
Deciphering the Interplay between the Epithelial Barrier, Immune Cells, and Metabolic Mediators in Allergic Disease

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Epithelial Barrier Theory and Immune Regulation
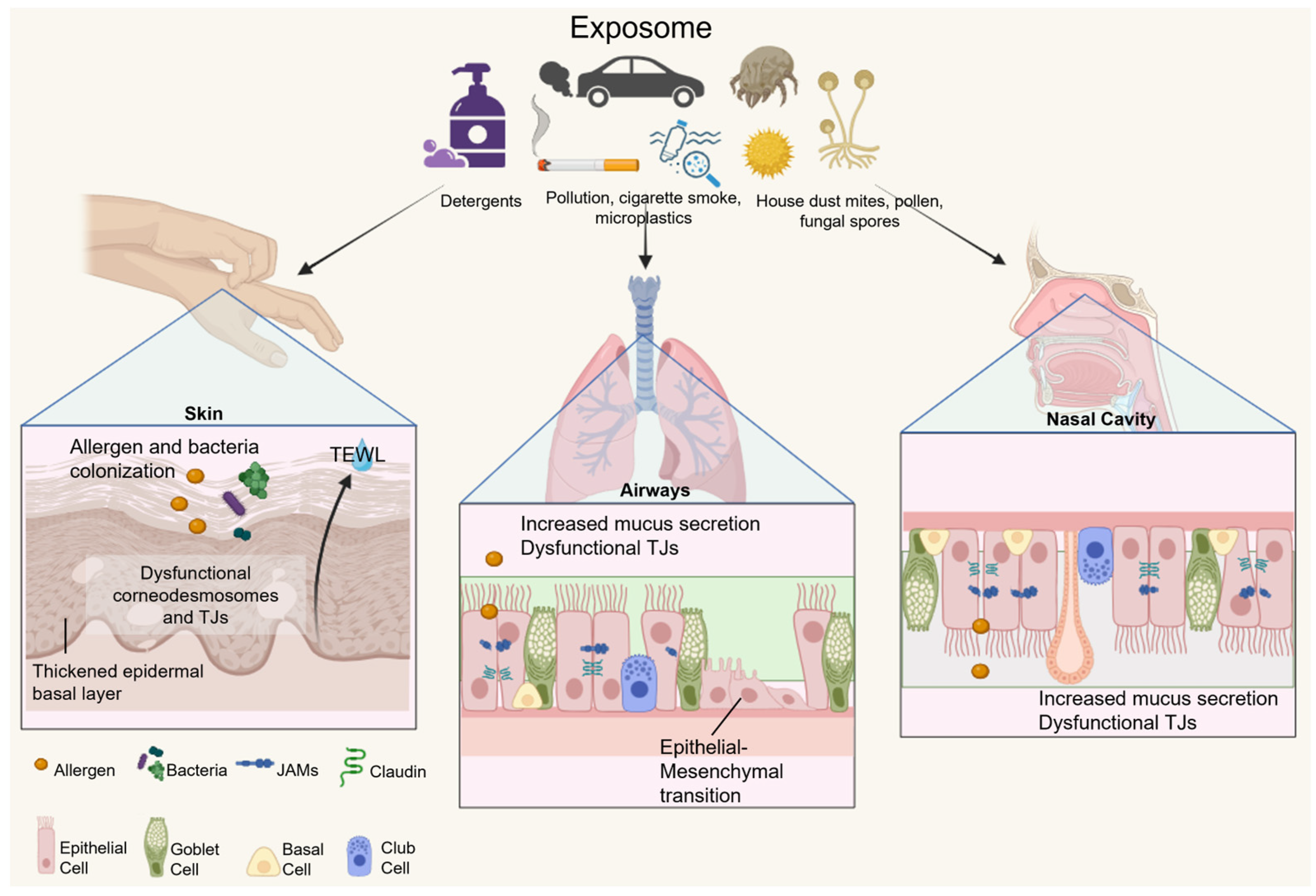
2.1. Epithelial Barrier Hypothesis
2.2. Characteristics and Functions of the Epithelial Barrier
3. Metabolism and Cell Effector Function
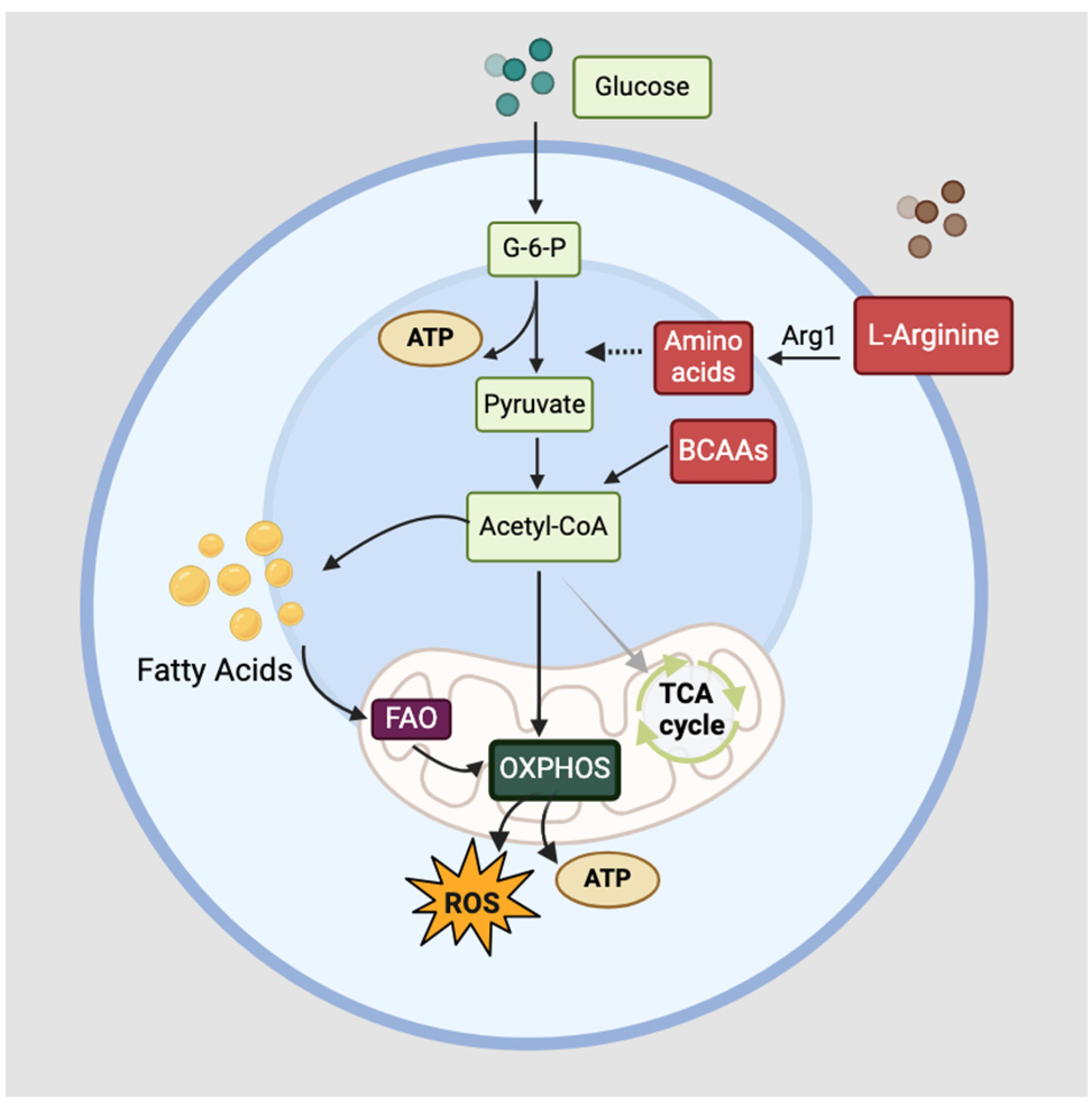
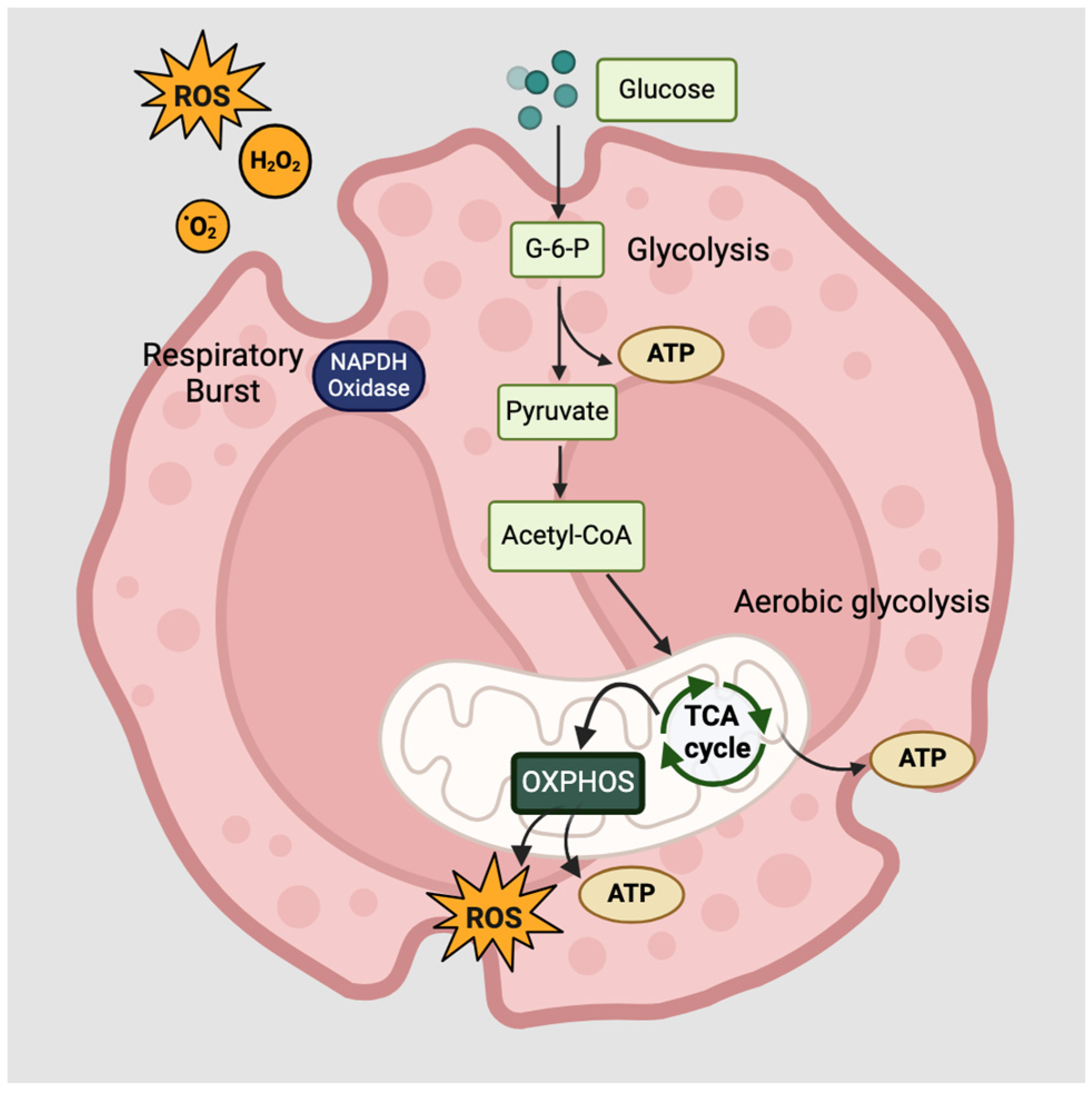
3.1. Cellular Metabolism
3.2. Functions of Innate Lymphoid Cells and Eosinophils
4. Lipid Metabolism in Allergic Diseases
5. Metabolic features of allergies
5.1. Atopic Dermatitis
5.2. Allergic Asthma
5.3. Allergic Rhinitis
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.; Xie, Y.; Tang, S.; Zhang, A. Small molecule metabolites: Discovery of biomarkers and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [PubMed]
- Baker, S.A.; Rutter, J. Metabolites as signalling molecules. Nat. Rev. Mol. Cell Biol. 2023, 24, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xu, W.; Zhao, S. Regulatory roles of metabolites in cell signaling networks. J. Genet. Genom. 2013, 40, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Samitas, K.; Carter, A.; Kariyawasam, H.H.; Xanthou, G. Upper and lower airway remodelling mechanisms in asthma, allergic rhinitis and chronic rhinosinusitis: The one airway concept revisited. Allergy 2018, 73, 993–1002. [Google Scholar] [CrossRef]
- Qin, Z.; Chen, Y.; Wang, Y.; Xu, Y.; Liu, T.; Mu, Q.; Huang, C. Immunometabolism in the pathogenesis of asthma. Immunology 2024, 171, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Vitte, J.; Vibhushan, S.; Bratti, M.; Montero-Hernandez, J.E.; Blank, U. Allergy, Anaphylaxis, and Nonallergic Hypersensitivity: IgE, Mast Cells, and Beyond. Med. Princ. Pract. 2022, 31, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, Y.; Zhang, H.; Hu, L.; Liu, J.; Wang, L.; Wang, T.; Zhang, H.; Cong, L.; Wang, Q. Pathogenesis of allergic diseases and implications for therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 138. [Google Scholar] [CrossRef]
- Falcon, R.M.G.; Caoili, S.E.C. Immunologic, genetic, and ecological interplay of factors involved in allergic diseases. Front. Allergy 2023, 4, 1215616. [Google Scholar] [CrossRef]
- Leon, B. Understanding the development of Th2 cell-driven allergic airway disease in early life. Front. Allergy 2023, 3, 1080153. [Google Scholar] [CrossRef]
- Goretzki, A.; Lin, Y.J.; Schülke, S. Immune metabolism in allergies, does it matter?—A review of immune metabolic basics and adaptations associated with the activation of innate immune cells in allergy. Allergy 2021, 76, 3314–3331. [Google Scholar] [CrossRef]
- Michaeloudes, C.; Bhavsar, P.K.; Mumby, S.; Xu, B.; Hui, C.K.M.; Chung, K.F.; Adcock, I.M. Role of Metabolic Reprogramming in Pulmonary Innate Immunity and Its Impact on Lung Diseases. J. Innate Immun. 2020, 12, 31–46. [Google Scholar] [CrossRef]
- Purohit, V.; Wagner, A.; Yosef, N.; Kuchroo, V.K. Systems-based approaches to study immunometabolism. Cell. Mol. Immunol. 2022, 19, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.; Rahman, A.; Mohammed, A.R. Application of genomics, proteomics and metabolomics in drug discovery, development and clinic. Ther. Deliv. 2013, 4, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Kucuksezer, U.C.; Ozdemir, C.; Yazici, D.; Pat, Y.; Mitamura, Y.; Li, M.; Sun, N.; D’Avino, P.; Bu, X.; Zhu, X.; et al. The epithelial barrier theory: Development and exacerbation of allergic and other chronic inflammatory diseases. Asia Pac. Allergy 2023, 13, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.; Weber, K.S.; Johnson, S.M. Exposome and Immunity Training: How Pathogen Exposure Order Influences Innate Immune Cell Lineage Commitment and Function. Int. J. Mol. Sci. 2020, 21, 8462. [Google Scholar] [CrossRef] [PubMed]
- Losol, P.; Sokolowska, M.; Hwang, Y.K.; Ogulur, I.; Mitamura, Y.; Yazici, D.; Pat, Y.; Radzikowska, U.; Ardicli, S.; Yoon, J.E.; et al. Epithelial Barrier Theory: The Role of Exposome, Microbiome, and Barrier Function in Allergic Diseases. Allergy Asthma Immunol. Res. 2023, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.E.; Lodge, C.J.; Lowe, A.J.; Burgess, J.A.; Matheson, M.C.; Dharmage, S.C. Exposure to ‘farming’ and objective markers of atopy: A systematic review and meta-analysis. Clin. Exp. Allergy 2015, 45, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Illi, S.; Depner, M.; Genuneit, J.; Horak, E.; Loss, G.; Strunz-Lehner, C.; Büchele, G.; Boznanski, A.; Danielewicz, H.; Cullinan, P.; et al. Protection from childhood asthma and allergy in Alpine farm environments-the GABRIEL Advanced Studies. J. Allergy Clin. Immunol. 2012, 129, 1470–1477.e6. [Google Scholar] [CrossRef]
- Kääriö, H.; Huttunen, K.; Karvonen, A.M.; Schaub, B.; von Mutius, E.; Pekkanen, J.; Hirvonen, M.R.; Roponen, M. Exposure to a farm environment is associated with T helper 1 and regulatory cytokines at age 4.5 years. Clin. Exp. Allergy 2016, 46, 71–77. [Google Scholar] [CrossRef]
- Deckers, J.; Marsland, B.J.; von Mutius, E. Protection against allergies: Microbes, immunity, and the farming effect. Eur. J. Immunol. 2021, 51, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, M. Trained immunity in allergic asthma. J. Allergy Clin. Immunol. 2023, 151, 1471–1473. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat. Rev. Immunol. 2021, 21, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, Y.; Ogulur, I.; Pat, Y.; Rinaldi, A.O.; Ardicli, O.; Cevhertas, L.; Brüggen, M.C.; Traidl-Hoffman, C.; Akdis, M.; Akdis, C.A. Dysregulation of the epithelial barrier by environmental and other exogenous factors. Contact Dermat. 2021, 85, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Yazici, D.; Ogulur, I.; Kucukkase, O.; Li, M.; Ronaldi, A.O.; Pat, Y.; Wallimann, A.; Wawrocki, S.; Sözener, Z.C.; Buyuktiryaki, B.; et al. Epithelial barrier hypothesis and the development of allergic and autoimmune diseases. Allergo J. Int. 2022, 31, 91–102. [Google Scholar] [CrossRef]
- Pat, Y.; Yazici, D.; D’Avino, P.; Li, M.; Ardicli, S.; Ardicli, O.; Mitamura, Y.; Akdis, M.; Dhir, R.; Nadeau, K.; et al. Recent advances in the epithelial barrier theory. Int. Immunol. 2024, 36, 211–222. [Google Scholar] [CrossRef]
- Mijač, S.; Banić, I.; Genc, A.M.; Lipej, M.; Turkalj, M. The Effects of Environmental Exposure on Epigenetic Modifications in Allergic Diseases. Medicina 2024, 60, 110. [Google Scholar] [CrossRef]
- Sözener, Z.C.; Cevhertas, L.; Nadeau, K.; Akdis, M.; Akdis, C.A. Environmental factors in epithelial barrier dysfunction. J. Allergy Clin. Immunol. 2020, 145, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Sozener, Z.C.; Yücel, Ü.Ö.; Altiner, S.; Oztürk, B.O.; Cerci, P.; Türk, M.; Akin, B.G.; Akdis, M.; Yilmaz, I.; Ozdemir, C.; et al. The External Exposome and Allergies: From the Perspective of the Epithelial Barrier Hypothesis. Front. Allergy 2022, 3, 887672. [Google Scholar]
- Zuo, L.; Kuo, W.T.; Turner, J.R. Tight Junctions as Targets and Effectors of Mucosal Immune Homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 327–340. [Google Scholar] [CrossRef]
- Hellings, P.W.; Steelant, B. Epithelial barriers in allergy and asthma. J. Allergy Clin. Immunol. 2020, 145, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, L.; Li, P.; Pang, K.; Liu, H.; Tian, L. Epithelial Barrier in the Nasal Mucosa, Related Risk Factors and Diseases. Int. Arch. Allergy Immunol. 2023, 184, 481–501. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Puddicombe, S.M.; Field, S.; Haywood, J.; Broughton-Head, V.; Puxeddu, I.; Haitchi, H.M.; Vernon-Wilson, E.; Sammut, D.; Bedke, N.; et al. Defective epithelial barrier function in asthma. J. Allergy Clin. Immunol. 2011, 128, 549–556. [Google Scholar] [CrossRef]
- Yu, L.C.; Wang, J.T.; Wei, S.C.; Ni, Y.H. Host-microbial interactions and regulation of intestinal epithelial barrier function: From physiology to pathology. World J. Gastrointest. Pathophysiol. 2012, 3, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Barbaro, M.R.; Fuschi, D.; Palombo, M.; Falangone, F.; Cremon, C.; Marasco, G.; Stanghellini, V. Inflammatory and Microbiota-Related Regulation of the Intestinal Epithelial Barrier. Front. Nutr. 2021, 8, 718356. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Magalhaes, J.G.; Tattoli, I.; Girardin, S.E. The intestinal epithelial barrier: How to distinguish between the microbial flora and pathogens. Semin. Immunol. 2007, 19, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hornef, M.W.; Dupont, A. The intestinal epithelium as guardian of gut barrier integrity. Cell. Microbiol. 2015, 17, 1561–1569. [Google Scholar] [CrossRef]
- Edelblum, K.L.; Turner, J.R. Chapter 12—Epithelial cells: Structure, transport, and barrier function. In Mucosal Immunology, 4th ed.; Mestecky, J., Strober, W., Russell, M., Kelsall, B., Cheroutre, H., Lambrecht, B., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 1, pp. 187–210. [Google Scholar]
- Lu, H.F.; Zhou, Y.C.; Yang, L.T.; Zhou, Q.; Wang, X.J.; Qiu, S.Q.; Cheng, B.H.; Zeng, X.H. Involvement and repair of epithelial barrier dysfunction in allergic diseases. Front. Immunol. 2024, 15, 1348272. [Google Scholar] [CrossRef]
- Goleva, E.; Berdyshev, E.; Leung, D.Y. Epithelial barrier repair and prevention of allergy. J. Clin. Investig. 2019, 129, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- van Smeden, J.; Janssens, M.; Gooris, G.S.; Bouwstra, J.A. The important role of stratum corneum lipids for the cutaneous barrier function. Biochim. Biophys. Acta. 2014, 1841, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 2867. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Woodfolk, J.A. Skin barrier defects in atopic dermatitis. Curr. Allergy Asthma Rep. 2014, 14, 433. [Google Scholar] [CrossRef]
- Rath, E.; Haller, D. Intestinal epithelial cell metabolism at the interface of microbial dysbiosis and tissue injury. Mucosal Immunol. 2022, 15, 595–604. [Google Scholar] [CrossRef]
- Ganesan, S.; Comstock, A.T.; Sajjan, U.S. Barrier function of airway tract epithelium. Tissue Barriers 2013, 1, e24997. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, E.; Rodriguez-Coira, J.; Delgado-Dolset, M.I.; Gomez-Casado, C.; Barber, D.; Escribese, M.M. Epithelial Barrier: Protector and Trigger of Allergic Disorders. J. Investig. Allergol. Clin. Immunol. 2022, 32, 81–96. [Google Scholar] [CrossRef]
- Laidlaw, T.M.; Mullol, J.; Woessner, K.M.; Amin, N.; Mannent, L.P. Chronic Rhinosinusitis with Nasal Polyps and Asthma. J. Allergy Clin. Immunol. Pract. 2021, 9, 1133–1141. [Google Scholar] [CrossRef]
- Johnson, J.R.; Roos, A.; Berg, T.; Nord, M.; Fuxe, J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS ONE 2011, 6, e16175. [Google Scholar] [CrossRef]
- Heijink, I.H.; Postma, D.S.; Noordhoek, J.A.; Broekema, M.; Kapus, A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am. J. Respir. Cell. Mol. Biol. 2010, 42, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef] [PubMed]
- Mogilenko, D.A.; Sergushichev, A.; Artyomov, M.N. Systems Immunology Approaches to Metabolism. Annu. Rev. Immunol. 2023, 41, 317–342. [Google Scholar] [CrossRef] [PubMed]
- Wolowczuk, I.; Verwaerde, C.; Viltart, O.; Delanoye, A.; Delacre, M.; Pot, B.; Grangette, C. Feeding our immune system: Impact on metabolism. J. Immunol. Res. 2008, 2008, 639803. [Google Scholar] [CrossRef]
- Sun, L.; Yang, X.; Yuan, Z.; Wang, H. Metabolic Reprogramming in Immune Response and Tissue Inflammation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1990–2001. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Hortová-Kohoutková, M.; Lázniçková, P.; Friç, J. How immune-cell fate and function are determined by metabolic pathway choice: The bioenergetics underlying the immune response. Bioessays 2021, 43, e2000067. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, X. Metabolic Reprogramming in Resting and Activated Immune Cells. Metabolomics 2017, 7, 188. [Google Scholar]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2021, 21, 363–381. [Google Scholar] [CrossRef]
- Olenchock, B.A.; Rathmell, J.C.; Vander Heiden, M.G. Biochemical Underpinnings of Immune Cell Metabolic Phenotypes. Immunity 2017, 46, 703–713. [Google Scholar] [CrossRef]
- Chi, H. Immunometabolism at the intersection of metabolic signaling, cell fate, and systems immunology. Cell. Mol. Immunol. 2022, 19, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Raval, F.M.; Nikolajczyk, B.S. The Bidirectional Relationship between Metabolism and Immune Responses. Discoveries 2013, 1, e6. [Google Scholar] [CrossRef]
- Zhang, X.; Zink, F.; Hezel, F.; Vogt, J.; Wachter, U.; Wepler, M.; Loconte, M.; Kranz, C.; Hellmann, A.; Mizaikoff, B.; et al. Metabolic substrate utilization in stress-induced immune cells. Intensive Care Med. Exp. 2020, 8 (Suppl. S1), 28. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sirohi, K.; Verma, M.; McKay, J.; Michalec, L.; Sripada, A.; Danhorn, T.; Rollins, D.; Good, J.; Gorska, M.M.; et al. Optimal identification of human conventional and nonconventional (CRTH2−IL7Rα−) ILC2s using additional surface markers. J. Allergy Clin. Immunol. 2020, 146, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N. Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef] [PubMed]
- Herbert, D.R.; Douglas, B.; Zullo, K. Group 2 Innate Lymphoid Cells (ILC2): Type 2 Immunity and Helminth Immunity. Int. J. Mol. Sci. 2019, 20, 2276. [Google Scholar] [CrossRef] [PubMed]
- Konya, V.; Mjösberg, J. Lipid mediators as regulators of human ILC2 function in allergic diseases. Immunol. Lett. 2016, 179, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, I.; Steer, C.A.; Takei, F. Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol. 2015, 36, 189–195. [Google Scholar] [CrossRef]
- Goretzki, A.; Zimmermann, J.; Rainer, H.; Lin, Y.J.; Schülke, S. Immune Metabolism in TH2 Responses: New Opportunities to Improve Allergy Treatment—Disease-Specific Findings (Part 1). Curr. Allergy Asthma Rep. 2023, 23, 29–40. [Google Scholar] [CrossRef]
- Surace, L.; Doisne, J.M.; Croft, C.A.; Thaller, A.; Escoll, P.; Marie, S.; Petrosemoli, N.; Guillemot, V.; Dardalhon, V.; Topazio, D.; et al. Dichotomous metabolic networks govern human ILC2 proliferation and function. Nat. Immunol. 2021, 22, 1367–1374. [Google Scholar] [CrossRef]
- Pelletier, A.; Stockmann, C. The Metabolic Basis of ILC Plasticity. Front. Immunol. 2022, 13, 858051. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, C.; Berti, A.; Cottini, M. The emerging roles of eosinophils: Implications for the targeted treatment of eosinophilic-associated inflammatory conditions. Curr. Res. Immunol. 2022, 3, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.C.; Van Dyken, S.J.; von Moltke, J.; Cheng, L.E.; Mohapatra, A.; Molofsky, A.B.; Thornton, E.E.; Krummel, M.F.; Chawla, A.; Liang, H.E.; et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013, 502, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Davoine, F.; Lacy, P. Eosinophil cytokines, chemokines, and growth factors: Emerging roles in immunity. Front. Immunol. 2014, 5, 570. [Google Scholar] [CrossRef] [PubMed]
- Fettrelet, T.; Gigon, L.; Karaulov, A.; Yousefi, S.; Simon, H.U. The Enigma of Eosinophil Degranulation. Int. J. Mol. Sci. 2021, 22, 7091. [Google Scholar] [CrossRef] [PubMed]
- Lacy, P. Chapter 12—Eosinophil cytokines in allergy. In Cytokine Effector Functions in Tissues; Foti, M., Locati, M., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 173–218. [Google Scholar]
- Martin, L.B.; Kita, H.; Leiferman, K.M.; Gleich, G.J. Eosinophils in allergy: Role in disease, degranulation, and cytokines. Int. Arch. Allergy Immunol. 1996, 109, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Gigon, L.; Fettrelet, T.; Yousefi, S.; Simon, D.; Simon, H.U. Eosinophils from A to Z. Allergy 2023, 78, 1810–1846. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Vincent, E.E.; Felix, L.C.; Cronin, J.G.; Scott, L.M.; Hole, P.S.; Lacy, P.; Thornton, C.A. Interleukin-5 drives glycolysis and reactive oxygen species-dependent citric acid cycling by eosinophils. Allergy 2020, 75, 1361–1370. [Google Scholar] [CrossRef]
- Wu, W.; Samoszuk, M.K.; Comhair, S.A.; Thomassen, M.J.; Farver, C.F.; Dweik, R.A.; Kavuru, M.S.; Erzurum, S.C.; Hazen, S.L. Eosinophils generate brominating oxidants in allergen-induced asthma. J. Clin. Investig. 2000, 105, 1455–1463. [Google Scholar] [CrossRef]
- Gaurav, R.; Bewtra, A.K.; Agrawal, D.K. Chloride Channel 3 Channels in the Activation and Migration of Human Blood Eosinophils in Allergic Asthma. Am. J. Respir. Cell Mol. Biol. 2015, 53, 235–245. [Google Scholar] [CrossRef]
- Yousefi, S.; Simon, D.; Simon, H.U. Eosinophil extracellular DNA traps: Molecular mechanisms and potential roles in disease. Curr. Opin. Immunol. 2012, 24, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Le Pham, D.; Lee, D.H.; Lee, S.H.; Kim, S.H.; Park, H.S. Biological function of eosinophil extracellular traps in patients with severe eosinophilic asthma. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Silveira, J.S.; Antunes, G.L.; Kaiber, D.B.; da Costa, M.S.; Marques, E.P.; Ferreira, F.S.; Gassen, R.B.; Breda, R.V.; Wyse, A.T.S.; Pitrez, P.; et al. Reactive oxygen species are involved in eosinophil extracellular traps release and in airway inflammation in asthma. J. Cell. Physiol. 2019, 234, 23633–23646. [Google Scholar] [CrossRef] [PubMed]
- Schauberger, E.; Peinhaupt, M.; Cazares, T.; Lindsley, A.W. Lipid Mediators of Allergic Disease: Pathways, Treatments, and Emerging Therapeutic Targets. Curr. Allergy Asthma Rep. 2016, 16, 48. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T. The roles of lipid mediators in type I hypersensitivity. J. Pharmacol. Sci. 2021, 147, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Miyata, J.; Fukunaga, K.; Kawashima, Y.; Ohara, O.; Kawana, A.; Asano, K.; Arita, M. Dysregulated metabolism of polyunsaturated fatty acids in eosinophilic allergic diseases. Prostaglandins Other Lipid Mediat. 2020, 150, 106477. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef] [PubMed]
- Di Gennaro, A.; Haeggström, J.Z. The leukotrienes: Immune-modulating lipid mediators of disease. Adv. Immunol. 2012, 116, 51–92. [Google Scholar]
- Sasaki, F.; Yokomizo, T. The leukotriene receptors as therapeutic targets of inflammatory diseases. Int. Immunol. 2019, 31, 607–615. [Google Scholar] [CrossRef]
- Jo-Watanabe, A.; Okuno, T.; Yokomizo, T. The Role of Leukotrienes as Potential Therapeutic Targets in Allergic Disorders. Int. J. Mol. Sci. 2019, 20, 3580. [Google Scholar] [CrossRef]
- Liu, M.; Yokomizo, T. The role of leukotrienes in allergic diseases. Allergol. Int. 2015, 64, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Steinke, J.W.; Borish, L. Leukotriene receptors in rhinitis and sinusitis. Curr. Allergy Asthma Rep. 2004, 4, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Voisin, T.; Perner, C.; Messou, M.A.; Shiers, S.; Ualiyeva, S.; Kanaoka, Y.; Price, T.J.; Sokol, C.L.; Bankova, L.G.; Austen, K.F.; et al. The CysLT2R receptor mediates leukotriene C4-driven acute and chronic itch. Proc. Natl. Acad. Sci. USA 2021, 118, e2022087118. [Google Scholar] [CrossRef]
- Bankova, L.G.; Boyce, J.A. A new spin on mast cells and cysteinyl leukotrienes: Leukotriene E4 activates mast cells in vivo. J. Allergy Clin. Immunol. 2018, 142, 1056–1057. [Google Scholar] [CrossRef]
- Salimi, M.; Stöger, L.; Liu, W.; Go, S.; Pavord, I.; Klenerman, P.; Ogg, G.; Xue, L. Cysteinyl leukotriene E4 activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D2 and epithelial cytokines. J. Allergy Clin. Immunol. 2017, 140, 1090–1100.e11. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R., Jr.; Tang, L.O.; Chu, S.J.; Tsao, S.M.; Chiang, G.K.; Jones, F.; Jonas, M.; Pae, C.; Wang, H.; Chi, E.Y. A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am. J. Respir. Crit. Care Med. 2002, 165, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Oyesola, O.O.; Tait Wojno, E.D. Prostaglandin regulation of type 2 inflammation: From basic biology to therapeutic interventions. Eur. J. Immunol. 2021, 51, 2399–2416. [Google Scholar] [CrossRef]
- Peebles, R.S., Jr. Prostaglandins in asthma and allergic diseases. Pharmacol. Ther. 2019, 193, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Murata, T. Regulation of vascular permeability in anaphylaxis. Br. J. Pharmacol. 2018, 175, 2538–2542. [Google Scholar] [CrossRef]
- Kupczyk, M.; Kuna, P. Targeting the PGD2/CRTH2/DP1 Signaling Pathway in Asthma and Allergic Disease: Current Status and Future Perspectives. Drugs 2017, 77, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Duffney, P.F.; Falsetta, M.L.; Rackow, A.R.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Key roles for lipid mediators in the adaptive immune response. J. Clin. Investig. 2018, 128, 2724–2731. [Google Scholar] [CrossRef]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, R.; Rezaiemanesh, A.; Mortazavi, S.H.; Karaji, A.G.; Salari, F. Immunoresolvents in asthma and allergic diseases: Review and update. J. Cell. Physiol. 2019, 234, 8579–8596. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Menon, C.; Hoffstad, O.; Bilker, W.; Leclerc, P.; Margolis, D.J. The prevalence of atopic triad in children with physician-confirmed atopic dermatitis. J. Am. Acad. Dermatol. 2008, 58, 68–73. [Google Scholar] [CrossRef]
- Peng, W.; Novak, N. Pathogenesis of atopic dermatitis. Clin. Exp. Allergy 2015, 45, 566–574. [Google Scholar] [CrossRef]
- Luger, T.; Amagai, M.; Dreno, B.; Dagnelie, M.A.; Liao, W.; Kabashima, K.; Schikowski, T.; Proksch, E.; Elias, P.M.; Simon, M.; et al. Atopic dermatitis: Role of the skin barrier, environment, microbiome, and therapeutic agents. J. Dermatol. Sci. 2021, 102, 142–157. [Google Scholar] [CrossRef]
- Afghani, J.; Traidl-Hoffmann, C.; Schmitt-Kopplin, P.; Reiger, M.; Mueller, C. An Overview of the Latest Metabolomics Studies on Atopic Eczema with New Directions for Study. Int. J. Mol. Sci. 2022, 23, 8791. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Sato, W.J.; Kelly, A.; Ganguli-Indra, G.; Indra, A.K. Epidermal Lipids: Key Mediators of Atopic Dermatitis Pathogenesis. Trends Mol. Med. 2019, 25, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Pavel, P.; Leman, G.; Hermann, M.; Ploner, C.; Eichmann, T.O.; Minzaghi, D.; Radner, F.P.W.; Del Frari, B.; Gruber, R.; Dubrac, S. Peroxisomal Fatty Acid Oxidation and Glycolysis Are Triggered in Mouse Models of Lesional Atopic Dermatitis. JID Innov. 2021, 1, 100033. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, G.; Liu, X.; Shao, Y.; Gao, P.; Xin, C.; Cui, Z.; Zhao, X.; Xu, G. Serum metabolomics study and eicosanoid analysis of childhood atopic dermatitis based on liquid chromatography-mass spectrometry. J. Proteome Res. 2014, 13, 5715–5723. [Google Scholar] [CrossRef] [PubMed]
- Töröcsik, D.; Weise, C.; Gericke, J.; Szegedi, A.; Lucas, R.; Mihaly, J.; Worm, M.; Rühl, R. Transcriptomic and lipidomic profiling of eicosanoid/docosanoid signalling in affected and non-affected skin of human atopic dermatitis patients. Exp. Dermatol. 2019, 28, 177–189. [Google Scholar] [CrossRef]
- Nicolaou, A. Eicosanoids in skin inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 131–138. [Google Scholar] [CrossRef]
- Hamers, A.; Primus, C.P.; Whitear, C.; Kumar, N.A.; Masucci, M.; Montalvo Moreira, S.A.; Rathod, K.; Chen, J.; Bubb, K.; Colas, R.; et al. 20-hydroxyeicosatetraenoic acid (20-HETE) is a pivotal endogenous ligand for TRPV1-mediated neurogenic inflammation in the skin. Br. J. Pharmacol. 2022, 179, 1450–1469. [Google Scholar] [CrossRef] [PubMed]
- Kiezel-Tsugunova, M.; Kendall, A.C.; Nicolaou, A. Fatty acids and related lipid mediators in the regulation of cutaneous inflammation. Biochem. Soc. Trans. 2018, 46, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Herrmann, N.; Maintz, L.; Nümm, T.J.; Welchowski, T.; Claus, R.A.; Gräler, M.H.; Bieber, T. Serum sphingosine-1-phosphate is elevated in atopic dermatitis and associated with severity. Allergy 2021, 76, 2592–2595. [Google Scholar] [CrossRef]
- Kleuser, B.; Bäumer, W. Sphingosine 1-Phosphate as Essential Signaling Molecule in Inflammatory Skin Diseases. Int. J. Mol. Sci. 2023, 24, 1456. [Google Scholar] [CrossRef]
- Wedman, P.A.; Aladhami, A.; Chumanevich, A.P.; Fuseler, J.W.; Oskeritzian, C.A. Mast cells and sphingosine-1-phosphate underlie prelesional remodeling in a mouse model of eczema. Allergy 2018, 73, 405–415. [Google Scholar] [CrossRef]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Ottas, A.; Fishman, D.; Okas, T.L.; Püssa, T.; Toomik, P.; Märtson, A.; Kingo, K.; Soomets, U. Blood serum metabolome of atopic dermatitis: Altered energy cycle and the markers of systemic inflammation. PLoS ONE 2017, 12, e0188580. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.Z.; Deng, J.; Parthasarathy, V.; Lee, K.K.; Pritchard, T.; Guo, S.; Zhang, C.; Kwatra, M.M.; Le, A.; Kwatra, S.G. Integrated plasma metabolomic and cytokine analysis reveals a distinct immunometabolic signature in atopic dermatitis. Front. Immunol. 2024, 15, 1354128. [Google Scholar] [CrossRef] [PubMed]
- Oyoshi, M.K.; He, R.; Kanaoka, Y.; ElKhal, A.; Kawamoto, S.; Lewis, C.N.; Austen, K.F.; Geha, R.S. Eosinophil-derived leukotriene C4 signals via type 2 cysteinyl leukotriene receptor to promote skin fibrosis in a mouse model of atopic dermatitis. Proc. Natl. Acad. Sci. USA 2012, 109, 4992–4997. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, S.; Mehta, H.; Bissonnette, R.; Sarfati, M. Increased frequencies of baso-phils, type 2 innate lymphoid cells and Th2 cells in skin of patients with atopic dermatitis but not psoriasis. J. Dermatol. Sci. 2017, 88, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Alkon, N.; Bauer, W.M.; Krausgruber, T.; Goh, I.; Griss, J.; Nguyen, V.; Reininger, B.; Bangert, C.; Staud, C.; Brunner, P.M.; et al. Single-cell analysis reveals innate lymphoid cell lineage infidelity in atopic dermatitis. J. Allergy Clin. Immunol. 2022, 149, 624–639. [Google Scholar] [CrossRef]
- Chang, J.E.; Doherty, T.A.; Baum, R.; Broide, D. Prostaglandin D2 regulates human type 2 innate lymphoid cell chemotaxis. J. Allergy Clin. Immunol. 2014, 133, 899–901.e3. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.; Jeffery, P.K.; Busse, W.W.; Johnson, M.; Vignola, A.M. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am. J. Respir. Crit. Care Med. 2000, 161, 1720–1745. [Google Scholar] [CrossRef] [PubMed]
- Banno, A.; Reddy, A.T.; Lakshmi, S.P.; Reddy, R.C. Bidirectional interaction of airway epithelial remodeling and inflammation in asthma. Clin. Sci. 2020, 134, 1063–1079. [Google Scholar] [CrossRef]
- Varricchi, G.; Ferri, S.; Pepys, J.; Poto, R.; Spadaro, G.; Nappi, E.; Paoletti, G.; Virchow, J.C.; Heffler, E.; Canonica, W.G. Biologics and airway remodeling in severe asthma. Allergy 2022, 77, 3538–3552. [Google Scholar] [CrossRef]
- Kubo, M. Innate and adaptive type 2 immunity in lung allergic inflammation. Immunol. Rev. 2017, 278, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Bian, X.; Zhai, Y.; Li, C.; Li, N.; Hao, C.; Huang, H.; Luo, W.; Huang, Z.; Liao, C.; et al. Metabolomics reveals a correlation between hydroxyeicosatetraenoic acids and allergic asthma: Evidence from three years’ immunotherapy. Pediatr. Allergy Immunol. 2021, 32, 1654–1662. [Google Scholar] [CrossRef]
- Gai, X.Y.; Zhang, L.J.; Chang, C.; Guo, C.L.; Abulikemu, M.; Li, W.X.; Wang, J.; Yao, W.Z.; Zhang, X. Metabolomic Analysis of Serum Glycerophospholipid Levels in Eosinophilic and Neutrophilic Asthma. Biomed. Environ. Sci. 2019, 32, 96–106. [Google Scholar] [PubMed]
- Kelly, R.S.; Virkud, Y.; Giorgio, R.; Celedón, J.C.; Weiss, S.T.; Lasky-Su, J. Metabolomic profiling of lung function in Costa-Rican children with asthma. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Chen, M.; Bao, Y.L.; Xu, C.D.; Qin, Q.Z.; Zhang, W.X.; He, Y.T.; Shao, Q. Sputum metabolomic profiling of bronchial asthma based on quadruple time-of-flight mass spectrometry. Int. J. Clin. Exp. Pathol. 2017, 10, 10363–10373. [Google Scholar]
- Seng, J.A.; Nealon, J.R.; Blanksby, S.J.; Mitchell, T.W. Distribution of Glycerophospholipids in the Adult Human Lens. Biomolecules 2018, 8, 156. [Google Scholar] [CrossRef]
- Pascoe, C.D.; Jha, A.; Ryu, M.H.; Ragheb, M.; Vaghasiya, J.; Basu, S.; Stelmack, G.L.; Srinathan, S.; Kidane, B.; Kindrachuk, J.; et al. Allergen inhalation generates pro-inflammatory oxidised phosphatidylcholine associated with airway dysfunction. Eur. Respir. J. 2021, 57, 2000839. [Google Scholar] [CrossRef]
- Sano, A.; Sano, H.; Iwanaga, T.; Tohda, Y. Functional role of phosphatidylcholine-specific phospholipase C in regulating leukotriene synthesis and degranulation in human eosinophils. Eur. J. Pharmacol. 2020, 884, 173353. [Google Scholar] [CrossRef]
- Ammit, A.J.; Hastie, A.T.; Edsall, L.C.; Hoffman, R.K.; Amrani, Y.; Krymskaya, V.P.; Kane, S.A.; Peters, S.P.; Penn, R.B.; Spiegel, S.; et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. 2001, 15, 1212–1214. [Google Scholar] [CrossRef]
- Costello, R.W.; Maloney, M.; Atiyeh, M.; Gleich, G.; Walsh, M.T. Mechanism of sphingosine 1-phosphate- and lysophosphatidic acid-induced up-regulation of adhesion molecules and eosinophil chemoattractant in nerve cells. Int. J. Mol. Sci. 2011, 12, 3237–3249. [Google Scholar] [CrossRef]
- Fuerst, E.; Foster, H.R.; Ward, J.P.; Corrigan, C.J.; Cousins, D.J.; Woszczek, G. Sphingosine-1-phosphate induces pro-remodelling response in airway smooth muscle cells. Allergy 2014, 69, 1531–1539. [Google Scholar] [CrossRef]
- Nishiuma, T.; Nishimura, Y.; Okada, T.; Kuramoto, E.; Kotani, Y.; Jahangeer, S.; Nakamura, S. Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1085–L1093. [Google Scholar] [CrossRef]
- Montuschi, P.; Santini, G.; Valente, S.; Mondino, C.; Macagno, F.; Cattani, P.; Zini, G.; Mores, N. Liquid chromatography-mass spectrometry measurement of leukotrienes in asthma and other respiratory diseases. J Chromatogr B Analyt Technol Biomed Life Sci. 2014, 964, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Akuthota, P.; Kanaoka, Y.; Weller, P.F. Airway eosinophil migration into lymph nodes in mice depends on leukotriene C4. Allergy. 2017, 72, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Gauvreau, G.M.; Parameswaran, K.N.; Watson, R.M.; O’Byrne, P.M. Inhaled leukotriene E(4), but not leukotriene D(4), increased airway inflammatory cells in subjects with atopic asthma. Am. J. Respir. Crit. Care Med. 2001, 164, 1495–1500. [Google Scholar] [CrossRef]
- Neves, J.S.; Radke, A.L.; Weller, P.F. Cysteinyl leukotrienes acting via granule membrane-expressed receptors elicit secretion from within cell-free human eosinophil granules. J. Allergy Clin. Immunol. 2010, 125, 477–482. [Google Scholar] [CrossRef]
- Pelaia, C.; Crimi, C.; Vatrella, A.; Busceti, M.T.; Gaudio, A.; Garofalo, E.; Bruni, A.; Terracciano, R.; Pelaia, G. New treatments for asthma: From the pathogenic role of prostaglandin D2 to the therapeutic effects of fevipiprant. Pharmacol. Res. 2020, 155, 104490. [Google Scholar] [CrossRef]
- Tang, M.; Da, X.; Xu, Z.; Zhao, X.; Zhou, H. UHPLC/MS-based metabolomics of asthmatic mice reveals metabolic changes in group 2 innate lymphoid cells. Int. Immunopharmacol. 2024, 130, 111775. [Google Scholar] [CrossRef] [PubMed]
- LeSuer, W.E.; Kienzl, M.; Ochkur, S.I.; Schicho, R.; Doyle, A.D.; Wright, B.L.; Rank, M.A.; Krupnick, A.S.; Kita, H.; Jacobsen, E.A. Eosinophils promote effector functions of lung group 2 innate lymphoid cells in allergic airway inflammation in mice. J. Allergy Clin. Immunol. 2023, 152, 469–485.e10. [Google Scholar] [CrossRef]
- Pawankar, R.; Mori, S.; Ozu, C.; Kimura, S. Overview on the pathomechanisms of allergic rhinitis. Asia Pac. Allergy. 2011, 3, 157–167. [Google Scholar] [CrossRef]
- Liva, G.A.; Karatzanis, A.D.; Prokopakis, E.P. Review of Rhinitis: Classification, Types, Pathophysiology. J. Clin. Med. 2021, 10, 3183. [Google Scholar] [CrossRef] [PubMed]
- Nur Husna, S.M.; Tan, H.T.; Md Shukri, N.; Mohd Ashari, N.S.; Wong, K.K. Nasal Epithelial Barrier Integrity and Tight Junctions Disruption in Allergic Rhinitis: Overview and Pathogenic Insights. Front. Immunol. 2021, 12, 663626. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.C.; Wang, T.S.; Wang, J.; Ma, Z.J.; Pu, S.B. Serum metabolomics study of patients with allergic rhinitis. Biomed. Chromatogr. 2020, 34, e4739. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, J.; Liu, Y.; Zhang, N.; Holtappels, G.; Lin, P.; Liu, S.; Bachert, C. Inflammatory profiles in nasal mucosa of patients with persistent vs intermittent allergic rhinitis. Allergy 2010, 65, 1149–1157. [Google Scholar] [CrossRef]
- Mackle, T.; Gendy, S.S.; Walsh, M.; McConn-Walsh, R.; Costello, R.W.; Walsh, M.T. Role of sphingosine 1-phosphate receptor expression in eosinophils of patients with allergic rhinitis, and effect of topical nasal steroid treatment on this receptor expression. J. Laryngol. Otol. 2008, 122, 1309–1317. [Google Scholar] [CrossRef]
- Tojima, I.; Shimizu, T. Group 2 innate lymphoid cells and eosinophilic chronic rhinosinusitis. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Bandeira-Melo, C.; Bozza, P.T.; Weller, P.F. The cellular biology of eosinophil eicosanoid formation and function. J. Allergy Clin. Immunol. 2002, 109, 393–400. [Google Scholar] [CrossRef]
- Powell, W.S.; Rokach, J. The eosinophil chemoattractant 5-oxo-ETE and the OXE receptor. Prog. Lipid Res. 2013, 52, 651–665. [Google Scholar] [CrossRef]
- Lin, L.; Dai, F.; Wei, J.; Chen, Z. Biological Roles of 5-Oxo-6,8,11,14-Eicosatetraenoic Acid and the OXE Receptor in Allergic Diseases: Collegium Internationale Allergologicum Update 2024. Int. Arch. Allergy Immunol. 2024, 185, 301–310. [Google Scholar] [CrossRef]
- Kowal, K.; Gielicz, A.; Sanak, M. The effect of allergen-induced bronchoconstriction on concentration of 5-oxo-ETE in exhaled breath condensate of house dust mite-allergic patients. Clin. Exp. Allergy 2017, 47, 1253–1262. [Google Scholar] [CrossRef]
- Schjødt, M.S.; Gürdeniz, G.; Chawes, B. The Metabolomics of Childhood Atopic Diseases: A Comprehensive Pathway-Specific Review. Metabolites 2020, 10, 511. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Park, Y.M.; Yoo, H.J.; Hong, S.J. Metabolomic pathways in food allergy. Pediatr. Allergy Immunol. 2024, 35, e14133. [Google Scholar] [CrossRef] [PubMed]
- van Ginkel, C.D.; Flokstra-de Blok, B.M.; Kollen, B.J.; Kukler, J.; Koppelman, G.H.; Dubois, A.E. Loss-of-function variants of the filaggrin gene are associated with clinical reactivity to foods. Allergy 2015, 70, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, S.W.; Hong, S. Regulation of Allergic Immune Responses by Microbial Metabolites. Immune Netw. 2018, 18, e15. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Alashkar Alhamwe, B.; Santner-Nanan, B.; Miethe, S.; Harb, H.; Renz, H.; Potaczek, D.P.; Nanan, R.K. Short-Chain Fatty Acids Augment Differentiation and Function of Human Induced Regulatory T Cells. Int. J. Mol. Sci. 2022, 23, 5740. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, S.H.; Yoon, H.J.; Paik, D.J.; Kim, J.M.; Youn, J. Bacillus-derived poly-γ-glutamic acid attenuates allergic airway inflammation through a Toll-like receptor-4-dependent pathway in a murine model of asthma. Clin. Exp. Allergy 2011, 41, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Michaudel, C.; Sokol, H. The Gut Microbiota at the Service of Immunometabolism. Cell Metab. 2020, 32, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Sturm, E.M.; Schratl, P.; Schuligoi, R.; Konya, V.; Sturm, G.J.; Lippe, I.T.; Peskar, B.A.; Heinemann, A. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J. Immunol. 2008, 181, 7273–7283. [Google Scholar] [CrossRef] [PubMed]
- Stepanovska, B.; Huwiler, A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Thieme, M.; Zillikens, D.; Sadik, C.D. Sphingosine-1-phosphate modulators in inflammatory skin diseases—Lining up for clinical translation. Exp. Dermatol. 2017, 26, 206–210. [Google Scholar] [CrossRef]
- Karmouty-Quintana, H.; Siddiqui, S.; Hassan, M.; Tsuchiya, K.; Risse, P.A.; Xicota-Vila, L.; Marti-Solano, M.; Martin, J.G. Treatment with a sphingosine-1-phosphate analog inhibits airway remodeling following repeated allergen exposure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L736–L745. [Google Scholar] [CrossRef] [PubMed]
- Jutel, M.; Agache, I.; Zemelka-Wiacek, M.; Akdis, M.; Chivato, T.; Del Giacco, S.; Gajdanowicz, P.; Gracia, I.E.; Klimek, L.; Lauerma, A.; et al. Nomenclature of allergic diseases and hypersensitivity reactions: Adapted to modern needs: An EAACI position paper. Allergy 2023, 78, 2851–2874. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kan, L.L.-Y.; Li, P.; Hon, S.S.-M.; Lai, A.Y.-T.; Li, A.; Wong, K.C.-Y.; Huang, D.; Wong, C.-K. Deciphering the Interplay between the Epithelial Barrier, Immune Cells, and Metabolic Mediators in Allergic Disease. Int. J. Mol. Sci. 2024, 25, 6913. https://doi.org/10.3390/ijms25136913
Kan LL-Y, Li P, Hon SS-M, Lai AY-T, Li A, Wong KC-Y, Huang D, Wong C-K. Deciphering the Interplay between the Epithelial Barrier, Immune Cells, and Metabolic Mediators in Allergic Disease. International Journal of Molecular Sciences. 2024; 25(13):6913. https://doi.org/10.3390/ijms25136913
Chicago/Turabian StyleKan, Lea Ling-Yu, Peiting Li, Sharon Sze-Man Hon, Andrea Yin-Tung Lai, Aixuan Li, Katie Ching-Yau Wong, Danqi Huang, and Chun-Kwok Wong. 2024. "Deciphering the Interplay between the Epithelial Barrier, Immune Cells, and Metabolic Mediators in Allergic Disease" International Journal of Molecular Sciences 25, no. 13: 6913. https://doi.org/10.3390/ijms25136913