
Metabolic Endotoxemia: From the Gut to Neurodegeneration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Metabolic Endotoxemia
2.1. Factors Promoting ME Development
2.1.1. High-Fat Diet
2.1.2. Fatty Acids
2.1.3. Gut Microbiota
2.1.4. Obesity
3. Endotoxin Transfer from the Gut to Circulation
3.1. Transcellular Endotoxin Translocation through the Intestinal Epithelium
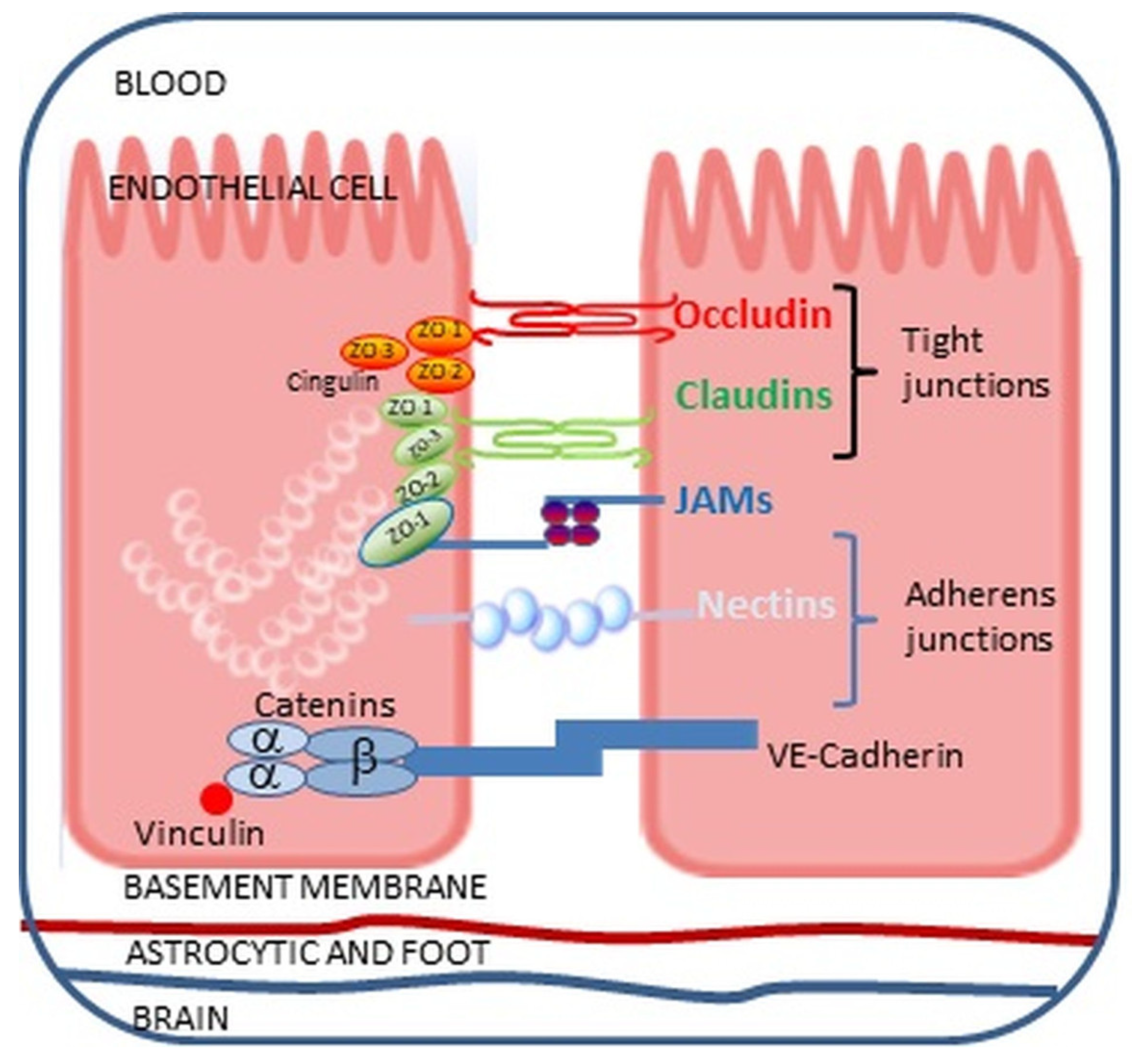
3.2. Paracellular Endotoxin Transport through the Intestinal Epithelium
3.3. Endotoxin Neutralization in the blood
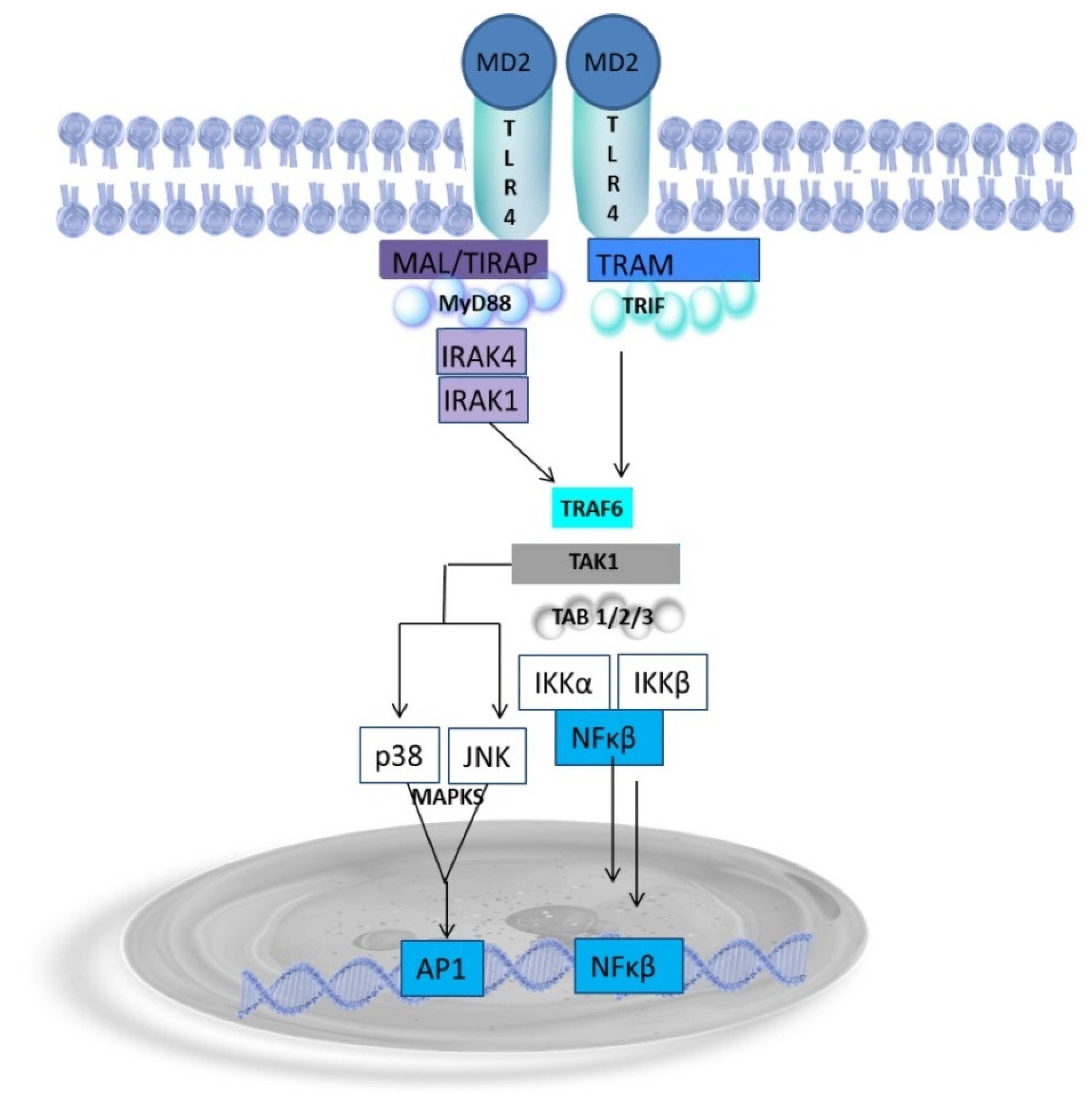
4. Endotoxin Structure Dictates the Host Immune Response
Endotoxin as a Potent Inductor of Proinflammatory Response
5. Can Endotoxin Cross the Blood–Brain Barrier?
The Impact of Endotoxin on the Blood–Brain Barrier
6. Metabolic Endotoxemia-Related Microglia Activation
7. Role of Endotoxin in Neurodegenerative Diseases
7.1. Endotoxin in Alzheimer’s Disease (AD)
7.2. Endotoxin in Parkinson’s Disease (PD)
7.3. Endotoxin in Amyotrophic Lateral Sclerosis (ALS)
7.4. Endotoxin in Huntington’s Disease (HD)
8. Endotoxin-Induced Epigenetic Regulation of the Microglial Phenotype
8.1. DNA Methylation
8.2. MicroRNAs
8.3. Histone Modifications
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brown, G.C. The Endotoxin Hypothesis of Neurodegeneration. J. Neuroinflamm. 2019, 16, 180. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.D. The Gut Microbiome and Its Role in Obesity. Nutr. Today 2016, 51, 167–174. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.; Ramakers, B.P.; van Alfen, N.; van der Werf, S.P.; Fick, W.F.; Hoedemaekers, C.W.; Verbeek, M.M.; Schoonhoven, L.; van der Hoeven, J.G.; Pickkers, P. Endotoxemia-Induced Inflammation and the Effect on the Human Brain. Crit. Care 2010, 14, R81. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, M.; Tousif, A.H.; Sonali, S.; Vichitra, C.; Sunanda, T.; Praveenraj, S.S.; Ray, B.; Gorantla, V.R.; Rungratanawanich, W.; Mahalakshmi, A.M.; et al. Role of Endogenous Lipopolysaccharides in Neurological Disorders. Cells 2022, 11, 4038. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277. [Google Scholar] [CrossRef] [PubMed]
- Boutagy, N.E.; McMillan, R.P.; Frisard, M.I.; Hulver, M.W. Metabolic Endotoxemia with Obesity: Is It Real and Is It Relevant? Biochimie 2016, 124, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.M. The Deadly Quartet. Upper-Body Obesity, Glucose Intolerance, Hypertriglyceridemia, and Hypertension. Arch. Intern. Med. 1989, 149, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Scarano, F.; Nucera, S.; Scicchitano, M.; Bosco, F.; Ruga, S.; Zito, M.C.; et al. From Metabolic Syndrome to Neurological Diseases: Role of Autophagy. Front. Cell Dev. Biol. 2021, 9, 651021. [Google Scholar] [CrossRef]
- Nádházi, Z.; Takáts, A.; Offenmüller, K.; Bertók, L. Plasma Endotoxin Level of Healthy Donors. Acta Microbiol. Immunol. Hung. 2002, 49, 151–157. [Google Scholar] [CrossRef]
- Lasselin, J.; Lekander, M.; Benson, S.; Schedlowski, M.; Engler, H. Sick for Science: Experimental Endotoxemia as a Translational Tool to Develop and Test New Therapies for Inflammation-Associated Depression. Mol. Psychiatry 2021, 26, 3672–3683. [Google Scholar] [CrossRef] [PubMed]
- Engler, H.; Brendt, P.; Wischermann, J.; Wegner, A.; Röhling, R.; Schoemberg, T.; Meyer, U.; Gold, R.; Peters, J.; Benson, S.; et al. Selective Increase of Cerebrospinal Fluid IL-6 during Experimental Systemic Inflammation in Humans: Association with Depressive Symptoms. Mol. Psychiatry 2017, 22, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Reichenberg, A.; Yirmiya, R.; Schuld, A.; Kraus, T.; Haack, M.; Morag, A.; Pollmächer, T. Cytokine-Associated Emotional and Cognitive Disturbances in Humans. Arch. Gen. Psychiatry 2001, 58, 445. [Google Scholar] [CrossRef] [PubMed]
- Grigoleit, J.-S.; Kullmann, J.S.; Wolf, O.T.; Hammes, F.; Wegner, A.; Jablonowski, S.; Engler, H.; Gizewski, E.; Oberbeck, R.; Schedlowski, M. Dose-Dependent Effects of Endotoxin on Neurobehavioral Functions in Humans. PLoS ONE 2011, 6, e28330. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, N.I.; Berkman, E.T.; Inagaki, T.K.; Rameson, L.T.; Mashal, N.M.; Irwin, M.R. Inflammation-Induced Anhedonia: Endotoxin Reduces Ventral Striatum Responses to Reward. Biol. Psychiatry 2010, 68, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Lasselin, J.; Elsenbruch, S.; Lekander, M.; Axelsson, J.; Karshikoff, B.; Grigoleit, J.-S.; Engler, H.; Schedlowski, M.; Benson, S. Mood Disturbance during Experimental Endotoxemia: Predictors of State Anxiety as a Psychological Component of Sickness Behavior. Brain Behav. Immun. 2016, 57, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Sandiego, C.M.; Gallezot, J.D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.F.; Matuskey, D.; Lee, J.Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging Robust Microglial Activation after Lipopolysaccharide Administration in Humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef] [PubMed]
- Draper, A.; Koch, R.M.; Van Der Meer, J.W.; Apps, M.A.; Pickkers, P.; Husain, M.; Van Der Schaaf, M.E. Effort but Not Reward Sensitivity Is Altered by Acute Sickness Induced by Experimental Endotoxemia in Humans. Neuropsychopharmacology 2018, 43, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Yirmiya, R.; Goshen, I. Immune Modulation of Learning, Memory, Neural Plasticity and Neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, J.S.; Grigoleit, J.-S.; Wolf, O.T.; Engler, H.; Oberbeck, R.; Elsenbruch, S.; Forsting, M.; Schedlowski, M.; Gizewski, E.R. Experimental Human Endotoxemia Enhances Brain Activity during Social Cognition. Soc. Cogn. Affect. Neurosci. 2014, 9, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Kealy, J.; Murray, C.; Griffin, E.W.; Belen Lopez-Rodriguez, A.; Healy, D.; Silva Tortorelli, L.; Lowry, J.P.; Otto Watne, L.; Cunningham, C. Neurobiology of Disease Acute Inflammation Alters Brain Energy Metabolism in Mice and Humans: Role in Suppressed Spontaneous Activity, Impaired Cognition, and Delirium. J. Neurosci. 2020, 40, 5681–5696. [Google Scholar] [CrossRef] [PubMed]
- Teeling, J.L.; Felton, L.M.; Deacon, R.M.J.; Cunningham, C.; Rawlins, J.N.P.; Perry, V.H. Sub-Pyrogenic Systemic Inflammation Impacts on Brain and Behavior, Independent of Cytokines. Brain Behav. Immun. 2007, 21, 836–850. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation Induced by Lipopolysaccharide Causes Cognitive Impairment in Mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [PubMed]
- Kurita, N.; Yamashiro, K.; Kuroki, T.; Tanaka, R.; Urabe, T.; Ueno, Y.; Miyamoto, N.; Takanashi, M.; Shimura, H.; Inaba, T.; et al. Metabolic Endotoxemia Promotes Neuroinflammation after Focal Cerebral Ischemia. J. Cerebral Blood Flow Metab. 2020, 40, 2505–2520. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.I. Nutritional Management of Metabolic Endotoxemia: A Clinical Review. Altern. Ther. Health Med. 2017, 23, 42–54. [Google Scholar] [PubMed]
- Lyte, J.M.; Gabler, N.K.; Hollis, J.H. Postprandial Serum Endotoxin in Healthy Humans Is Modulated by Dietary Fat in a Randomized, Controlled, Cross-over Study. Lipids Health Dis. 2016, 15, 186. [Google Scholar] [CrossRef] [PubMed]
- Kallio, K.A.E.; Hätönen, K.A.; Lehto, M.; Salomaa, V.; Männistö, S.; Pussinen, P.J. Endotoxemia, Nutrition, and Cardiometabolic Disorders. Acta Diabetol. 2015, 52, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Herieka, M.; Erridge, C. High-fat Meal Induced Postprandial Inflammation. Mol. Nutr. Food Res. 2014, 58, 136–146. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in Plasma Endotoxin Concentrations and the Expression of Toll-like Receptors and Suppressor of Cytokine Signaling-3 in Mononuclear Cells after a High-Fat, High-Carbohydrate Meal: Implications for Insulin Resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated With Endotoxemia That Originates From the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [PubMed]
- Cândido, T.L.N.; da Silva, L.E.; Tavares, J.F.; Conti, A.C.M.; Rizzardo, R.A.G.; Gonçalves Alfenas, R.d.C. Effects of Dietary Fat Quality on Metabolic Endotoxaemia: A Systematic Review. Br. J. Nutr. 2020, 124, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Oscarsson, J.; Hurt-Camejo, E. Omega-3 Fatty Acids Eicosapentaenoic Acid and Docosahexaenoic Acid and Their Mechanisms of Action on Apolipoprotein B-Containing Lipoproteins in Humans: A Review. Lipids Health Dis. 2017, 16, 149. [Google Scholar] [CrossRef]
- Seufert, A.L.; Hickman, J.W.; Traxler, S.K.; Peterson, R.M.; Waugh, T.A.; Lashley, S.J.; Shulzhenko, N.; Napier, R.J.; Napier, B.A. Enriched Dietary Saturated Fatty Acids Induce Trained Immunity via Ceramide Production That Enhances Severity of Endotoxemia and Clearance of Infection. Elife 2022, 11, e76744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Y.; Franco, D.A.; Schwartz, E.; D’Souza, K.; Karnick, S.; Reaven, P.D. HDL Inhibits Saturated Fatty Acid Mediated Augmentation of Innate Immune Responses in Endothelial Cells by a Novel Pathway. Atherosclerosis 2017, 259, 83–96. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Laugerette, F.; Féart, C. Metabolic Endotoxemia: A Potential Underlying Mechanism of the Relationship between Dietary Fat Intake and Risk for Cognitive Impairments in Humans? Nutrients 2019, 11, 1887. [Google Scholar] [CrossRef] [PubMed]
- Wit, M.; Trujillo-Viera, J.; Strohmeyer, A.; Klingenspor, M.; Hankir, M.; Sumara, G. When Fat Meets the Gut—Focus on Intestinal Lipid Handling in Metabolic Health and Disease. EMBO Mol. Med. 2022, 14, e14742. [Google Scholar] [CrossRef] [PubMed]
- Deopurkar, R.; Ghanim, H.; Friedman, J.; Abuaysheh, S.; Sia, C.L.; Mohanty, P.; Viswanathan, P.; Chaudhuri, A.; Dandona, P. Differential Effects of Cream, Glucose, and Orange Juice on Inflammation, Endotoxin, and the Expression of Toll-like Receptor-4 and Suppressor of Cytokine Signaling-3. Diabetes Care 2010, 33, 991–997. [Google Scholar] [CrossRef]
- Burton-Freeman, B. Dietary Fiber and Energy Regulation. J. Nutr. 2000, 130, 272S–275S. [Google Scholar] [CrossRef]
- Mo, Z.; Huang, S.; Burnett, D.J.; Rutledge, J.C.; Hwang, D.H. Endotoxin May Not Be the Major Cause of Postprandial Inflammation in Adults Who Consume a Single High-Fat or Moderately High-Fat Meal. J. Nutr. 2020, 150, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Kobyliak, N.; Virchenko, O.; Falalyeyeva, T. Pathophysiological Role of Host Microbiota in the Development of Obesity. Nutr. J. 2015, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human Gut Microbes Associated with Obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human Gut Microbiota in Obesity and after Gastric Bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Lutz, B.; Ruiz de Azua, I. The Microbiome and Gut Endocannabinoid System in the Regulation of Stress Responses and Metabolism. Front. Cell Neurosci. 2022, 16, 867267. [Google Scholar] [CrossRef] [PubMed]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, J.C.; Matheus, V.A.; Oliveira, R.B.; Tada, S.F.S.; Collares-Buzato, C.B. High-Fat Diet Induces Disruption of the Tight Junction-Mediated Paracellular Barrier in the Proximal Small Intestine Before the Onset of Type 2 Diabetes and Endotoxemia. Dig. Dis. Sci. 2021, 66, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Mandard, S.; Tan, N.S.; Escher, P.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Characterization of the Fasting-Induced Adipose Factor FIAF, a Novel Peroxisome Proliferator-Activated Receptor Target Gene. J. Biol. Chem. 2000, 275, 28488–28493. [Google Scholar] [CrossRef] [PubMed]
- Hafidi, M.E.; Buelna-Chontal, M.; Sánchez-Muñoz, F.; Carbó, R. Adipogenesis: A Necessary but Harmful Strategy. Int. J. Mol. Sci. 2019, 20, 3657. [Google Scholar] [CrossRef] [PubMed]
- Zu, L.; He, J.; Jiang, H.; Xu, C.; Pu, S.; Xu, G. Bacterial Endotoxin Stimulates Adipose Lipolysis via Toll-like Receptor 4 and Extracellular Signal-Regulated Kinase Pathway. J. Biol. Chem. 2009, 284, 5915–5926. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; El Ghoch, M.; Colao, A.; Hassapidou, M.; Yumuk, V.; Busetto, L. European Guidelines for Obesity Management in Adults with a Very Low-Calorie Ketogenic Diet: A Systematic Review and Meta-Analysis. Obes. Facts 2021, 14, 222–245. [Google Scholar] [CrossRef]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms Underlying the Resistance to Diet-Induced Obesity in Germ-Free Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef]
- Pérez-Hernández, E.G.; Delgado-Coello, B.; Luna-Reyes, I.; Mas-Oliva, J. New Insights into Lipopolysaccharide Inactivation Mechanisms in Sepsis. Biomed. Pharmacother. 2021, 141, 111890. [Google Scholar] [CrossRef] [PubMed]
- Cario, E.; Podolsky, D.K. Differential Alteration in Intestinal Epithelial Cell Expression of Toll-Like Receptor 3 (TLR3) and TLR4 in Inflammatory Bowel Disease. Infect. Immun. 2000, 68, 7010–7017. [Google Scholar] [CrossRef]
- Ashton, T.; Young, I.S.; Davison, G.W.; Rowlands, C.C.; McEneny, J.; Van Blerk, C.; Jones, E.; Peters, J.R.; Jackson, S.K. Exercise-Induced Endotoxemia: The Effect of Ascorbic Acid Supplementation. Free Radic. Biol. Med. 2003, 35, 284–291. [Google Scholar] [CrossRef]
- de Punder, K.; Pruimboom, L. Stress Induces Endotoxemia and Low-Grade Inflammation by Increasing Barrier Permeability. Front. Immunol. 2015, 6, 223. [Google Scholar] [CrossRef]
- Guerville, M.; Boudry, G. Gastrointestinal and Hepatic Mechanisms Limiting Entry and Dissemination of Lipopolysaccharide into the Systemic Circulation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 311, G1–G15. [Google Scholar] [CrossRef]
- Akiba, Y.; Maruta, K.; Takajo, T.; Narimatsu, K.; Said, H.; Kato, I.; Kuwahara, A.; Kaunitz, J.D. Lipopolysaccharides Transport during Fat Absorption in Rodent Small Intestine. Am. J. Physiol.-Gastrointest. Liver Physiol. 2020, 318, G1070–G1087. [Google Scholar] [CrossRef]
- McDole, J.R.; Wheeler, L.W.; McDonald, K.G.; Wang, B.; Konjufca, V.; Knoop, K.A.; Newberry, R.D.; Miller, M.J. Goblet Cells Deliver Luminal Antigen to CD103 + Dendritic Cells in the Small Intestine. Nature 2012, 483, 345–349. [Google Scholar] [CrossRef]
- Knoop, K.A.; Gustafsson, J.K.; McDonald, K.G.; Kulkarni, D.H.; Kassel, R.; Newberry, R.D. Antibiotics Promote the Sampling of Luminal Antigens and Bacteria via Colonic Goblet Cell Associated Antigen Passages. Gut Microbes 2017, 8, 400–411. [Google Scholar] [CrossRef]
- Hollander, D.; Kaunitz, J.D. The “Leaky Gut”: Tight Junctions but Loose Associations? Dig. Dis. Sci. 2020, 65, 1277–1287. [Google Scholar] [CrossRef]
- Matikainen, S.; Nyman, T.A.; Cypryk, W. Function and Regulation of Noncanonical Caspase-4/5/11 Inflammasome. J. Immunol. 2020, 204, 3063–3069. [Google Scholar] [CrossRef]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B.; Lippens, L.; De Scheerder, M.-A.; Miinalainen, I.; Rappu, P.; De Geest, B.G.; et al. Increased Levels of Systemic LPS-Positive Bacterial Extracellular Vesicles in Patients with Intestinal Barrier Dysfunction. Gut 2020, 69, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-S.; Zhou, H.-N.; He, S.-S.; Xue, M.-Y.; Li, T.; Liu, L.-M. Research Advances in Pericyte Function and Their Roles in Diseases. Chin. J. Traumatol. 2020, 23, 89–95. [Google Scholar] [CrossRef]
- Li, W.; Deng, M.; Loughran, P.A.; Yang, M.; Lin, M.; Yang, C.; Gao, W.; Jin, S.; Li, S.; Cai, J.; et al. LPS Induces Active HMGB1 Release From Hepatocytes Into Exosomes Through the Coordinated Activities of TLR4 and Caspase-11/GSDMD Signaling. Front. Immunol. 2020, 11, 229. [Google Scholar] [CrossRef]
- Von Moltke, J.; Trinidad, N.J.; Moayeri, M.; Kintzer, A.F.; Wang, S.B.; Van Rooijen, N.; Brown, C.R.; Krantz, B.A.; Leppla, S.H.; Gronert, K.; et al. Rapid Induction of Inflammatory Lipid Mediators by the Inflammasome in Vivo. Nature 2012, 490, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Sharma, A.K.; Ismail, N. Non-Canonical Inflammasome Pathway: The Role of Cell Death and Inflammation in Ehrlichiosis. Cells 2023, 12, 2597. [Google Scholar] [CrossRef]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons Promote Intestinal Absorption of Lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef]
- Shen, Y.-H.A.; Nahas, R. Complementary and Alternative Medicine for Treatment of Irritable Bowel Syndrome. Can. Fam. Physician 2009, 55, 143–148. [Google Scholar]
- Buckley, A.; Turner, J.R. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef] [PubMed]
- Nighot, M.; Al-Sadi, R.; Guo, S.; Rawat, M.; Nighot, P.; Watterson, M.D.; Ma, T.Y. Lipopolysaccharide-Induced Increase in Intestinal Epithelial Tight Permeability Is Mediated by Toll-Like Receptor 4/Myeloid Differentiation Primary Response 88 (MyD88) Activation of Myosin Light Chain Kinase Expression. Am. J. Pathol. 2017, 187, 2698–2710. [Google Scholar] [CrossRef] [PubMed]
- Read, T.E.; Harris, H.W.; Grunfeld, C.; Feingold, K.R.; Kane, J.P.; Rapp, J.H. The Protective Effect of Serum Lipoproteins against Bacterial Lipopolysaccharide. Eur. Heart J. 1993, 14 (Suppl. K), 125–129. [Google Scholar] [PubMed]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a Receptor for Complexes of Lipopolysaccharide (LPS) and LPS Binding Protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G. Peptide Antibiotics and Their Role in Innate Immunity. Annu. Rev. Immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, J.; Milošević, A.; Milin, Č. Gas Chromatographic Determination of Fatty Acids Contained in Different Lipid Classes after Their Separation by Solid-Phase Extraction. J. Chromatogr. A 2002, 976, 47–54. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Harm, S.; Schildböck, C.; Strobl, K.; Hartmann, J. An in Vitro Study on Factors Affecting Endotoxin Neutralization in Human Plasma Using the Limulus Amebocyte Lysate Test. Sci. Rep. 2021, 11, 4192. [Google Scholar] [CrossRef]
- Pulido, J.N.; Afessa, B.; Masaki, M.; Yuasa, T.; Gillespie, S.; Herasevich, V.; Brown, D.R.; Oh, J.K. Clinical Spectrum, Frequency, and Significance of Myocardial Dysfunction in Severe Sepsis and Septic Shock. Mayo Clin. Proc. 2012, 87, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, S.; Zhang, X.; Zhang, X.; Dong, H.; Qian, Y. IL-17A Is Implicated in Lipopolysaccharide-Induced Neuroinflammation and Cognitive Impairment in Aged Rats via Microglial Activation. J. Neuroinflamm. 2015, 12, 165. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, C.; Ullrich, H.; Hohe, R.; Berg, A.; Baumstark, M.W.; Frey, I.; Northoff, H.; Flegel, W.A. Low Density Lipoproteins Inhibit Endotoxin Activation of Monocytes. Arterioscler. Thromb. 1992, 12, 341–347. [Google Scholar] [CrossRef]
- Cavaillon, J.M.; Haeffner-Cavaillon, N. Signals Involved in Interleukin 1 Synthesis and Release by Lipopolysaccharide-Stimulated Monocytes/Macrophages. Cytokine 1990, 2, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Dinges, M.M.; Schlievert, P.M. Comparative Analysis of Lipopolysaccharide-Induced Tumor Necrosis Factor Alpha Activity in Serum and Lethality in Mice and Rabbits Pretreated with the Staphylococcal Superantigen Toxic Shock Syndrome Toxin 1. Infect. Immun. 2001, 69, 7169–7172. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.M.; Zimmermann, K. Lipopolysaccharides in Food, Food Supplements, and Probiotics: Should We Be Worried? Eur. J. Microbiol. Immunol. 2018, 8, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Gilliam, E.A.; Li, L. Innate Immune Programing by Endotoxin and Its Pathological Consequences. Front. Immunol. 2015, 5, 680. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Chen, J.; Ernst, R.K.; Wang, X. Influence of Lipid a Acylation Pattern on Membrane Permeability and Innate Immune Stimulation. Mar. Drugs 2013, 11, 3197–3208. [Google Scholar] [CrossRef] [PubMed]
- Mazgaeen, L.; Gurung, P. Recent Advances in Lipopolysaccharide Recognition Systems. Int. J. Mol. Sci. 2020, 21, 379. [Google Scholar] [CrossRef] [PubMed]
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, N.; Fernandez, R.C. Recognition of Lipid A Variants by the TLR4-MD-2 Receptor Complex. Front. Cell. Infect. Microbiol. 2013, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Steimle, A.; Autenrieth, I.B.; Frick, J.-S. Structure and Function: Lipid A Modifications in Commensals and Pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef]
- Gorelik, A.; Illes, K.; Nagar, B. Crystal Structure of the Mammalian Lipopolysaccharide Detoxifier. Proc. Natl. Acad. Sci. USA 2018, 115, E896–E905. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Barra, N.G.; Cavallari, J.F.; Henriksbo, B.D.; Schertzer, J.D. Metabolic Endotoxemia Is Dictated by the Type of Lipopolysaccharide. Cell Rep. 2021, 36, 109691. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2021, 11, 594150. [Google Scholar] [CrossRef] [PubMed]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed]
- Fenton, M.J.; Golenbock, D.T. LPS-Binding Proteins and Receptors. J. Leukoc. Biol. 1998, 64, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Taban, Q.; Mumtaz, P.T.; Masoodi, K.Z.; Haq, E.; Ahmad, S.M. Scavenger Receptors in Host Defense: From Functional Aspects to Mode of Action. Cell Commun. Signal. 2022, 20, 2. [Google Scholar] [CrossRef]
- Kim, S.-W.; Oh, S.-A.; Seol, S.-I.; Davaanyam, D.; Lee, J.-K. Cytosolic HMGB1 Mediates LPS-Induced Autophagy in Microglia by Interacting with NOD2 and Suppresses Its Proinflammatory Function. Cells 2022, 11, 2410. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Mori, Y.; Hagiwara, K.; Suzuki, T.; Sakakibara, T.; Kikuchi, T.; Igarashi, T.; Ebina, M.; Abe, T.; Miyazaki, J.; et al. Increased Susceptibility to LPS-Induced Endotoxin Shock in Secretory Leukoprotease Inhibitor (SLPI)-Deficient Mice. J. Exp. Med. 2003, 197, 669–674. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 Trafficking and Its Influence on LPS-Induced pro-Inflammatory Signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Lizundia, R.; Sauter, K.S.; Taylor, G.; Werling, D. Host Species-Specific Usage of the TLR4-LPS Receptor Complex. Innate Immun. 2008, 14, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Sakai, J.; Cammarota, E.; Wright, J.A.; Cicuta, P.; Gottschalk, R.A.; Li, N.; Fraser, I.D.C.; Bryant, C.E. Lipopolysaccharide-Induced NF-ΚB Nuclear Translocation Is Primarily Dependent on MyD88, but TNFα Expression Requires TRIF and MyD88. Sci. Rep. 2017, 7, 1428. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.L.; Suzuki, K. The pattern recognition receptors and lipopolysaccharides (LPS)-induced systemic inflammation. Int. J. Res. Stud. Med. Health Sci. 2017, 2, 1–7. [Google Scholar]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D.; Balka, K.R.; Gloria, Y.C.; Rao, V.R.; Latz, E.; Masters, S.L. Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) Plays a Dual Role in Myddosome Formation and Toll-like Receptor Signaling. J. Biol. Chem. 2018, 293, 15195–15207. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 Controls the LPS-Induced Endocytosis of Toll-like Receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.-F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.-M.; Minogue, E.; Curham, L.; Tyrrell, H.; Gavigan, P.; Hind, W.; Downer, E.J. MyD88-Dependent and -Independent Signalling via TLR3 and TLR4 Are Differentially Modulated by Δ9-Tetrahydrocannabinol and Cannabidiol in Human Macrophages. J. Neuroimmunol. 2020, 343, 577217. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR Signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kumazawa, Y.; Inoue, J. Liposomal Lipopolysaccharide Initiates TRIF-Dependent Signaling Pathway Independent of CD14. PLoS ONE 2013, 8, e60078. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, Y.; Wang, J.; Dong, L.; Liu, S.; Li, S.; Wu, Q. Purinergic Signaling: A Gatekeeper of Blood-Brain Barrier Permeation. Front. Pharmacol. 2023, 14, 1112758. [Google Scholar] [CrossRef] [PubMed]
- Mapunda, J.A.; Tibar, H.; Regragui, W.; Engelhardt, B. How Does the Immune System Enter the Brain? Front. Immunol. 2022, 13, 805657. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, X. The Crosstalk between the Blood–Brain Barrier Dysfunction and Neuroinflammation after General Anaesthesia. Curr. Issues Mol. Biol. 2022, 44, 5700–5717. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Engelhardt, B. Immune Cell Trafficking across the Blood-Brain Barrier in the Absence and Presence of Neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular Mechanisms of Alzheimer’s Neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Pachter, J.S.; de Vries, H.E.; Fabry, Z. The Blood-Brain Barrier and Its Role in Immune Privilege in the Central Nervous System. J. Neuropathol. Exp. Neurol. 2003, 62, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood–Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2019, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Young, A.P.; Denovan-Wright, E.M. The Dynamic Role of Microglia and the Endocannabinoid System in Neuroinflammation. Front. Pharmacol. 2022, 12, 806417. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The Blood-Brain Barrier in Alzheimer’s Disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Verheggen, I.C.M.; de Jong, J.J.A.; van Boxtel, M.P.J.; Postma, A.A.; Verhey, F.R.J.; Jansen, J.F.A.; Backes, W.H. Permeability of the Windows of the Brain: Feasibility of Dynamic Contrast-Enhanced MRI of the Circumventricular Organs. Fluids Barriers CNS 2020, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Galea, I. The Blood–Brain Barrier in Systemic Infection and Inflammation. Cell Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-Induced Blood-Brain Barrier Disruption: Roles of Cyclooxygenase, Oxidative Stress, Neuroinflammation, and Elements of the Neurovascular Unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Robinson, S.M. Minimal Penetration of Lipopolysaccharide across the Murine Blood–Brain Barrier. Brain Behav. Immun. 2010, 24, 102–109. [Google Scholar] [CrossRef]
- Wang, P.; You, S.-W.; Yang, Y.-J.; Wei, X.-Y.; Wang, Y.-Z.; Wang, X.; Hao, D.-J.; Kuang, F.; Shang, L.-X. Systemic Injection of Low-Dose Lipopolysaccharide Fails to Break down the Blood–Brain Barrier or Activate the TLR4-MyD88 Pathway in Neonatal Rat Brain. Int. J. Mol. Sci. 2014, 15, 10101–10115. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Han, J.; Suk, K. Protective Effects of Complement Component 8 Gamma Against Blood-Brain Barrier Breakdown. Front. Physiol. 2021, 12, 671250. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.F.; Bodart-Santos, V.; De Felice, F.G.; Ferreira, S.T. Neuroprotective Actions of Glucagon-Like Peptide-1 (GLP-1) Analogues in Alzheimer’s and Parkinson’s Diseases. CNS Drugs 2019, 33, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-Negative Bacterial Molecules Associate with Alzheimer Disease Pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cong, L.; Lukiw, W.J. Lipopolysaccharide (LPS) Accumulates in Neocortical Neurons of Alzheimer’s Disease (AD) Brain and Impairs Transcription in Human Neuronal-Glial Primary Co-Cultures. Front. Aging Neurosci. 2017, 9, 407. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide Associates with Amyloid Plaques, Neurons and Oligodendrocytes in Alzheimer’s Disease Brain: A Review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef]
- Vargas-Caraveo, A.; Sayd, A.; Maus, S.R.; Caso, J.R.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C. Lipopolysaccharide Enters the Rat Brain by a Lipoprotein-Mediated Transport Mechanism in Physiological Conditions. Sci. Rep. 2017, 7, 13113. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Luo, Z.; He, S.; Zhang, L.; Li, Y. Blood-Brain Barrier Disruption by Lipopolysaccharide and Sepsis-Associated Encephalopathy. Front. Cell Infect. Microbiol. 2021, 11, 768108. [Google Scholar] [CrossRef] [PubMed]
- Varatharaj, A.; Galea, I. The Blood-Brain Barrier in Systemic Inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Lee, W.; Jung, Y.-S. Chrysin Attenuates VCAM-1 Expression and Monocyte Adhesion in Lipopolysaccharide-Stimulated Brain Endothelial Cells by Preventing NF-ΚB Signaling. Int. J. Mol. Sci. 2017, 18, 1424. [Google Scholar] [CrossRef] [PubMed]
- Pfalzgraff, A.; Correa, W.; Heinbockel, L.; Schromm, A.B.; Lübow, C.; Gisch, N.; Martinez-de-Tejada, G.; Brandenburg, K.; Weindl, G. LPS-Neutralizing Peptides Reduce Outer Membrane Vesicle-Induced Inflammatory Responses. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1503–1513. [Google Scholar] [CrossRef]
- Nonaka, S.; Kadowaki, T.; Nakanishi, H. Secreted Gingipains from Porphyromonas Gingivalis Increase Permeability in Human Cerebral Microvascular Endothelial Cells through Intracellular Degradation of Tight Junction Proteins. Neurochem. Int. 2022, 154, 105282. [Google Scholar] [CrossRef]
- Ha, J.Y.; Choi, S.-Y.; Lee, J.H.; Hong, S.-H.; Lee, H.-J. Delivery of Periodontopathogenic Extracellular Vesicles to Brain Monocytes and Microglial IL-6 Promotion by RNA Cargo. Front. Mol. Biosci. 2020, 7, 596366. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V.A.K. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Galka, F.; Sun, N.W.; Kusch, H.; Engelmann, S.; Hecker, M.; Schmeck, B.; Hippenstiel, S.; Uhlin, B.E.; Steinert, M. Proteomic Characterization of the Whole Secretome of Legionella Pneumophila and Functional Analysis of Outer Membrane Vesicles. Infect. Immun. 2008, 76, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Caruana, J.C.; Walper, S.A. Bacterial Membrane Vesicles as Mediators of Microbe–Microbe and Microbe–Host Community Interactions. Front. Microbiol. 2020, 11, 432. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Leukocyte–Endothelial-Cell Interactions in Leukocyte Transmigration and the Inflammatory Response. Trends Immunol. 2003, 24, 326–333. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, S.J.; Kho, D.T.; Wiltshire, R.; Nelson, V.; Rotimi, O.; Johnson, R.; Angel, C.E.; Graham, E.S. Pro-Inflammatory TNFα and IL-1β Differentially Regulate the Inflammatory Phenotype of Brain Microvascular Endothelial Cells. J. Neuroinflamm. 2015, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Weber, M.T.; Xiao, R.; Cullen, D.K.; Meaney, D.F.; Stewart, W.; Smith, D.H. Mechanical Disruption of the Blood–Brain Barrier Following Experimental Concussion. Acta Neuropathol. 2018, 135, 711–726. [Google Scholar] [CrossRef]
- Bowyer, J.F.; Sarkar, S.; Burks, S.M.; Hess, J.N.; Tolani, S.; O’Callaghan, J.P.; Hanig, J.P. Microglial Activation and Responses to Vasculature That Result from an Acute LPS Exposure. Neurotoxicology 2020, 77, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Nagyoszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.K.; Haskó, J.; Krizbai, I.A. Expression and Regulation of Toll-like Receptors in Cerebral Endothelial Cells. Neurochem. Int. 2010, 57, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.M.; Kim, J.M.; Park, T.Y.; Baik, E.J.; Lee, S.H. Fructose-1,6-Bisphosphate Ameliorates Lipopolysaccharide-Induced Dysfunction of Blood-Brain Barrier. Arch. Pharm. Res. 2013, 36, 1149–1159. [Google Scholar] [CrossRef]
- Veszelka, S.; Pásztói, M.; Farkas, A.E.; Krizbai, I.; Dung, N.T.K.; Niwa, M.; Ábrahám, C.S.; Deli, M.A. Pentosan Polysulfate Protects Brain Endothelial Cells against Bacterial Lipopolysaccharide-Induced Damages. Neurochem. Int. 2007, 50, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Huang, W.; Mo, X.; Chen, Y.; Wu, X. LPS Induces Occludin Dysregulation in Cerebral Microvascular Endothelial Cells via MAPK Signaling and Augmenting MMP-2 Levels. Oxid. Med. Cell Longev. 2015, 2015, 120641. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.-H.; Lim, J.-H.; Kim, J.-H. Lipopolysaccharide Induces Matrix Metalloproteinase-9 Expression via a Mitochondrial Reactive Oxygen Species-P38 Kinase-Activator Protein-1 Pathway in Raw 264.7 Cells. J. Immunol. 2004, 173, 6973–6980. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse Roles of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Neuroinflammation and Cerebral Ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Mun-Bryce, S.; Lukes, A.; Wallace, J.; Lukes-Marx, M.; Rosenberg, G.A. Stromelysin-1 and Gelatinase A Are Upregulated before TNF-α in LPS-Stimulated Neuroinflammation. Brain Res. 2002, 933, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Versele, R.; Sevin, E.; Gosselet, F.; Fenart, L.; Candela, P. TNF-α and IL-1β Modulate Blood-Brain Barrier Permeability and Decrease Amyloid-β Peptide Efflux in a Human Blood-Brain Barrier Model. Int. J. Mol. Sci. 2022, 23, 10235. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Teng, T.; Li, R.; Simonyi, A.; Sun, G.Y.; Lee, J.C. TNFα Alters Occludin and Cerebral Endothelial Permeability: Role of P38MAPK. PLoS ONE 2017, 12, e0170346. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Chang, S.L. Meta-Analysis of the Effects of Palmitic Acid on Microglia Activation and Neurodegeneration. NeuroImmune Pharmacol. Ther. 2023, 2, 281–291. [Google Scholar] [CrossRef]
- Wu, F.; Liu, L.; Zhou, H. Endothelial Cell Activation in Central Nervous System Inflammation. J. Leukoc. Biol. 2017, 101, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Hong, J.S.; Crews, F.T. NADPH Oxidase and Aging Drive Microglial Activation, Oxidative Stress, and Dopaminergic Neurodegeneration Following Systemic LPS Administration. Glia 2013, 61, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Wiesnet, M.; Renz, D.; Schaper, W. H2O2 Induces Paracellular Permeability of Porcine Brain-Derived Microvascular Endothelial Cells by Activation of the P44/42 MAP Kinase Pathway. Eur. J. Cell Biol. 2005, 84, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, H.; Soma, G.-I. Low-Dose Lipopolysaccharide as an Immune Regulator for Homeostasis Maintenance in the Central Nervous System through Transformation to Neuroprotective Microglia. Neural Regen. Res. 2021, 16, 1928. [Google Scholar] [CrossRef] [PubMed]
- Tauber, A.I. Metchnikoff and the Phagocytosis Theory. Nat. Rev. Mol. Cell Biol. 2003, 4, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Wendeln, A.C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate Immune Memory in the Brain Shapes Neurological Disease Hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Cunningham, C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019, 40, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kan, J.; Tang, M.; Zhu, Y.; Hu, B. Lipopolysaccharide Preconditioning Restricts Microglial Overactivation and Alleviates Inflammation-Induced Depressive-like Behavior in Mice. Brain Sci. 2023, 13, 549. [Google Scholar] [CrossRef] [PubMed]
- Koss, K.; Churchward, M.A.; Tsui, C.; Todd, K.G. In Vitro Priming and Hyper-Activation of Brain Microglia: An Assessment of Phenotypes. Mol. Neurobiol. 2019, 56, 6409–6425. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Tangney, C.C. Dietary Fat Composition and Dementia Risk. Neurobiol. Aging 2014, 35, S59–S64. [Google Scholar] [CrossRef] [PubMed]
- Lajqi, T.; Lang, G.P.; Haas, F.; Williams, D.L.; Hudalla, H.; Bauer, M.; Groth, M.; Wetzker, R.; Bauer, R. Memory-Like Inflammatory Responses of Microglia to Rising Doses of LPS: Key Role of PI3Kγ. Front. Immunol. 2019, 10, 2492. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, O.; Coleman, M.P.; Durrant, C.S. Lipopolysaccharide-Induced Neuroinflammation Induces Presynaptic Disruption through a Direct Action on Brain Tissue Involving Microglia-Derived Interleukin 1 Beta. J. Neuroinflamm. 2019, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a P38-MAPK Pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef]
- Van Schependom, J.; D’haeseleer, M. Advances in Neurodegenerative Diseases. J. Clin. Med. 2023, 12, 1709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Wang, C.; Wang, X. TET (Ten-Eleven Translocation) Family Proteins: Structure, Biological Functions and Applications. Signal Transduct. Target. Ther. 2023, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Chen, J.; Li, Y. Microglia and Astrocytes Underlie Neuroinflammation and Synaptic Susceptibility in Autism Spectrum Disorder. Front. Neurosci. 2023, 17, 1125428. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lu, C.; Leszek, J.; Zhang, J. Design and Development of Nanomaterial-Based Drug Carriers to Overcome the Blood–Brain Barrier by Using Different Transport Mechanisms. Int. J. Mol. Sci. 2021, 22, 10118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, Y.; Zhu, Y.; Sun, D.; Wang, S.; Weng, J.; Zhu, Y.; Peng, W.; Yu, B.; Jiang, Y. Microglia-Specific Transcriptional Repression of Interferon-Regulated Genes after Prolonged Stress in Mice. Neurobiol. Stress. 2022, 21, 100495. [Google Scholar] [CrossRef]
- Gardner, L.E.; White, J.D.; Eimerbrink, M.J.; Boehm, G.W.; Chumley, M.J. Imatinib Methanesulfonate Reduces Hyperphosphorylation of Tau Following Repeated Peripheral Exposure to Lipopolysaccharide. Neuroscience 2016, 331, 72–77. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau Pathology by the Microglial Fractalkine Receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-A.; Jeong, J.-J.; Yoo, S.-Y.; Kim, D.-H. Gut Microbiota Lipopolysaccharide Accelerates Inflamm-Aging in Mice. BMC Microbiol. 2016, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Wang, T.L. MicroRNAs of Microglia: Wrestling with Central Nervous System Disease. Neural Regen. Res. 2018, 13, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment through Enhancement of Beta-Amyloid Generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Fainstein, N.; Elad, A.; Lachish, M.; Goldfarb, S.; Einstein, O.; Ben-Hur, T. Microbial Pathogens Induce Neurodegeneration in Alzheimer’s Disease Mice: Protection by Microglial Regulation. J. Neuroinflamm. 2022, 19, 5. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.S.; Kranjac, D.; Alonzo, C.A.; Haase, J.H.; Cedillos, R.O.; McLinden, K.A.; Boehm, G.W.; Chumley, M.J. Prolonged Elevation in Hippocampal Aβ and Cognitive Deficits Following Repeated Endotoxin Exposure in the Mouse. Behav. Brain Res. 2012, 229, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Camacho, M.; Williams-Gray, C.H. The Endotoxin Hypothesis of Parkinson’s Disease. Mov. Disord. 2023, 38, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Li, J.; Qin, Q.; Wang, D.; Zhao, J.; An, K.; Mao, Z.; Min, Z.; Xiong, Y.; Li, J.; et al. A Systematic Review and Meta-Analysis of Inflammatory Biomarkers in Parkinson’s Disease. NPJ Park. Dis. 2023, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Walker, D.I.; Lill, C.M.; Bloem, B.R.; Darweesh, S.K.L.; Pinto-Pacheco, B.; McNeil, B.; Miller, G.W.; Heath, A.K.; Frissen, M.; et al. Lipopolysaccharide-Binding Protein and Future Parkinson’s Disease Risk: A European Prospective Cohort. J. Neuroinflamm. 2023, 20, 170. [Google Scholar] [CrossRef] [PubMed]
- Shannon, K.M. Gut-Derived Sterile Inflammation and Parkinson’s Disease. Front. Neurol. 2022, 13, 831090. [Google Scholar] [CrossRef] [PubMed]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease among Patients with Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Wijeyekoon, R.S.; Kronenberg-Versteeg, D.; Scott, K.M.; Hayat, S.; Kuan, W.L.; Evans, J.R.; Breen, D.P.; Cummins, G.; Jones, J.L.; Clatworthy, M.R.; et al. Peripheral Innate Immune and Bacterial Signals Relate to Clinical Heterogeneity in Parkinson’s Disease. Brain Behav. Immun. 2020, 87, 473–488. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased Intestinal Permeability Correlates with Sigmoid Mucosa Alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef]
- de Waal, G.M.; Engelbrecht, L.; Davis, T.; de Villiers, W.J.S.; Kell, D.B.; Pretorius, E. Correlative Light-Electron Microscopy Detects Lipopolysaccharide and Its Association with Fibrin Fibres in Parkinson’s Disease, Alzheimer’s Disease and Type 2 Diabetes Mellitus. Sci. Rep. 2018, 8, 16798. [Google Scholar] [CrossRef] [PubMed]
- Gayle, D.A.; Ling, Z.; Tong, C.; Landers, T.; Lipton, J.W.; Carvey, P.M. Lipopolysaccharide (LPS)-Induced Dopamine Cell Loss in Culture: Roles of Tumor Necrosis Factor-α, Interleukin-1β, and Nitric Oxide. Dev. Brain Res. 2002, 133, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, D.M.; Perez-Otano, I.; Sun, V.; Mullis Sawin, S.B.; Chan, J.; Wu, G.-C.; Hudson, P.M.; Kong, L.-Y.; Hong, J.-S.; McMillian, M.K. Glia-Dependent Neurotoxicity and Neuroprotection in Mesencephalic Cultures. Brain Res. 1995, 704, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Bodea, L.-G.; Wang, Y.; Linnartz-Gerlach, B.; Kopatz, J.; Sinkkonen, L.; Musgrove, R.; Kaoma, T.; Muller, A.; Vallar, L.; Di Monte, D.A.; et al. Neurodegeneration by Activation of the Microglial Complement-Phagosome Pathway. J. Neurosci. 2014, 34, 8546–8556. [Google Scholar] [CrossRef] [PubMed]
- Keizman, D.; Rogowski, O.; Berliner, S.; Ish-Shalom, M.; Maimon, N.; Nefussy, B.; Artamonov, I.; Drory, V.E. Low-Grade Systemic Inflammation in Patients with Amyotrophic Lateral Sclerosis. Acta Neurol. Scand. 2009, 119, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Zhao, W.; Neal, D.W.; Thonhoff, J.R.; Thome, A.D.; Faridar, A.; Wen, S.; Wang, J.; Appel, S.H. Elevated Acute Phase Proteins Reflect Peripheral Inflammation and Disease Severity in Patients with Amyotrophic Lateral Sclerosis. Sci. Rep. 2020, 10, 15295. [Google Scholar] [CrossRef] [PubMed]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–24. [Google Scholar] [CrossRef]
- Steinberg, N.; Galleguillos, D.; Zaidi, A.; Horkey, M.; Sipione, S. Naïve Huntington’s Disease Microglia Mount a Normal Response to Inflammatory Stimuli but Display a Partially Impaired Development of Innate Immune Tolerance That Can Be Counteracted by Ganglioside GM1. J. Neuroinflamm. 2023, 20, 276. [Google Scholar] [CrossRef] [PubMed]
- Valadão, P.A.C.; Santos, K.B.S.; Ferreira e Vieira, T.H.; Macedo e Cordeiro, T.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s Disease: A Few New Twists on an Old Tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef] [PubMed]
- Donley, D.W.; Nelson, R.; Gigley, J.P.; Fox, J.H. Mutant Huntingtin Protein Alters the Response of Microglial Cells to Inflammatory Stimuli. bioRxiv 2019, 550913. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Gubert, C.; Kong, G.; Costello, C.; Adams, C.D.; Masson, B.A.; Qin, W.; Choo, J.; Narayana, V.K.; Rogers, G.; Renoir, T.; et al. Dietary Fibre Confers Therapeutic Effects in a Preclinical Model of Huntington’s Disease. Brain Behav. Immun. 2024, 116, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Biswas, S.S.; Mishra, J.; Navik, U.; Kandimalla, R.; Reddy, P.H.; Bhatti, G.K.; Bhatti, J.S. Gut Microbiota Dysbiosis and Huntington’s Disease: Exploring the Gut-Brain Axis and Novel Microbiota-Based Interventions. Life Sci. 2023, 328, 121882. [Google Scholar] [CrossRef] [PubMed]
- Wronka, D.; Karlik, A.; Misiorek, J.O.; Przybyl, L. What the Gut Tells the Brain—Is There a Link between Microbiota and Huntington’s Disease? Int. J. Mol. Sci. 2023, 24, 4477. [Google Scholar] [CrossRef] [PubMed]
- Wasser, C.I.; Mercieca, E.C.; Kong, G.; Hannan, A.J.; McKeown, S.J.; Glikmann-Johnston, Y.; Stout, J.C. Gut Dysbiosis in Huntington’s Disease: Associations among Gut Microbiota, Cognitive Performance and Clinical Outcomes. Brain Commun. 2020, 2, fcaa110. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, B. Epigenetics: The Science of Change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, C.-C.; Liu, X.-Y.; Deng, X.-N.; Tian, Q.; Du, Y.-J. Epigenetic Modulation of Microglia Function and Phenotypes in Neurodegenerative Diseases. Neural Plast. 2021, 2021, 9912686. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Trotta, T.; Antonietta Panaro, M.; Cianciulli, A.; Mori, G.; Di Benedetto, A.; Porro, C. Microglia-Derived Extracellular Vesicles in Alzheimer’s Disease: A Double-Edged Sword. Biochem. Pharmacol. 2018, 148, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sci. 2022, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.R.; O’Connell, K.M.S.; Kaczorowski, C.C. Gene-by-Environment Interactions in Alzheimer’s Disease and Parkinson’s Disease. Neurosci. Biobehav. Rev. 2019, 103, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ciernia, A.V.; Link, V.M.; Careaga, M.; LaSalle, J.M.; Ashwood, P. Genetic Variants Drive Altered Epigenetic Regulation of Endotoxin Response in BTBR Macrophages. Brain Behav. Immun. 2020, 89, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Liu, Y. DNA Methylation in Human Diseases. Genes. Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Shahal, T.; Gabrieli, T.; Gilat, N.; Torchinsky, D.; Michaeli, Y.; Vogel, V.; Ebenstein, Y. Global Modulation in DNA Epigenetics during Pro-Inflammatory Macrophage Activation. Epigenetics 2019, 14, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.M.; Lawson, M.A.; Johnson, R.W. Aging and Peripheral Lipopolysaccharide Can Modulate Epigenetic Regulators and Decrease IL-1β Promoter DNA Methylation in Microglia. Neurobiol. Aging 2016, 47, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schüffer, W.; et al. Role of the Toll-Like Receptor 4 in Neuroinflammation in Alzheimer’s Disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, C.P.; McEwen, L.M.; Kohli, V.; Wolfson, C.; Bowdish, D.M.E.; Raina, P.; Kobor, M.S.; Balion, C. The Relation between DNA Methylation Patterns and Serum Cytokine Levels in Community-Dwelling Adults: A Preliminary Study. BMC Genet. 2017, 18, 57. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the Development of MiRNA Bioinformatics Tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lan, S.; Shi, X.-J.; Fan, F.-C.; Liu, Q.-S.; Cong, L.; Cheng, Y. Systematic Review and Meta-Analysis on MicroRNAs in Amyotrophic Lateral Sclerosis. Brain Res. Bull. 2023, 194, 82–89. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Li, Z.; Yang, M.; Yu, W.; Luo, R.; Zhou, J.; He, J.; Chen, Q.; Song, Z.; Cheng, S. Non-Coding RNA in Microglia Activation and Neuroinflammation in Alzheimer’s Disease. J. Inflamm. Res. 2023, 16, 4165–4211. [Google Scholar] [CrossRef] [PubMed]
- Quinn, E.M.; Wang, J.; Redmond, H.P. The Emerging Role of MicroRNA in Regulation of Endotoxin Tolerance. J. Leukoc. Biol. 2012, 91, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Nahid, M.A.; Satoh, M.; Chan, E.K.L. Mechanistic Role of MicroRNA-146a in Endotoxin-Induced Differential Cross-Regulation of TLR Signaling. J. Immunol. 2011, 186, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-ΚB-Dependent Induction of MicroRNA MiR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sun, J.; Shao, X.; Feng, J.; Yan, J.; Qin, Y. Inhibition of MicroRNA-155 Modulates Endotoxin Tolerance by Upregulating Suppressor of Cytokine Signaling 1 in Microglia. Exp. Ther. Med. 2018, 15, 4709–4716. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Gomes, C.; Vaz, A.R.; Brites, D. Exploring New Inflammatory Biomarkers and Pathways during LPS-Induced M1 Polarization. Mediat. Inflamm. 2016, 2016, 6986175. [Google Scholar] [CrossRef]
- Venkatesh, S.; Workman, J.L. Histone Exchange, Chromatin Structure and the Regulation of Transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef]
- Wu, C.; Li, A.; Hu, J.; Kang, J. Histone Deacetylase 2 Is Essential for LPS-Induced Inflammatory Responses in Macrophages. Immunol. Cell Biol. 2019, 97, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Santana, D.A.; Smith, M.d.A.C.; Chen, E.S. Histone Modifications in Alzheimer’s Disease. Genes 2023, 14, 347. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Saadat, A. Neurodegeneration and Epigenetics: A Review. Neurología 2023, 38, e62–e68. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Roberts, K.L.; Niyongere, S.A.; Cong, Y.; Elson, C.O.; Benveniste, E.N. Molecular Mechanism of Lipopolysaccharide-Induced SOCS-3 Gene Expression in Macrophages and Microglia. J. Immunol. 2007, 179, 5966–5976. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Yuan, J.; Li, N.; Pei, S.; Xu, J.; Luo, X.; Mao, C.; Liu, J.; Yu, T.; et al. Macrophage/Microglial Ezh2 Facilitates Autoimmune Inflammation through Inhibition of Socs3. J. Exp. Med. 2018, 215, 1365–1382. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chmielarz, M.; Sobieszczańska, B.; Środa-Pomianek, K. Metabolic Endotoxemia: From the Gut to Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 7006. https://doi.org/10.3390/ijms25137006
Chmielarz M, Sobieszczańska B, Środa-Pomianek K. Metabolic Endotoxemia: From the Gut to Neurodegeneration. International Journal of Molecular Sciences. 2024; 25(13):7006. https://doi.org/10.3390/ijms25137006
Chicago/Turabian StyleChmielarz, Mateusz, Beata Sobieszczańska, and Kamila Środa-Pomianek. 2024. "Metabolic Endotoxemia: From the Gut to Neurodegeneration" International Journal of Molecular Sciences 25, no. 13: 7006. https://doi.org/10.3390/ijms25137006