

The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases


Abstract
1. Introduction
2. NAD+ Metabolism in the Kidney and Sirtuins
3. SIRT3: Structure, Expression and Function in the Renal Tissue
4. SIRT3 Deficiency in Renal Diseases and Pharmacological Interventions
5. SIRT3 and Renal Cell Adaptation to Metabolic Challenges
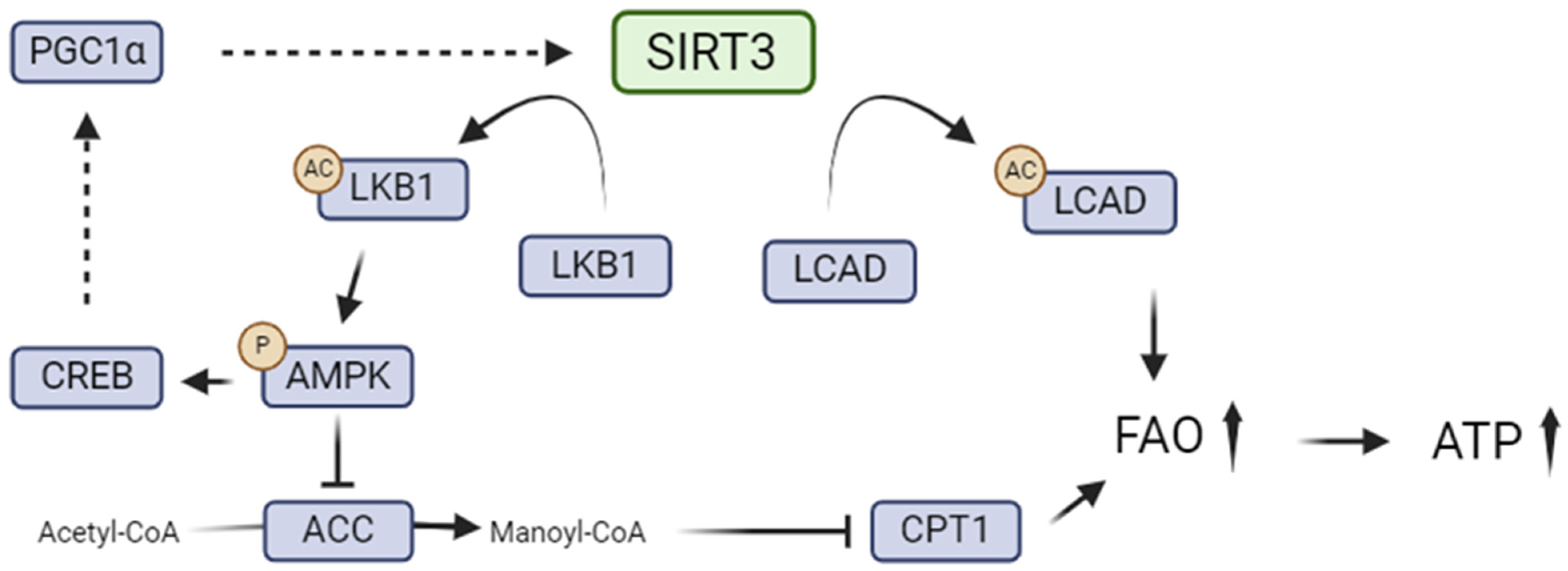
5.1. SIRT3 and Renal Lipid Metabolism
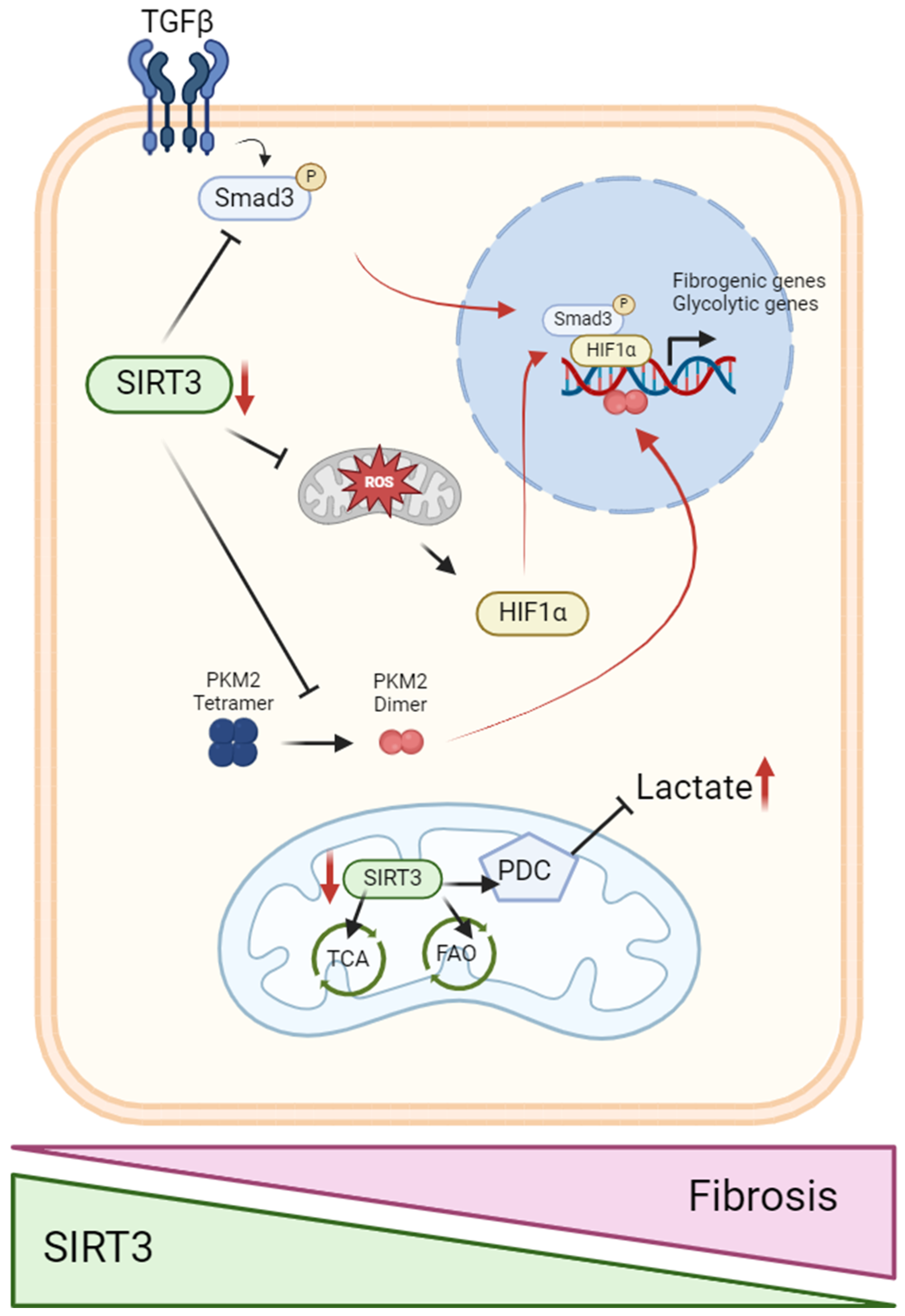
5.2. SIRT3 in the Metabolic Switch from Fatty Acid Oxidation to Glycolysis
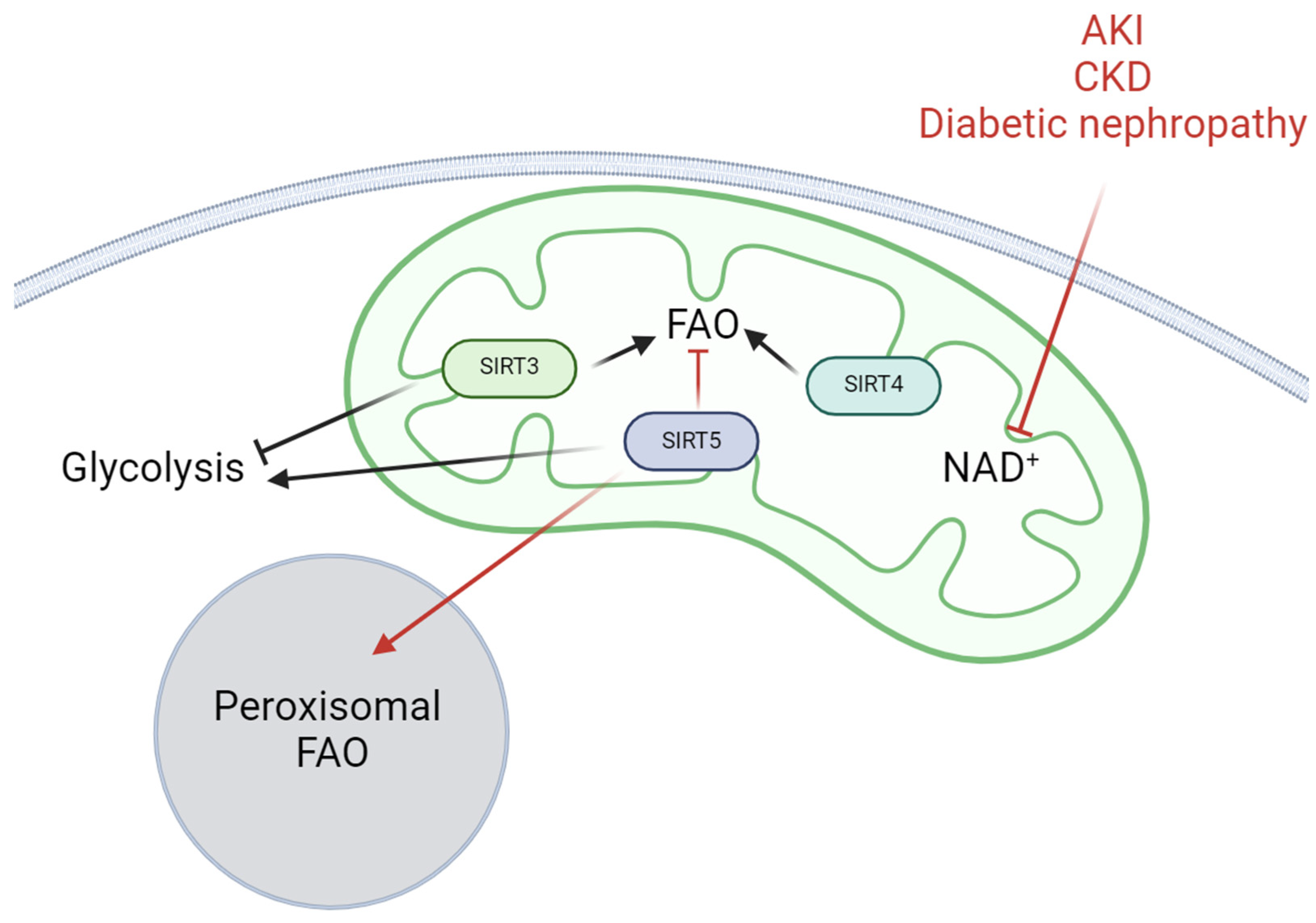
6. Other Mitochondrial Sirtuins That Regulate Renal Cell Metabolism
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doke, T.; Susztak, K. The Multifaceted Role of Kidney Tubule Mitochondrial Dysfunction in Kidney Disease Development. Trends Cell Biol. 2022, 32, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lin, P.-H. Mitochondria Damage and Kidney Disease. Adv. Exp. Med. Biol. 2017, 982, 529–551. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and Cubilin in Proximal Tubule Protein Reabsorption: From Experimental Models to Human Disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, Y.J.; Han, H.J. Regulatory Mechanisms of Na+/Glucose Cotransporters in Renal Proximal Tubule Cells. Kidney Int. 2007, 72, S27–S35. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial Energetics in the Kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Lewington, A.J.; Cerdá, J.; Mehta, R.L. Raising Awareness of Acute Kidney Injury: A Global Perspective of a Silent Killer. Kidney Int. 2013, 84, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Lameire, N.H.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.; Kellum, J.A.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W.; et al. Acute Kidney Injury: An Increasing Global Concern. Lancet Lond. Engl. 2013, 382, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Case, J.; Khan, S.; Khalid, R.; Khan, A. Epidemiology of Acute Kidney Injury in the Intensive Care Unit. Crit. Care Res. Pract. 2013, 2013, 479730. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute Kidney Injury. Lancet Lond. Engl. 2019, 394, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Koza, Y. Acute Kidney Injury: Current Concepts and New Insights. J. Inj. Violence Res. 2016, 8, 58–62. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar] [PubMed]
- Kellum, J.A.; Lameire, N. Diagnosis, Evaluation, and Management of Acute Kidney Injury: A KDIGO Summary (Part 1). Crit. Care 2013, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Belayev, L.Y.; Palevsky, P.M. The Link Between AKI and CKD. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Inker, L.A.; Astor, B.C.; Fox, C.H.; Isakova, T.; Lash, J.P.; Peralta, C.A.; Kurella Tamura, M.; Feldman, H.I. KDOQI US Commentary on the 2012 KDIGO Clinical Practice Guideline for the Evaluation and Management of CKD. Am. J. Kidney Dis. 2014, 63, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Lean, M.E. A Clinical Perspective of Obesity, Metabolic Syndrome and Cardiovascular Disease. JRSM Cardiovasc. Dis. 2016, 5, 2048004016633371. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Borrelli, S.; Minutolo, R.; Chiodini, P.; De Nicola, L.; Conte, G. A Systematic Review and Meta-Analysis Suggests Obesity Predicts Onset of Chronic Kidney Disease in the General Population. Kidney Int. 2017, 91, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.L.; Kalantar-Zadeh, K.; Ma, J.Z.; Quarles, L.D.; Kovesdy, C.P. Association of Body Mass Index with Outcomes in Patients with CKD. J. Am. Soc. Nephrol. JASN 2014, 25, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, F.; Caron, N.; Mathew, A.V.; Declèves, A.-E. Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 7994. [Google Scholar] [CrossRef]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial Dysfunction and the AKI-to-CKD Transition. Am. J. Physiol.-Ren. Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Gewin, L.S. Sugar or Fat? Renal Tubular Metabolism Reviewed in Health and Disease. Nutrients 2021, 13, 1580. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, S.; Ueda, S.; Imamura, H.; Mori, K.; Asanuma, K.; Yanagita, M.; Nakagawa, T. Glycolysis, but Not Mitochondria, Responsible for Intracellular ATP Distribution in Cortical Area of Podocytes. Sci. Rep. 2015, 5, 18575. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yuan, Y.; Bai, M.; Ding, G.; Jia, Z.; Huang, S.; Zhang, A. PGC-1α Overexpression Protects against Aldosterone-Induced Podocyte Depletion: Role of Mitochondria. Oncotarget 2016, 7, 12150–12162. [Google Scholar] [CrossRef] [PubMed]
- Audzeyenka, I.; Bierżyńska, A.; Lay, A.C. Podocyte Bioenergetics in the Development of Diabetic Nephropathy: The Role of Mitochondria. Endocrinology 2022, 163, bqab234. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017, 66, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.-X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698.e7. [Google Scholar] [CrossRef] [PubMed]
- Dumas, S.J.; Meta, E.; Borri, M.; Luo, Y.; Li, X.; Rabelink, T.J.; Carmeliet, P. Phenotypic Diversity and Metabolic Specialization of Renal Endothelial Cells. Nat. Rev. Nephrol. 2021, 17, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Strzyz, P. A Metabolic Switch of Fate. Nat. Rev. Mol. Cell Biol. 2018, 19, 211. [Google Scholar] [CrossRef] [PubMed]
- Caja, S.; Enríquez, J.A. Mitochondria in Endothelial Cells: Sensors and Integrators of Environmental Cues. Redox Biol. 2017, 12, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective Fatty Acid Oxidation in Renal Tubular Epithelial Cells Has a Key Role in Kidney Fibrosis Development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, S.; Ivanova, A.; Osaki, Y.; Funk, S.; Yang, H.; Viquez, O.; Delgado, R.; Lu, D.; Mignemi, M.P.; Tonello, J.; et al. Tubular CPT1A Deletion Minimally Affects Aging and Chronic Kidney Injury. JCI Insight 2024, 9, e171961. [Google Scholar] [CrossRef]
- Menzies, K.J.; Hood, D.A. The Role of SirT1 in Muscle Mitochondrial Turnover. Mitochondrion 2012, 12, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ Homeostasis in Health and Disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ Metabolism and Its Roles in Cellular Processes during Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De Novo NAD+ Biosynthetic Impairment in Acute Kidney Injury in Humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD+ Homeostasis in Renal Health and Disease. Nat. Rev. Nephrol. 2020, 16, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Morevati, M.; Fang, E.F.; Mace, M.L.; Kanbay, M.; Gravesen, E.; Nordholm, A.; Egstrand, S.; Hornum, M. Roles of NAD+ in Acute and Chronic Kidney Diseases. Int. J. Mol. Sci. 2023, 24, 137. [Google Scholar] [CrossRef] [PubMed]
- Bignon, Y.; Rinaldi, A.; Nadour, Z.; Poindessous, V.; Nemazanyy, I.; Lenoir, O.; Fohlen, B.; Weill-Raynal, P.; Hertig, A.; Karras, A.; et al. Cell Stress Response Impairs de Novo NAD+ Biosynthesis in the Kidney. JCI Insight 2022, 7, e153019. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Kitada, M.; Xu, J.; Monno, I.; Koya, D. CD38 Inhibition by Apigenin Ameliorates Mitochondrial Oxidative Stress through Restoration of the Intracellular NAD+/NADH Ratio and Sirt3 Activity in Renal Tubular Cells in Diabetic Rats. Aging 2020, 12, 11325–11336. [Google Scholar] [CrossRef] [PubMed]
- Hopp, A.-K.; Grüter, P.; Hottiger, M.O. Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells 2019, 8, 890. [Google Scholar] [CrossRef] [PubMed]
- Kumakura, S.; Sato, E.; Sekimoto, A.; Hashizume, Y.; Yamakage, S.; Miyazaki, M.; Ito, S.; Harigae, H.; Takahashi, N. Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease. Toxins 2021, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Kang, X.; Tan, L.; Ren, Y.; Qu, L.; Tang, J.; Liu, G.; Wang, S.; Xiong, Z.; Yang, L. Nicotinamide Mononucleotide Attenuates Renal Interstitial Fibrosis After AKI by Suppressing Tubular DNA Damage and Senescence. Front. Physiol. 2021, 12, 649547. [Google Scholar] [CrossRef] [PubMed]
- Morevati, M.; Egstrand, S.; Nordholm, A.; Mace, M.L.; Andersen, C.B.; Salmani, R.; Olgaard, K.; Lewin, E. Effect of NAD+ Boosting on Kidney Ischemia-Reperfusion Injury. PLoS ONE 2021, 16, e0252554. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and Sirtuins in Aging and Disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, M.S.; Sinclair, D.A. Slowing Ageing by Design: The Rise of NAD+ and Sirtuin-Activating Compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Mori, V.; Amici, A.; Mazzola, F.; Di Stefano, M.; Conforti, L.; Magni, G.; Ruggieri, S.; Raffaelli, N.; Orsomando, G. Metabolic Profiling of Alternative NAD Biosynthetic Routes in Mouse Tissues. PLoS ONE 2014, 9, e113939. [Google Scholar] [CrossRef] [PubMed]
- Chanvillard, L.; Tammaro, A.; Sorrentino, V. NAD+ Metabolism and Interventions in Premature Renal Aging and Chronic Kidney Disease. Cells 2022, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The Secret Life of NAD+: An Old Metabolite Controlling New Metabolic Signaling Pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Remuzzi, G.; Benigni, A. Sirtuins in Kidney Health and Disease. Nat. Rev. Nephrol. 2024, 20, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. JASN 2018, 29, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.A.; Kim, J.E.; Jo, M.; Ko, G.-J. The Role of Sirtuins in Kidney Diseases. Int. J. Mol. Sci. 2020, 21, 6686. [Google Scholar] [CrossRef] [PubMed]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD+ in Regulating Cellular and Metabolic Signaling Pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Pinho, R.; Gu, Y.; Radak, Z. The Role of SIRT3 in Exercise and Aging. Cells 2022, 11, 2596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, P.; Luo, J.; Ding, H.; Cao, H.; He, W.; Zen, K.; Zhou, Y.; Yang, J.; Jiang, L. Sirtuin 3 Regulates Mitochondrial Protein Acetylation and Metabolism in Tubular Epithelial Cells during Renal Fibrosis. Cell Death Dis. 2021, 12, 847. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.-R.; Kim, J.; Na, J.C.; Han, W.K. Mitochondrial Metabolic Reprogramming by SIRT3 Regulation Ameliorates Drug Resistance in Renal Cell Carcinoma. PLoS ONE 2022, 17, e0269432. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.-J.; Zhang, T.-N.; Chen, H.-H.; Yu, X.-F.; Lv, J.-L.; Liu, Y.-Y.; Liu, Y.-S.; Zheng, G.; Zhao, J.-Q.; Wei, Y.-F.; et al. The Sirtuin Family in Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; North, B.J.; Frye, R.A.; Ott, M.; Verdin, E. The Human Silent Information Regulator (Sir)2 Homologue hSIRT3 Is a Mitochondrial Nicotinamide Adenine Dinucleotide-Dependent Deacetylase. J. Cell Biol. 2002, 158, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. SIRT3, a Human SIR2 Homologue, Is an NAD-Dependent Deacetylase Localized to Mitochondria. Proc. Natl. Acad. Sci. USA 2002, 99, 13653–13658. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Kim, J.; Lee, C.-Y.; Kim, H. Expression of SIRT1 and SIRT3 Varies According to Age in Mice. Anat. Cell Biol. 2015, 48, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, A.; Fritz, K.S.; Petersen, D.R.; Gius, D. The Human Sirtuin Family: Evolutionary Divergences and Functions. Hum. Genom. 2011, 5, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, D.; Dato, S.; Cavalcante, P.; Covello, G.; Di Cianni, F.; Passarino, G.; Rose, G.; De Benedictis, G. Characterization of a Bidirectional Promoter Shared between Two Human Genes Related to Aging: SIRT3 and PSMD13. Genomics 2007, 89, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Neeli, P.K.; Gollavilli, P.N.; Mallappa, S.; Hari, S.G.; Kotamraju, S. A Novel metadherinΔ7 Splice Variant Enhances Triple Negative Breast Cancer Aggressiveness by Modulating Mitochondrial Function via NFĸB-SIRT3 Axis. Oncogene 2020, 39, 2088–2102. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a New Target of PGC-1alpha, Plays an Important Role in the Suppression of ROS and Mitochondrial Biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Hondares, E.; Villena, J.A.; Ribas, F.; Díaz-Delfín, J.; Giralt, M.; Iglesias, R.; Villarroya, F. Peroxisome Proliferator-Activated Receptor-Gamma Coactivator-1alpha Controls Transcription of the Sirt3 Gene, an Essential Component of the Thermogenic Brown Adipocyte Phenotype. J. Biol. Chem. 2011, 286, 16958–16966. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Swindell, W.R.; Laurent, G.; Vyas, S.; Bulyk, M.L.; Haigis, M.C. Nuclear Respiratory Factor 2 Induces SIRT3 Expression. Aging Cell 2015, 14, 818–825. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Lu, Z.; Joseph, J.J.; Carabenciov, D.; Dimond, C.C.; Pang, L.; Samsel, L.; McCoy, J.P.; Leclerc, J.; Nguyen, P.; et al. Characterization of the Murine SIRT3 Mitochondrial Localization Sequence and Comparison of Mitochondrial Enrichment and Deacetylase Activity of Long and Short SIRT3 Isoforms. J. Cell. Biochem. 2010, 110, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ameer, F.S.; Azhar, G.; Wei, J.Y. Alternative Splicing Increases Sirtuin Gene Family Diversity and Modulates Their Subcellular Localization and Function. Int. J. Mol. Sci. 2021, 22, 473. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hubbard, B.P.; Sinclair, D.A.; Tong, Q. Characterization of Murine SIRT3 Transcript Variants and Corresponding Protein Products. J. Cell. Biochem. 2010, 111, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wei, W.; Jiang, Y.; Peng, H.; Cai, J.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal Structures of Human SIRT3 Displaying Substrate-Induced Conformational Changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.; Li, H.; Jiang, H.; Long, Y.; Liu, D. Crystal Structures of SIRT3 Reveal That the A2-A3 Loop and A3-Helix Affect the Interaction with Long-Chain Acyl Lysine. FEBS Lett. 2016, 590, 3019–3028. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Morigi, M.; Pezzotta, A.; Corna, D.; Brizi, V.; Conti, S.; Zanchi, C.; Sangalli, F.; Trionfini, P.; Buttò, S.; et al. Post-Translational Modifications by SIRT3 de-2-Hydroxyisobutyrylase Activity Regulate Glycolysis and Enable Nephrogenesis. Sci. Rep. 2021, 11, 23580. [Google Scholar] [CrossRef] [PubMed]
- Pezzotta, A.; Perico, L.; Morigi, M.; Corna, D.; Locatelli, M.; Zoja, C.; Benigni, A.; Remuzzi, G.; Imberti, B. Low Nephron Number Induced by Maternal Protein Restriction Is Prevented by Nicotinamide Riboside Supplementation Depending on Sirtuin 3 Activation. Cells 2022, 11, 3316. [Google Scholar] [CrossRef]
- Bellizzi, D.; Rose, G.; Cavalcante, P.; Covello, G.; Dato, S.; De Rango, F.; Greco, V.; Maggiolini, M.; Feraco, E.; Mari, V.; et al. A Novel VNTR Enhancer within the SIRT3 Gene, a Human Homologue of SIR2, Is Associated with Survival at Oldest Ages. Genomics 2005, 85, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II Type 1 Receptor Promotes Longevity in Mice. J. Clin. Investig. 2009, 119, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.; Dato, S.; Altomare, K.; Bellizzi, D.; Garasto, S.; Greco, V.; Passarino, G.; Feraco, E.; Mari, V.; Barbi, C.; et al. Variability of the SIRT3 Gene, Human Silent Information Regulator Sir2 Homologue, and Survivorship in the Elderly. Exp. Gerontol. 2003, 38, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Bindu, S.; Pillai, V.B.; Samant, S.; Pan, Y.; Huang, J.-Y.; Gupta, M.; Nagalingam, R.S.; Wolfgeher, D.; Verdin, E.; et al. SIRT3 Blocks Aging-Associated Tissue Fibrosis in Mice by Deacetylating and Activating Glycogen Synthase Kinase 3β. Mol. Cell. Biol. 2016, 36, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Macconi, D.; Corna, D.; Cerullo, D.; Rottoli, D.; Remuzzi, G.; Benigni, A.; Zoja, C. Sirtuin 3 Deficiency Aggravates Kidney Disease in Response to High-Fat Diet through Lipotoxicity-Induced Mitochondrial Damage. Int. J. Mol. Sci. 2022, 23, 8345. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Holliday, M.; Sheikh-Hamad, D.; Li, Q.; Tong, Q.; Hamad, C.D.; Pan, J.S. Sirtuin-3 Mediates Sex Differences in Kidney Ischemia-Reperfusion Injury. Transl. Res. 2021, 235, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Matouk, A.I.; Awad, E.M.; Mousa, A.A.K.; Abdelhafez, S.M.N.; Fahmy, U.A.; El-Moselhy, M.A.; Abdel-Naim, A.B.; Anter, A. Dihydromyricetin Protects against Gentamicin-Induced Nephrotoxicity via Upregulation of Renal SIRT3 and PAX2. Life Sci. 2024, 336, 122318. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zou, W.; Liu, S.; Yang, Q.; Hu, T.; Zhu, W.; Tang, H.; Wang, C. Site 1 Protease Aggravates Acute Kidney Injury by Promoting Tubular Epithelial Cell Ferroptosis through SIRT3-SOD2-mtROS Signaling. FEBS J. 2024, 291, 1575–1592. [Google Scholar] [CrossRef]
- Deng, Z.; He, M.; Hu, H.; Zhang, W.; Zhang, Y.; Ge, Y.; Ma, T.; Wu, J.; Li, L.; Sun, M.; et al. Melatonin Attenuates Sepsis-Induced Acute Kidney Injury by Promoting Mitophagy through SIRT3-Mediated TFAM Deacetylation. Autophagy 2024, 20, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, H.; Hu, J.; Li, H.; Guo, S.; Chen, B.; Liu, C.; Wang, G.; Zhou, F. Magnesium Isoglycyrrhizinate Reduces the Target-Binding Amount of Cisplatin to Mitochondrial DNA and Renal Injury through SIRT3. Int. J. Mol. Sci. 2022, 23, 13093. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Zhang, Y.; Xiong, Y.; Zhu, Z.; Wang, L.; Liu, X. Mitochondria-Targeted Antioxidant Mitoquinone Maintains Mitochondrial Homeostasis through the Sirt3-Dependent Pathway to Mitigate Oxidative Damage Caused by Renal Ischemia/Reperfusion. Oxid. Med. Cell. Longev. 2022, 2022, 2213503. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; He, S.; Lan, J.; Zhu, W. Honokiol Ameliorates Cisplatin-Induced Acute Kidney Injury via Inhibition of Mitochondrial Fission. Br. J. Pharmacol. 2022, 179, 3886–3904. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-M.; Li, X.-L.; Zhu, Y.; Shi, R.; Wang, Z.-J.; Xiao, J.-P.; Wang, D.-G. Diosmin Ameliorates Renal Fibrosis through Inhibition of Inflammation by Regulating SIRT3-Mediated NF-κB P65 Nuclear Translocation. BMC Complement. Med. Ther. 2024, 24, 29. [Google Scholar] [CrossRef] [PubMed]
- Myakala, K.; Wang, X.X.; Shults, N.V.; Krawczyk, E.; Jones, B.A.; Yang, X.; Rosenberg, A.Z.; Ginley, B.; Sarder, P.; Brodsky, L.; et al. NAD Metabolism Modulates Inflammation and Mitochondria Function in Diabetic Kidney Disease. J. Biol. Chem. 2023, 299, 104975. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, L.; Jin, B.; Wu, Y.; Xu, L.; Chang, X.; Hu, L.; Wang, G.; Huang, Y.; Song, L.; et al. Metrnl Alleviates Lipid Accumulation by Modulating Mitochondrial Homeostasis in Diabetic Nephropathy. Diabetes 2023, 72, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhai, J.; Zhang, T.; He, L.; Ma, S.; Zuo, Q.; Zhang, G.; Wang, Y.; Guo, Y. Canagliflozin Ameliorates Epithelial-Mesenchymal Transition in High-Salt Diet-Induced Hypertensive Renal Injury through Restoration of Sirtuin 3 Expression and the Reduction of Oxidative Stress. Biochem. Biophys. Res. Commun. 2023, 653, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Park, W.; Jin, J.; Kim, W.; Park, S.K.; Kang, K.P. Sirtuin 3 Activation by Honokiol Decreases Unilateral Ureteral Obstruction-Induced Renal Inflammation and Fibrosis via Regulation of Mitochondrial Dynamics and the Renal NF-κB-TGF-Β1/Smad Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 402. [Google Scholar] [CrossRef] [PubMed]
- Doke, T.; Mukherjee, S.; Mukhi, D.; Dhillon, P.; Abedini, A.; Davis, J.G.; Chellappa, K.; Chen, B.; Baur, J.A.; Susztak, K. NAD+ Precursor Supplementation Prevents mtRNA/RIG-I-Dependent Inflammation during Kidney Injury. Nat. Metab. 2023, 5, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Bause, A.S.; Haigis, M.C. SIRT3 Regulation of Mitochondrial Oxidative Stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Storder, J.; Renard, P.; Arnould, T. Update on the Role of Sirtuin 3 in Cell Differentiation: A Major Metabolic Target That Can Be Pharmacologically Controlled. Biochem. Pharmacol. 2019, 169, 113621. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) Regulates Skeletal Muscle Metabolism and Insulin Signaling via Altered Mitochondrial Oxidation and Reactive Oxygen Species Production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; He, J.; Xu, Y.; Zuo, Y.; Zhou, W.; Yue, Z.; Shao, X.; Cheng, J.; Wang, T.; Mou, S. AMPK Activation Coupling SENP1-Sirt3 Axis Protects against Acute Kidney Injury. Mol. Ther. 2023, 31, 3052–3066. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Gao, Y.; Cheng, D.; Zhang, H.; Zhang, W.; Shen, Y.; Huang, Q.; An, X.; Wang, B.; Yu, Z.; et al. Notoginsenoside Fc Ameliorates Renal Tubular Injury and Mitochondrial Damage in Acetaminophen-Induced Acute Kidney Injury Partly by Regulating SIRT3/SOD2 Pathway. Front. Med. 2023, 9, 1055252. [Google Scholar] [CrossRef] [PubMed]
- Wongmekiat, O.; Lailerd, N.; Kobroob, A.; Peerapanyasut, W. Protective Effects of Purple Rice Husk against Diabetic Nephropathy by Modulating PGC-1α/SIRT3/SOD2 Signaling and Maintaining Mitochondrial Redox Equilibrium in Rats. Biomolecules 2021, 11, 1224. [Google Scholar] [CrossRef] [PubMed]
- Peerapanyasut, W.; Kobroob, A.; Palee, S.; Chattipakorn, N.; Wongmekiat, O. Activation of Sirtuin 3 and Maintenance of Mitochondrial Integrity by N-Acetylcysteine Protects Against Bisphenol A-Induced Kidney and Liver Toxicity in Rats. Int. J. Mol. Sci. 2019, 20, 267. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Wang, Z.; Zhang, R.; Chen, S.; Bu, X.; Lu, J. 3-MCPD Induced Mitochondrial Damage of Renal Cells Via the Rhythmic Protein BMAL1 Targeting SIRT3/SOD2. J. Agric. Food Chem. 2023, 71, 14351–14364. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, H.; Tan, B.; Yi, Q.; Sun, Y.; Xiang, H.; Chen, T.; Liu, H.; Xie, Q.; Wang, L.; et al. SIRT3 Promotes Metabolic Maturation of Human iPSC-Derived Cardiomyocytes via OPA1-Controlled Mitochondrial Dynamics. Free Radic. Biol. Med. 2023, 195, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Jian, Y.; Yang, Y.; Cheng, L.; Yang, X.; Liu, H.; Li, W.; Wan, Y.; Yang, D. Sirt3 Mitigates LPS-induced Mitochondrial Damage in Renal Tubular Epithelial Cells by Deacetylating YME1L1. Cell Prolif. 2022, 56, e13362. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Yang, J.; Li, Y.; Yuan, L.; Liu, F.; Yuan, Y.; Tang, X. Matrine Alleviates Cisplatin-Induced Acute Kidney Injury by Inhibiting Mitochondrial Dysfunction and Inflammation via SIRT3/OPA1 Pathway. J. Cell. Mol. Med. 2022, 26, 3702–3715. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhu, L.; Li, L.; Liu, J.; Chen, Y.; Cheng, J.; Peng, T.; Lu, Y. S-Sulfhydration of SIRT3 by Hydrogen Sulfide Attenuates Mitochondrial Dysfunction in Cisplatin-Induced Acute Kidney Injury. Antioxid. Redox Signal. 2019, 31, 1302–1319. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, J.; Li, X.; Liu, Z.; Han, Y.; Xu, X.; Li, X.; Tang, Y.; Liu, Y.; Yu, T.; et al. Sirt3 Modulate Renal Ischemia-Reperfusion Injury through Enhancing Mitochondrial Fusion and Activating the ERK-OPA1 Signaling Pathway. J. Cell. Physiol. 2019, 234, 23495–23506. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Sui, M.; Chen, R.; Lu, H.; Zhu, Y.; Zhang, L.; Zeng, L. SIRT3 Protects Kidneys from Ischemia-Reperfusion Injury by Modulating the DRP1 Pathway to Induce Mitochondrial Autophagy. Life Sci. 2021, 286, 120005. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, Q.; Tu, S.; Qin, W. SIRT3 Mediates Mitofusin 2 Ubiquitination and Degradation to Suppress Ischemia Reperfusion-Induced Acute Kidney Injury. Exp. Cell Res. 2021, 408, 112861. [Google Scholar] [CrossRef] [PubMed]
- Dittenhafer-Reed, K.E.; Richards, A.L.; Fan, J.; Smallegan, M.J.; Fotuhi Siahpirani, A.; Kemmerer, Z.A.; Prolla, T.A.; Roy, S.; Coon, J.J.; Denu, J.M. SIRT3 Mediates Multi-Tissue Coupling for Metabolic Fuel Switching. Cell Metab. 2015, 21, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 Deficiency and Mitochondrial Protein Hyperacetylation Accelerate the Development of the Metabolic Syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Rinaldo, P.; Rhead, W.J.; Tian, L.; Millington, D.S.; Vockley, J.; Hamm, D.A.; Brix, A.E.; Lindsey, J.R.; Pinkert, C.A.; et al. Targeted Disruption of Mouse Long-Chain Acyl-CoA Dehydrogenase Gene Reveals Crucial Roles for Fatty Acid Oxidation. Proc. Natl. Acad. Sci. USA 1998, 95, 15592–15597. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 Regulates Mitochondrial Fatty-Acid Oxidation by Reversible Enzyme Deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bharathi, S.S.; Beck, M.E.; Goetzman, E.S. The Fatty Acid Oxidation Enzyme Long-Chain Acyl-CoA Dehydrogenase Can Be a Source of Mitochondrial Hydrogen Peroxide. Redox Biol. 2019, 26, 101253. [Google Scholar] [CrossRef] [PubMed]
- Bharathi, S.S.; Zhang, Y.; Mohsen, A.-W.; Uppala, R.; Balasubramani, M.; Schreiber, E.; Uechi, G.; Beck, M.E.; Rardin, M.J.; Vockley, J.; et al. Sirtuin 3 (SIRT3) Protein Regulates Long-Chain Acyl-CoA Dehydrogenase by Deacetylating Conserved Lysines Near the Active Site. J. Biol. Chem. 2013, 288, 33837–33847. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.; Lazareth, H.; Poindessous, V.; Nemazanyy, I.; Sampaio, J.L.; Malpetti, D.; Bignon, Y.; Naesens, M.; Rabant, M.; Anglicheau, D.; et al. Impaired Fatty Acid Metabolism Perpetuates Lipotoxicity along the Transition to Chronic Kidney Injury. JCI Insight 2022, 7, e161783. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jia, P.; Fang, Y.; Jin, J.; Sun, Z.; Zhou, W.; Li, J.; Zhang, Y.; Wang, X.; Ren, T.; et al. Nuclear Farnesoid X Receptor Attenuates Acute Kidney Injury through Fatty Acid Oxidation. Kidney Int. 2022, 101, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, C.; Ye, Z.; Huang, J.; Li, Y.; Lai, W.; Peng, H.; Lou, T. Sirt3 Modulates Fatty Acid Oxidation and Attenuates Cisplatin-induced AKI in Mice. J. Cell. Mol. Med. 2020, 24, 5109–5121. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Park, W.; Lee, S.; Kim, W.; Park, S.K.; Kang, K.P. Absence of Sirt3 Aggravates Cisplatin Nephrotoxicity via Enhanced Renal Tubular Apoptosis and Inflammation. Mol. Med. Rep. 2018, 18, 3665–3672. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, X.; Cui, S.; Wu, Y.; Zhang, Y.; Shen, X.; Xie, C.; Li, J. Dephosphorylation of AMP-Activated Protein Kinase Exacerbates Ischemia/Reperfusion-Induced Acute Kidney Injury via Mitochondrial Dysfunction. Kidney Int. 2022, 101, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-F.; Chen, H.-H.; Lin, H. Role of PPARα and Its Agonist in Renal Diseases. PPAR Res. 2010, 2010, 345098. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, T.; Bennion, B.G.; Stenson, E.K.; Lynn, J.C.; Otinga, C.; Djukovic, D.; Raftery, D.; Fei, L.; Wong, H.R.; Liles, W.C.; et al. PPARα Contributes to Protection against Metabolic and Inflammatory Derangements Associated with Acute Kidney Injury in Experimental Sepsis. Physiol. Rep. 2019, 7, e14078. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Xu, L.; Tao, X.; Yin, L.; Qi, Y.; Xu, Y.; Han, X.; Tang, Z.; Ma, X.; Liu, K.; et al. Protective Effects of Dioscin against Fructose-Induced Renal Damage via Adjusting Sirt3-Mediated Oxidative Stress, Fibrosis, Lipid Metabolism and Inflammation. Toxicol. Lett. 2018, 284, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD Blocks Cardiac Hypertrophic Response via Activation of the SIRT3-LKB1-AMP-Activated Kinase Pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [PubMed]
- Declèves, A.-E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of Lipid Accumulation by AMK-Activated Kinase in High Fat Diet–Induced Kidney Injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Declèves, A.-E.; Mathew, A.V.; Cunard, R.; Sharma, K. AMPK Mediates the Initiation of Kidney Disease Induced by a High-Fat Diet. J. Am. Soc. Nephrol. JASN 2011, 22, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Rampanelli, E.; Orsó, E.; Ochodnicky, P.; Liebisch, G.; Bakker, P.J.; Claessen, N.; Butter, L.M.; van den Bergh Weerman, M.A.; Florquin, S.; Schmitz, G.; et al. Metabolic Injury-Induced NLRP3 Inflammasome Activation Dampens Phospholipid Degradation. Sci. Rep. 2017, 7, 2861. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Kimura, T.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.-Y.; Matsui, I.; Matsusaka, T.; et al. High-Fat Diet-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney. J. Am. Soc. Nephrol. JASN 2017, 28, 1534–1551. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, F.; Pierre, L.; Decarnoncle, M.; Jadot, I.; Martin, B.; Botton, O.; Caron, N.; Dehairs, J.; Swinnen, J.V.; Declèves, A.-E. Sex Differences in Obesity-Induced Renal Lipid Accumulation Revealed by Lipidomics: A Role of Adiponectin/AMPK Axis. Biol. Sex Differ. 2023, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Declèves, A.-E.; Mathew, A.V.; Armando, A.M.; Han, X.; Dennis, E.A.; Quehenberger, O.; Sharma, K. AMP-Activated Protein Kinase Activation Ameliorates Eicosanoid Dysregulation in High-Fat-Induced Kidney Disease in Mice. J. Lipid Res. 2019, 60, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, L.; Chen, R.; Lu, H.; Sui, M.; Zhu, Y.; Zeng, L. SIRT3 Protects Against Acute Kidney Injury via AMPK/mTOR-Regulated Autophagy. Front. Physiol. 2018, 9, 1526. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a Mitochondrial Sirtuin Deacetylase, Regulates Mitochondrial Function and Thermogenesis in Brown Adipocytes*. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Oliver, J.; Roca, P.; Sastre-Serra, J. SIRT3: Oncogene and Tumor Suppressor in Cancer. Cancers 2017, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Alsahli, M.; Gerich, J.E. Renal Glucose Metabolism in Normal Physiological Conditions and in Diabetes. Diabetes Res. Clin. Pract. 2017, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sha, Z.; Peng, H. Metabolic Reprogramming in Kidney Diseases: Evidence and Therapeutic Opportunities. Int. J. Nephrol. 2021, 2021, 5497346. [Google Scholar] [CrossRef] [PubMed]
- Gómez, H.; Kellum, J.A.; Ronco, C. Metabolic Reprogramming and Tolerance during Sepsis-Induced AKI. Nat. Rev. Nephrol. 2017, 13, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Stallons, L.J.; Schnellmann, R.G. Renal Cortical Hexokinase and Pentose Phosphate Pathway Activation through the EGFR/Akt Signaling Pathway in Endotoxin-Induced Acute Kidney Injury. Am. J. Physiol.—Ren. Physiol. 2014, 307, F435–F444. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. JASN 2016, 27, 3356–3367. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, Y.; Yamamoto, S.; Yano, F.; Orita, Y.; Fujiwara, Y.; Ueda, N.; Kamada, T.; Noguchi, T.; Tanaka, T. Changes in Activities and mRNA Levels of Glycolytic Enzymes of Ischemia-Reperfused Rat Kidney. Contrib. Nephrol. 1991, 95, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Ash, S.R.; Cuppage, F.E. Shift toward Anaerobic Glycolysis in the Regenerating Rat Kidney. Am. J. Pathol. 1970, 60, 385–402. [Google Scholar]
- Jensen, T.M.; Vistisen, D.; Fleming, T.; Nawroth, P.P.; Rossing, P.; Jørgensen, M.E.; Lauritzen, T.; Sandbaek, A.; Witte, D.R. Methylglyoxal Is Associated with Changes in Kidney Function among Individuals with Screen-Detected Type 2 Diabetes Mellitus. Diabet. Med. J. Br. Diabet. Assoc. 2016, 33, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Pang, Y.; Guo, Y.; Tian, L.; Liu, Y.; Shen, C.; Liu, M.; Meng, Y.; Cai, Z.; Wang, Y.; et al. Metabolic Reprogramming: A Novel Therapeutic Target in Diabetic Kidney Disease. Front. Pharmacol. 2022, 13, 970601. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Luo, J.; Zhang, Y.; Mao, X.; Wen, P.; Ding, H.; Xu, J.; Sun, Q.; He, W.; Dai, C.; et al. Tuberous Sclerosis 1 (Tsc1) Mediated mTORC1 Activation Promotes Glycolysis in Tubular Epithelial Cells in Kidney Fibrosis. Kidney Int. 2020, 98, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Li, Y.; Li, S.; Hu, X.; Wei, Q.; Dong, Z. Glucose Metabolism in Acute Kidney Injury and Kidney Repair. Front. Med. 2021, 8, 744122. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 Blocks the Cardiac Hypertrophic Response by Augmenting Foxo3a-Dependent Antioxidant Defense Mechanisms in Mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 Protein Deacetylates Isocitrate Dehydrogenase 2 (IDH2) and Regulates Mitochondrial Redox Status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Pennington, J.D.; Bisht, K.S.; Aykin-Burns, N.; Kim, H.-S.; Mishra, M.; Sun, L.; Nguyen, P.; Ahn, B.-H.; Leclerc, J.; et al. SIRT3 Interacts with the Daf-16 Homolog FOXO3a in the Mitochondria, as Well as Increases FOXO3a Dependent Gene Expression. Int. J. Biol. Sci. 2008, 4, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.S.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1α Destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 Activation in Hypoxic Tubules Prevents Chronic Kidney Disease. J. Clin. Investig. 2019, 129, 2374–2389. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia Promotes Fibrogenesis in Vivo via HIF-1 Stimulation of Epithelial-to-Mesenchymal Transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Li, J.; Kitada, M.; Fujita, H.; Yamada, Y.; Goodwin, J.E.; Kanasaki, K.; Koya, D. SIRT3 Deficiency Leads to Induction of Abnormal Glycolysis in Diabetic Kidney with Fibrosis. Cell Death Dis. 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; O’Neill, B.T.; Rardin, M.J.; Kleinridders, A.; Ilkeyeva, O.R.; Ussar, S.; Bain, J.R.; Lee, K.Y.; Verdin, E.M.; Newgard, C.B.; et al. Sirt3 Regulates Metabolic Flexibility of Skeletal Muscle Through Reversible Enzymatic Deacetylation. Diabetes 2013, 62, 3404–3417. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.-H.; Kim, H.-S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.-X.; Finkel, T. A Role for the Mitochondrial Deacetylase Sirt3 in Regulating Energy Homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.S.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate Dehydrogenase Is a Direct Target of Sirtuin 3 Deacetylase Activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef] [PubMed]
- Cimen, H.; Han, M.-J.; Yang, Y.; Tong, Q.; Koc, H.; Koc, E.C. Regulation of Succinate Dehydrogenase Activity by SIRT3 in Mammalian Mitochondria. Biochemistry 2010, 49, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Scott, I.; Lu, Z.; Pang, L.; Dimond, C.C.; Gius, D.; Sack, M.N. SIRT3 Is Regulated by Nutrient Excess and Modulates Hepatic Susceptibility to Lipotoxicity. Free Radic. Biol. Med. 2010, 49, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Li, J.; Takagaki, Y.; Kitada, M.; Goodwin, J.E.; Kanasaki, K.; Koya, D. Endothelial SIRT3 Regulates Myofibroblast Metabolic Shifts in Diabetic Kidneys. iScience 2021, 24, 102390. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium Structure and Function in Kidney Health and Disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Xie, L.; Feng, H.; Li, S.; Meng, G.; Liu, S.; Tang, X.; Ma, Y.; Han, Y.; Xiao, Y.; Gu, Y.; et al. SIRT3 Mediates the Antioxidant Effect of Hydrogen Sulfide in Endothelial Cells. Antioxid. Redox Signal. 2016, 24, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.; Dikalova, A. Mitochondrial Deacetylase Sirt3 in Vascular Dysfunction and Hypertension. Curr. Opin. Nephrol. Hypertens. 2022, 31, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Zoja, C.; Zanchi, C.; Corna, D.; Villa, S.; Bolognini, S.; Novelli, R.; Perico, L.; Remuzzi, G.; Benigni, A.; et al. Manipulating Sirtuin 3 Pathway Ameliorates Renal Damage in Experimental Diabetes. Sci. Rep. 2020, 10, 8418. [Google Scholar] [CrossRef] [PubMed]
- Pezzotta, A.; Perico, L.; Corna, D.; Morigi, M.; Remuzzi, G.; Benigni, A.; Imberti, B. Sirt3 Deficiency Promotes Endothelial Dysfunction and Aggravates Renal Injury. PLoS ONE 2023, 18, e0291909. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-R.; Zheng, Y.-J.; Zhang, Z.-B.; Shen, W.-L.; Li, X.-D.; Wei, T.; Ruan, C.-C.; Chen, X.-H.; Zhu, D.-L.; Gao, P.-J. Suppression of Endothelial-to-Mesenchymal Transition by SIRT (Sirtuin) 3 Alleviated the Development of Hypertensive Renal Injury. Hypertens. Dallas Tex 1979 2018, 72, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Haschler, T.N.; Horsley, H.; Balys, M.; Anderson, G.; Taanman, J.-W.; Unwin, R.J.; Norman, J.T. Sirtuin 5 Depletion Impairs Mitochondrial Function in Human Proximal Tubular Epithelial Cells. Sci. Rep. 2021, 11, 15510. [Google Scholar] [CrossRef] [PubMed]
- Yihan, L.; Xiaojing, W.; Ao, L.; Chuanjie, Z.; Haofei, W.; Yan, S.; Hongchao, H. SIRT5 Functions as a Tumor Suppressor in Renal Cell Carcinoma by Reversing the Warburg Effect. J. Transl. Med. 2021, 19, 521. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; Sas, K.; He, C.; Nair, V.; Giblin, W.; Inoki, A.; Zhang, H.; Yingbao, Y.; Hodgin, J.; Nelson, R.G.; et al. The Deacylase Sirtuin 5 Reduces Malonylation in Nonmitochondrial Metabolic Pathways in Diabetic Kidney Disease. J. Biol. Chem. 2023, 299, 102960. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Z.; Zhang, H.; Chen, H.; Hao, J.; Liu, H.; Li, X. Mitochondrial Metabolism and Targeted Treatment Strategies in Ischemic-Induced Acute Kidney Injury. Cell Death Discov. 2024, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tian, M.; Sun, R.; Zhang, M.; Zhou, L.; Jin, L.; Chen, L.; Zhou, W.; Duan, K.; Chen, Y.; et al. SIRT5 Inhibits Peroxisomal ACOX1 to Prevent Oxidative Damage and Is Downregulated in Liver Cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef] [PubMed]
- Fabbrizi, E.; Fiorentino, F.; Carafa, V.; Altucci, L.; Mai, A.; Rotili, D. Emerging Roles of SIRT5 in Metabolism, Cancer, and SARS-CoV-2 Infection. Cells 2023, 12, 852. [Google Scholar] [CrossRef]
- Chiba, T.; Peasley, K.D.; Cargill, K.R.; Maringer, K.V.; Bharathi, S.S.; Mukherjee, E.; Zhang, Y.; Holtz, A.; Basisty, N.; Yagobian, S.D.; et al. Sirtuin 5 Regulates Proximal Tubule Fatty Acid Oxidation to Protect against AKI. J. Am. Soc. Nephrol. JASN 2019, 30, 2384–2398. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lin, B.; Qiu, W.; Yu, B.; Li, J.; An, S.; Weng, L.; Li, Y.; Shi, M.; Chen, Z.; et al. ADENOSINE MONOPHOSPHATE–ACTIVATED PROTEIN KINASE PHOSPHORYLATION MEDIATED BY SIRTUIN 5 ALLEVIATES SEPTIC ACUTE KIDNEY INJURY. Shock 2023, 59, 477. [Google Scholar] [CrossRef] [PubMed]
- Mou, L.; Yang, L.; Hou, S.; Wang, B.; Wang, X.; Hu, L.; Deng, J.; Liu, J.; Chen, X.; Jiang, Y.; et al. Structure-Activity Relationship Studies of 2,4,5-Trisubstituted Pyrimidine Derivatives Leading to the Identification of a Novel and Potent Sirtuin 5 Inhibitor against Sepsis-Associated Acute Kidney Injury. J. Med. Chem. 2023, 66, 11517–11535. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, Y.; Li, Y.; Zhao, Y.; Jiang, H. Sirt5 Attenuates Cisplatin-Induced Acute Kidney Injury through Regulation of Nrf2/HO-1 and Bcl-2. BioMed Res. Int. 2019, 2019, 4745132. [Google Scholar] [CrossRef] [PubMed]
- Min, Z.; Gao, J.; Yu, Y. The Roles of Mitochondrial SIRT4 in Cellular Metabolism. Front. Endocrinol. 2019, 9, 783. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; de Boer, V.C.J.; Finley, L.W.S.; Sweeney, M.; Lu, H.; Schug, T.T.; Cen, Y.; Jeong, S.M.; Li, X.; Sauve, A.A.; et al. SIRT4 Represses Peroxisome Proliferator-Activated Receptor α Activity To Suppress Hepatic Fat Oxidation. Mol. Cell. Biol. 2013, 33, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Titus, A.S.; Banerjee, K.K.; George, S.; Lin, W.; Deota, S.; Saha, A.K.; Nakamura, K.; Gut, P.; Verdin, E.; et al. SIRT4 Regulates ATP Homeostasis and Mediates a Retrograde Signaling via AMPK. Aging 2013, 5, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cui, H.; Mei, C.; Cui, M.; He, Q.; Wang, Q.; Li, D.; Song, Y.; Li, J.; Chen, S.; et al. Sirtuin4 Alleviates Severe Acute Pancreatitis by Regulating HIF-1α/HO-1 Mediated Ferroptosis. Cell Death Dis. 2023, 14, 694. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, L.; Hua, F.; Zhang, C.; Zhang, C.; Mi, X.; Qin, N.; Wang, J.; Zhu, A.; Qin, Z.; et al. FOXM1-Activated SIRT4 Inhibits NF-κB Signaling and NLRP3 Inflammasome to Alleviate Kidney Injury and Podocyte Pyroptosis in Diabetic Nephropathy. Exp. Cell Res. 2021, 408, 112863. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Wo, C.; Yuan, Y.; Cao, H.; Tan, W.; Zhou, X.; Wang, D.; Chen, R.; Shi, M.; Zhang, F.; et al. miR-124-3p Improves Mitochondrial Function of Renal Tubular Epithelial Cells in Db/Db Mice. FASEB J. 2023, 37, e22794. [Google Scholar] [CrossRef]
- Murugasamy, K.; Munjal, A.; Sundaresan, N.R. Emerging Roles of SIRT3 in Cardiac Metabolism. Front. Cardiovasc. Med. 2022, 9, 850340. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xing, D.; Du, X.; Peng, T.; McFadden, J.W.; Wen, L.; Lei, H.; Dong, W.; Liu, G.; Wang, Z.; et al. Sirtuin 3 Improves Fatty Acid Metabolism in Response to High Nonesterified Fatty Acids in Calf Hepatocytes by Modulating Gene Expression. J. Dairy Sci. 2020, 103, 6557–6568. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, F.; Vlassembrouck, M.; Botton, O.; Zwakhals, T.; Decarnoncle, M.; Tassin, A.; Caron, N.; Declèves, A.-E. Delayed Exercise Training Improves Obesity-Induced Chronic Kidney Disease by Activating AMPK Pathway in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2020, 22, 350. [Google Scholar] [CrossRef] [PubMed]
- Lambona, C.; Zwergel, C.; Valente, S.; Mai, A. SIRT3 Activation a Promise in Drug Development? New Insights into SIRT3 Biology and Its Implications on the Drug Discovery Process. J. Med. Chem. 2024, 67, 1662–1689. [Google Scholar] [CrossRef] [PubMed]
- Suenkel, B.; Valente, S.; Zwergel, C.; Weiss, S.; Di Bello, E.; Fioravanti, R.; Aventaggiato, M.; Amorim, J.A.; Garg, N.; Kumar, S.; et al. Potent and Specific Activators for Mitochondrial Sirtuins Sirt3 and Sirt5. J. Med. Chem. 2022, 65, 14015–14031. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| AKI/CKD | Experimental Models | Intervention (Drug/Target) | SIRT3 Pathway | Mechanism | Pathways | Ref |
|---|---|---|---|---|---|---|
| AKI | Male Wistar rats administrated gentamicin | Dihydromyricetin | SIRT3 expression ↑ | Restored normal kidney function Improved renal histological changes Regeneration of renal tubular cells | NF-κB ↓ TNF-α ↓ Caspase-3 ↓ | [81] |
| AKI | Ischemia/reperfusion (I/R)-induced acute kidney injury (AKI) | Inhibition of site 1 protease (S1P) | SIRT3 expression ↑ | Attenuated tubular cell ferroptosis | SOD2 ↑ mtROS ↓ | [82] |
| AKI | CLP (cecal ligation and puncture treated)-induced septic mice In vitro: HK2 cells (human kidney cells) + LPS (lipopolysaccharide) | Melatonin | SIRT3 activity ↑ | Reduced mortality Enhanced mitophagic flux | SIRT3-dependant TFAM deacetylation | [83] |
| AKI | Cisplatin-induced nephrotoxicity in mice In vitro: HK2 cells + cisplatin | Magnesium isoglycyrrhizinate | SIRT3 expression ↑ | Protected mtDNA | NAD+ levels ↑ | [84] |
| AKI | Ischemia/reperfusion (I/R)-induced acute kidney injury (AKI) In vitro: HK2 cells in hypoxic conditions | MitoQ | SIRT3 expression ↑ | Improved mitochondrial function | mtROS ↓ | [85] |
| AKI | Cisplatin-induced nephrotoxicity in mice In vitro: HK2 cells + cisplatin | Honokiol | SIRT3 expression ↑ | Prevented mitochondrial fragmentation Decreased cell injury and death | AMPK activity ↑ Drp1 translocation in mitochondria ↓ | [86] |
| CKD | Ureteral obstruction (UUO) mouse model In vitro: HK2 cells + TGF-β1 | Diosmin | SIRT3 expression and activity ↑ | Inhibited fibrosis and inflammation | SIRT3-dependant reduced TGF-β1 | [87] |
| DKD | db/db mice | NR (nicotinamide riboside) | SIRT3 activity ↑ | Improved mitochondrial function | cGAS-STING pathway ↓ NAD+ levels ↑ | [88] |
| DKD | High-fat diet and streptozotocin mouse model In vitro: NRK-52 cells (rat kidney cells) + PA (Palmitic Acid) | Recombinant Metrnl (Meteorin-like protein) | SIRT3 expression ↑ | Alleviates renal injuries in diabetic mice | SIRT3-AMPK/UCP1 signaling axis ↑ | [89] |
| CKD | High-salt diet-induced hypertensive rats In vitro: HK2 cells + AngII (Angiotensin II) | Canagliflozin | SIRT3 expression ↑ | Improved EMT and renal injury | SIRT3-FOXO3 pathway ↑ | [90] |
| CKD | Ureteral obstruction (UUO) mouse model In vitro: NRK-49F cells + TGF-β1 | Honokiol | SIRT3 expression ↑ | Increased mitochondrial fusion Decreased inflammation | NF-κB/TGF-β1/Smad ↓ | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juszczak, F.; Arnould, T.; Declèves, A.-E. The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases. Int. J. Mol. Sci. 2024, 25, 6936. https://doi.org/10.3390/ijms25136936
Juszczak F, Arnould T, Declèves A-E. The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases. International Journal of Molecular Sciences. 2024; 25(13):6936. https://doi.org/10.3390/ijms25136936
Chicago/Turabian StyleJuszczak, Florian, Thierry Arnould, and Anne-Emilie Declèves. 2024. "The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases" International Journal of Molecular Sciences 25, no. 13: 6936. https://doi.org/10.3390/ijms25136936
APA StyleJuszczak, F., Arnould, T., & Declèves, A.-E. (2024). The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases. International Journal of Molecular Sciences, 25(13), 6936. https://doi.org/10.3390/ijms25136936