 | 133.8 | 133.7 |
| N6C12 | 134.1 | 133.9 | 134.0 | 133.8 | 132.6 | 131.6 | 133.8 | 133.7 |
| C12N5 | 136.6 | 136.2 | 137.3 | 137.2 | 137.5 | 137.8 | 136.6 | 136.2 |
| N5C9 | 136.6 | 136.2 | 136.1 | 135.4 | 135.9 | 135.0 | 136.6 | 136.2 |
| C9N4 | 134.1 | 133.9 | 135.1 | 135.5 | 134.0 | 133.9 | 133.8 | 133.7 |
| N4C8 | 134.1 | 133.9 | 132.9 | 132.4 | 134.0 | 133.9 | 133.8 | 133.7 |
| C8N3 | 136.6 | 136.2 | 136.7 | 136.4 | 135.9 | 135.0 | 136.6 | 136.2 |
| N3C5 | 136.6 | 136.2 | 136.7 | 136.4 | 137.5 | 137.8 | 136.6 | 136.2 |
| C5N1 | 134.1 | 133.9 | 132.9 | 132.4 | 132.6 | 131.6 | 133.8 | 133.7 |
| C–C bond lengths in 5-membered non-chelate rings (N2C1C2C3C4), pm | ||||||||
| C1C2 | 144.9 | 145.2 | 144.4 | 144.5 | 143.7 | 143.1 | 145.6 | 145.9 |
| C2C3 | 137.0 | 136.5 | 137.4 | 137.0 | 137.9 | 138.0 | 142.5 | 141.8 |
| C3C4 | 144.9 | 145.2 | 144.4 | 144.5 | 143.7 | 143.1 | 145.6 | 145.9 |
| C–C bond lengths in 5-membered non-chelate rings (N3C5C6C7C8), pm | ||||||||
| C5C6 | 144.9 | 145.2 | 146.4 | 147.0 | 146.2 | 146.7 | 145.6 | 145.9 |
| C6C7 | 137.0 | 136.5 | 142.1 | 141.3 | 142.2 | 141.4 | 142.5 | 141.8 |
| C7C8 | 144.9 | 145.2 | 146.4 | 147.0 | 146.1 | 146.6 | 145.6 | 145.9 |
| C–C bond lengths in 5-membered non-chelate rings (N5C9C10C11C12), pm | ||||||||
| C9C10 | 144.9 | 145.2 | 144.4 | 144.5 | 146.1 | 146.6 | 145.6 | 145.9 |
| C10C11 | 137.0 | 136.5 | 137.4 | 137.0 | 142.2 | 141.4 | 142.5 | 141.8 |
| C11C12 | 144.9 | 145.2 | 144.4 | 144.5 | 146.2 | 146.7 | 145.6 | 145.9 |
| Bond angles in BeN3 chelate node, deg | ||||||||
| N2Be1N5 | 110.8 | 110.2 | 111.5 | 111.0 | 111.3 | 110.8 | 111.0 | 110.3 |
| N5Be1N3 | 110.8 | 110.2 | 110.6 | 109.9 | 110.3 | 109.3 | 111.0 | 110.3 |
| N3Be1N2 | 110.8 | 110.2 | 110.6 | 109.9 | 111.3 | 110.8 | 111.0 | 110.3 |
| Bond angles sum (BAS), deg | 332.4 | 330.6 | 332.7 | 330.8 | 332.9 | 330.9 | 333.0 | 330.9 |
| Non-bond angles between N atoms in N3 grouping, deg | ||||||||
| N2N3N5 | 60.0 | 60.0 | 60.2 | 60.3 | 60.1 | 60.2 | 60.0 | 60.0 |
| N3N5N2 | 60.0 | 60.0 | 59.9 | 59.8 | 60.1 | 60.2 | 60.0 | 60.0 |
| N5N2N3 | 60.0 | 60.0 | 59.9 | 59.9 | 59.8 | 59.6 | 60.0 | 60.0 |
| Non-bond angles sum (NBAS), deg | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 |
| Bond angles in the 6-membered chelate ring (Be1N2C1N1C5N3), deg | ||||||||
| Be1N2C1 | 119.9 | 120.1 | 120.1 | 120.5 | 119.8 | 120.2 | 119.7 | 120.0 |
| N2C1N1 | 123.8 | 123.8 | 123.6 | 123.5 | 123.2 | 122.9 | 123.5 | 123.5 |
| C1N1C5 | 120.1 | 120.0 | 120.1 | 119.8 | 120.9 | 120.9 | 120.9 | 120.7 |
| N1C5N3 | 123.8 | 123.8 | 124.0 | 124.2 | 123.7 | 123.8 | 123.5 | 123.5 |
| C5N3Be1 | 119.9 | 120.1 | 119.8 | 119.9 | 119.4 | 119.4 | 119.7 | 120.0 |
| N3Be1N2 | 110.8 | 110.2 | 110.6 | 109.9 | 111.3 | 110.8 | 111.0 | 110.3 |
| Bond angles sum (BAS61), deg | 718.3 | 718.0 | 718.2 | 717.8 | 717.9 | 718.0 | 718.3 | 718.0 |
| Bond angles in the 6-membered chelate ring (Be1N2C4N6C12N5), deg | ||||||||
| Be1N2C4 | 119.9 | 120.1 | 119.6 | 119.8 | 119.8 | 120.2 | 119.7 | 120.0 |
| N2C4N6 | 123.8 | 123.8 | 123.6 | 123.4 | 123.2 | 122.9 | 123.5 | 123.5 |
| C4N6C12 | 120.1 | 120.0 | 120.8 | 121.0 | 120.9 | 120.9 | 120.9 | 120.7 |
| N6C12N5 | 123.8 | 123.8 | 123.5 | 123.5 | 123.7 | 123.8 | 123.5 | 123.5 |
| C12N5Be1 | 119.9 | 120.1 | 119.6 | 119.8 | 119.4 | 119.4 | 119.7 | 120.0 |
| N5Be1N2 | 110.8 | 110.2 | 111.5 | 111.0 | 111.3 | 110.8 | 111.0 | 110.3 |
| Bond angles sum (BAS62), deg | 718.3 | 718.0 | 718.6 | 718.5 | 717.9 | 718.0 | 718.3 | 718.0 |
| Bond angles in the 6-membered chelate ring (Be1N5C9N4C8N3), deg | ||||||||
| Be1N5C9 | 119.9 | 120.1 | 120.1 | 120.5 | 120.1 | 120.4 | 119.7 | 120.0 |
| N5C9N4 | 123.8 | 123.8 | 123.6 | 123.5 | 123.8 | 124.2 | 123.5 | 123.5 |
| C9N4C8 | 120.1 | 120.0 | 120.1 | 119.8 | 120.0 | 119.3 | 120.9 | 120.7 |
| N4C8N3 | 123.8 | 123.8 | 124.0 | 124.2 | 123.8 | 124.2 | 123.5 | 123.5 |
| C8N3Be1 | 119.9 | 120.1 | 119.8 | 119.9 | 120.0 | 120.4 | 119.7 | 120.0 |
| N3Be1N5 | 110.8 | 110.2 | 110.6 | 109.9 | 110.3 | 109.4 | 111.0 | 110.3 |
| Bond angles sum (BAS63), deg | 718.3 | 718.0 | 718.2 | 717.8 | 718.0 | 717.9 | 718.3 | 718.0 |
| Bond angles in the 5-membered ring (N2C1C2C3C4), deg | ||||||||
| N2C1C2 | 106.4 | 106.4 | 106.7 | 106.8 | 106.5 | 106.7 | 105.7 | 105.7 |
| C1C2C3 | 107.4 | 107.4 | 107.4 | 107.3 | 107.4 | 107.4 | 106.9 | 106.9 |
| C2C3C4 | 107.4 | 107.4 | 107.5 | 107.5 | 107.4 | 107.4 | 106.9 | 106.9 |
| C3C4N2 | 106.4 | 106.4 | 106.2 | 106.2 | 106.5 | 106.7 | 105.7 | 105.7 |
| C4N2C1 | 109.9 | 110.1 | 110.0 | 110.1 | 110.1 | 110.2 | 112.5 | 112.5 |
| Bond angles sum (BAS51), deg | 537.5 | 537.7 | 537.8 | 537.9 | 537.9 | 538.4 | 537.7 | 537.7 |
| Bond angles in the 5-membered ring (N3C5C6C7C8), deg | ||||||||
| N3C5C6 | 106.4 | 106.4 | 105.6 | 105.6 | 105.4 | 105.2 | 105.7 | 105.7 |
| C5C6C7 | 107.4 | 107.4 | 106.8 | 106.8 | 106.9 | 106.9 | 106.9 | 106.9 |
| C6C7C8 | 107.4 | 107.4 | 106.8 | 106.8 | 106.9 | 106.8 | 106.9 | 106.9 |
| C7C8N3 | 106.4 | 106.4 | 105.6 | 105.6 | 105.9 | 106.1 | 105.7 | 105.7 |
| C8N3C5 | 109.9 | 110.1 | 112.2 | 112.2 | 112.3 | 112.3 | 112.5 | 112.5 |
| Bond angles sum (BAS52), deg | 537.5 | 537.7 | 537.0 | 537.0 | 537.4 | 537.3 | 537.7 | 537.7 |
| Bond angles in the 5-membered ring (N5C9C10C11C12), deg | ||||||||
| N5C9C10 | 106.4 | 106.4 | 106.7 | 106.8 | 105.9 | 106.1 | 105.7 | 105.7 |
| C9C10C11 | 107.4 | 107.4 | 107.4 | 107.3 | 106.9 | 106.8 | 106.9 | 106.9 |
| C10C11C12 | 107.4 | 107.4 | 107.5 | 107.5 | 106.9 | 106.9 | 106.9 | 106.9 |
| C11C12N5 | 106.4 | 106.4 | 106.2 | 106.2 | 105.4 | 105.2 | 105.7 | 105.7 |
| C12N5C9 | 109.9 | 110.1 | 110.0 | 110.1 | 112.3 | 112.3 | 112.5 | 112.5 |
| Bond angles sum (BAS53), deg | 537.5 | 537.7 | 537.8 | 537.9 | 537.4 | 537.3 | 537.7 | 537.7 |
| DFT Method Level | Complex | |||
|---|---|---|---|---|
| [BeSP] | [BeMBSP] | [BeDBSP] | [BeTBSP] | |
| DFT B3PW91/TZVP | 0.47 | 1.52 | 1.72 | 0.88 |
| DFT M062X/def2TZVP | 0.40 | 1.54 | 2.06 | 0.81 |
| Complex | DFT Method Level | Effective Charge on Atom, in Electron Charge Units (ē) | <S**2> | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Be1 | N1 | N2 | N3 | N4 | N5 | N6 | |||
| [BeSP] | B3PW91/TZVP | +1.642 | −0.401 | −0.828 | −0.828 | −0.401 | −0.828 | −0.401 | 0.0000 |
| M062X/def2TZVP | +1.678 | −0.416 | −0.849 | −0.849 | −0.416 | −0.849 | −0.416 | 0.0000 | |
| [BeMBSP] | B3PW91/TZVP | +1.644 | −0.419 | −0.828 | −0.810 | −0.419 | −0.828 | −0.388 | 0.0000 |
| M062X/def2TZVP | +1.679 | −0.438 | −0.847 | −0.838 | −0.438 | −0.847 | −0.392 | 0.0000 | |
| [BeDBSP] | B3PW91/TZVP | +1.645 | −0.405 | −0.830 | −0.808 | −0.436 | −0.808 | −0.405 | 0.0000 |
| M062X/def2TZVP | +1.680 | −0.411 | −0.848 | −0.833 | −0.466 | −0.833 | −0.411 | 0.0000 | |
| [BeTBSP] | B3PW91/TZVP | +1.647 | −0.420 | −0.806 | −0.806 | −0.420 | −0.806 | −0.420 | 0.0000 |
| M062X/def2TZVP | +1.681 | −0.434 | −0.827 | −0.827 | −0.434 | −0.827 | −0.434 | 0.0000 | |
| Complex | ΔfH0, kJ/mol | S0, J/mol∙K | ΔfG0, kJ/mol |
|---|---|---|---|
| [BeSP] | 538.7 | 532.7 | 669.4 |
| [BeMBSP] | 565.8 | 624.2 | 708.1 |
| [BeDBSP] | 595.5 | 715.7 | 749.3 |
| [BeTBSP] | 629.2 | 807.3 | 794.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chachkov, D.V.; Mikhailov, O.V.; Girichev, G.V. First Examples of s-Metal Complexes with Subporphyrazine and Its Phenylene-Annulated Derivatives: DFT Calculations. Int. J. Mol. Sci. 2024, 25, 6897. https://doi.org/10.3390/ijms25136897
Chachkov DV, Mikhailov OV, Girichev GV. First Examples of s-Metal Complexes with Subporphyrazine and Its Phenylene-Annulated Derivatives: DFT Calculations. International Journal of Molecular Sciences. 2024; 25(13):6897. https://doi.org/10.3390/ijms25136897
Chicago/Turabian StyleChachkov, Denis V., Oleg V. Mikhailov, and Georgiy V. Girichev. 2024. "First Examples of s-Metal Complexes with Subporphyrazine and Its Phenylene-Annulated Derivatives: DFT Calculations" International Journal of Molecular Sciences 25, no. 13: 6897. https://doi.org/10.3390/ijms25136897
APA StyleChachkov, D. V., Mikhailov, O. V., & Girichev, G. V. (2024). First Examples of s-Metal Complexes with Subporphyrazine and Its Phenylene-Annulated Derivatives: DFT Calculations. International Journal of Molecular Sciences, 25(13), 6897. https://doi.org/10.3390/ijms25136897