
Chronic Epinephrine-Induced Endoplasmic Reticulum and Oxidative Stress Impairs Pancreatic β-Cells Function and Fate

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Glucose Stimulated Insulin Responsiveness
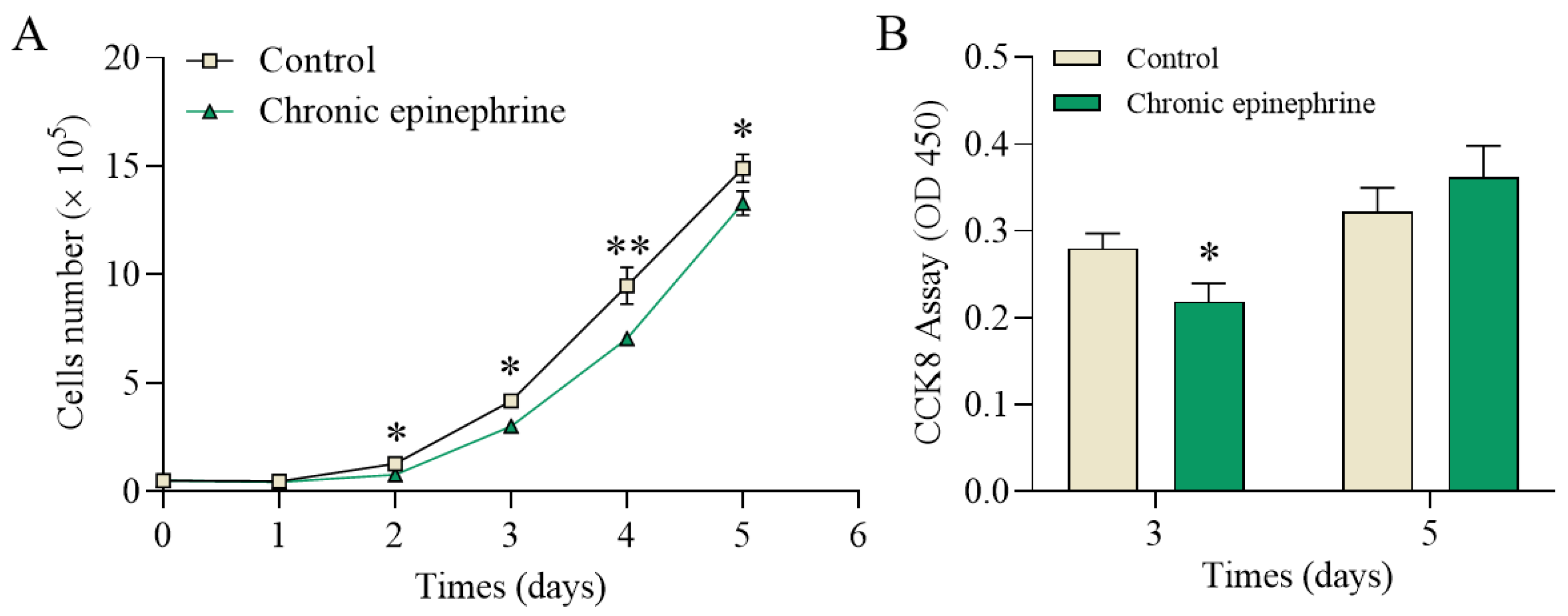
2.2. Cell Proliferation and Viability
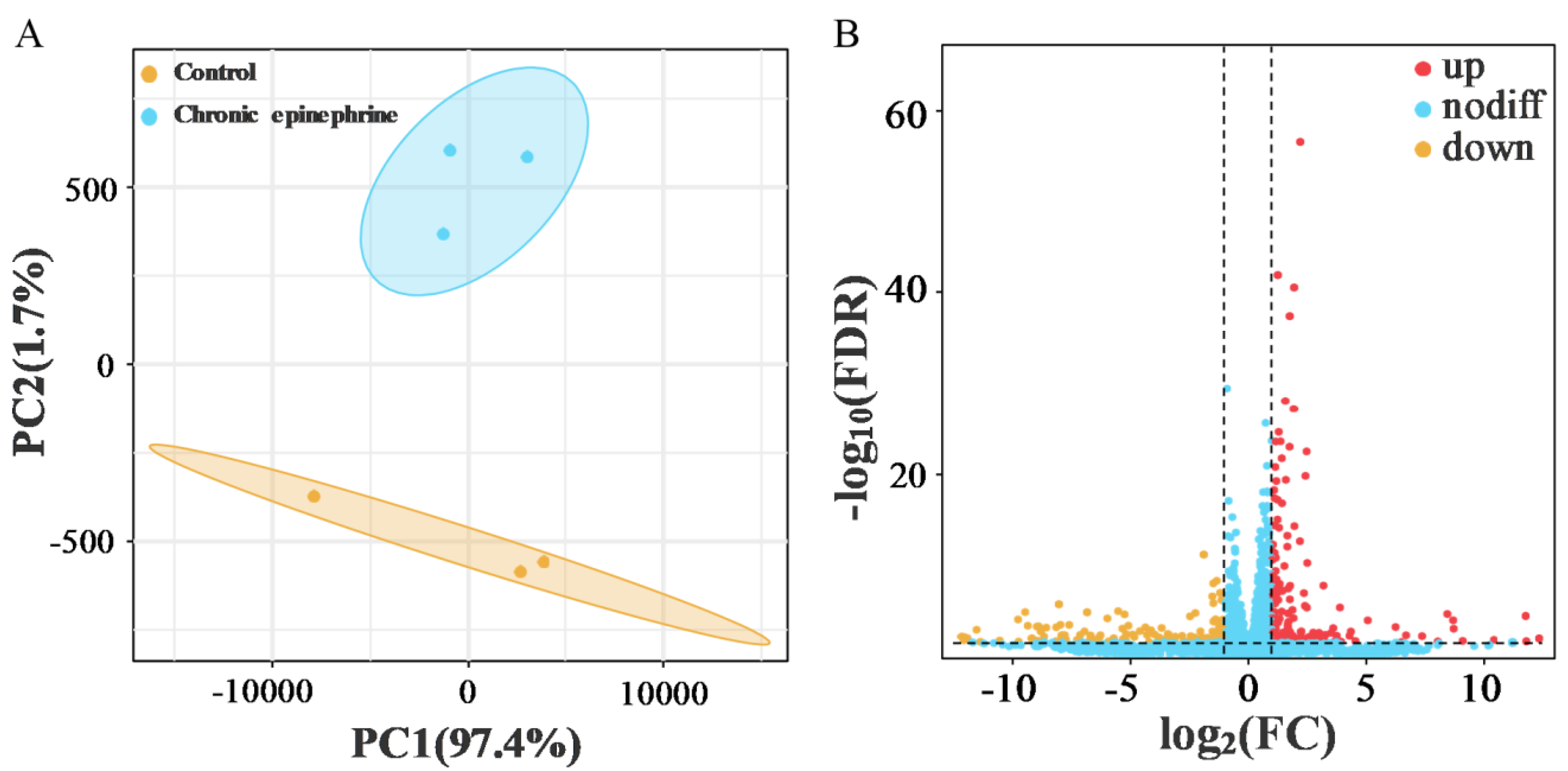
2.3. Differential Gene Expression of RNA-Seq
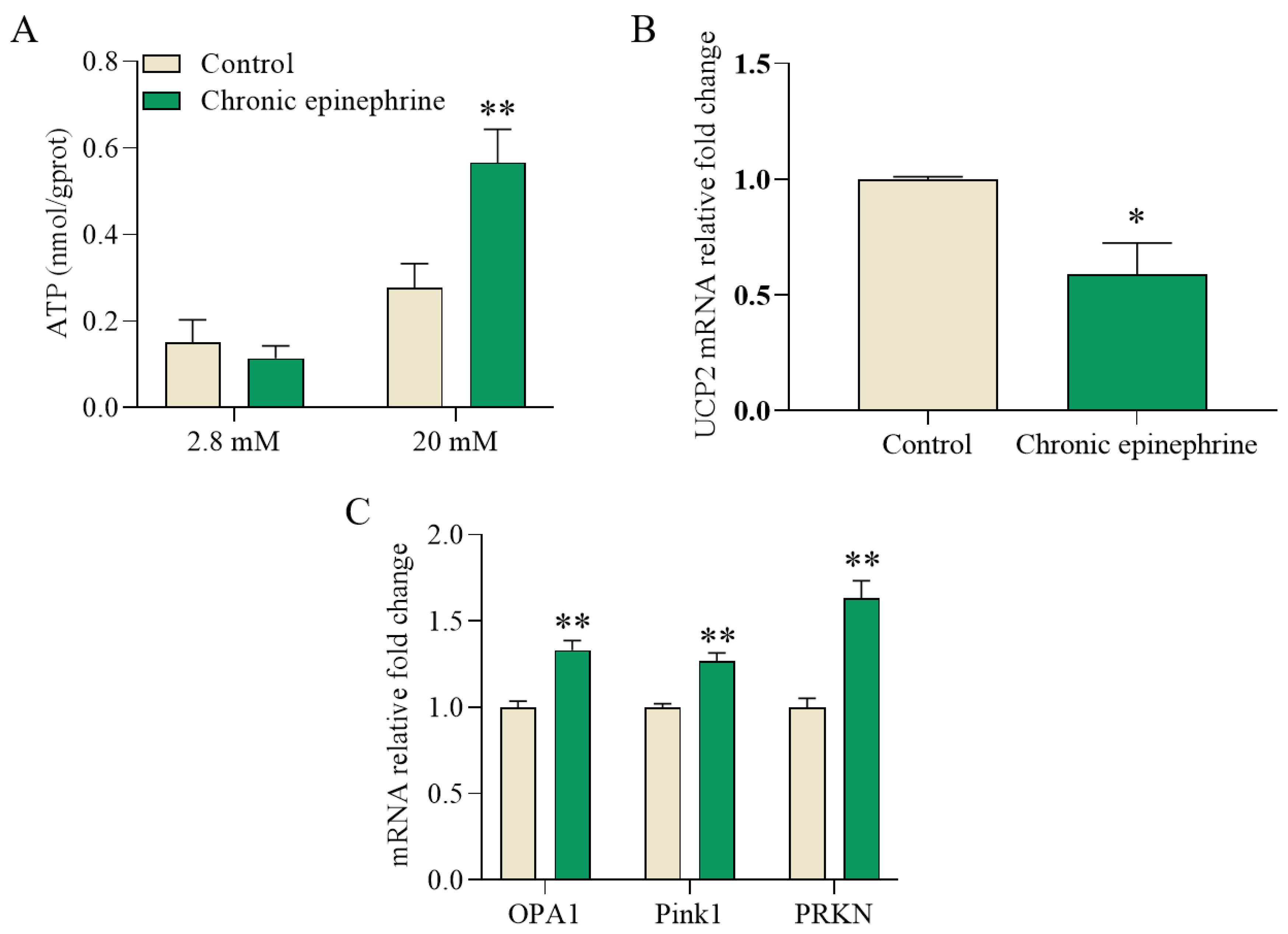
2.4. Impaired Mitochondria Signaling
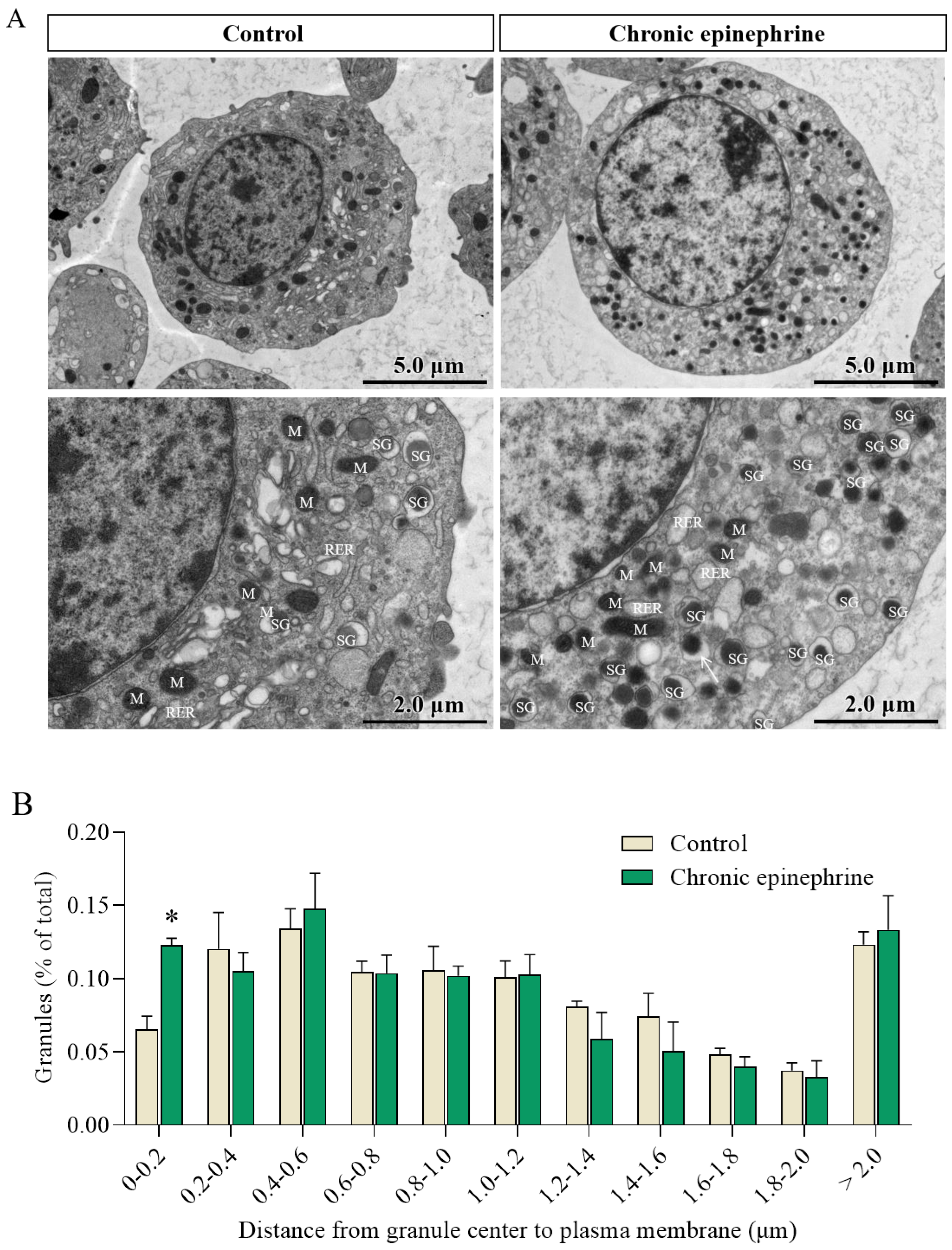
2.5. Ultrastructural Observation of MIN6
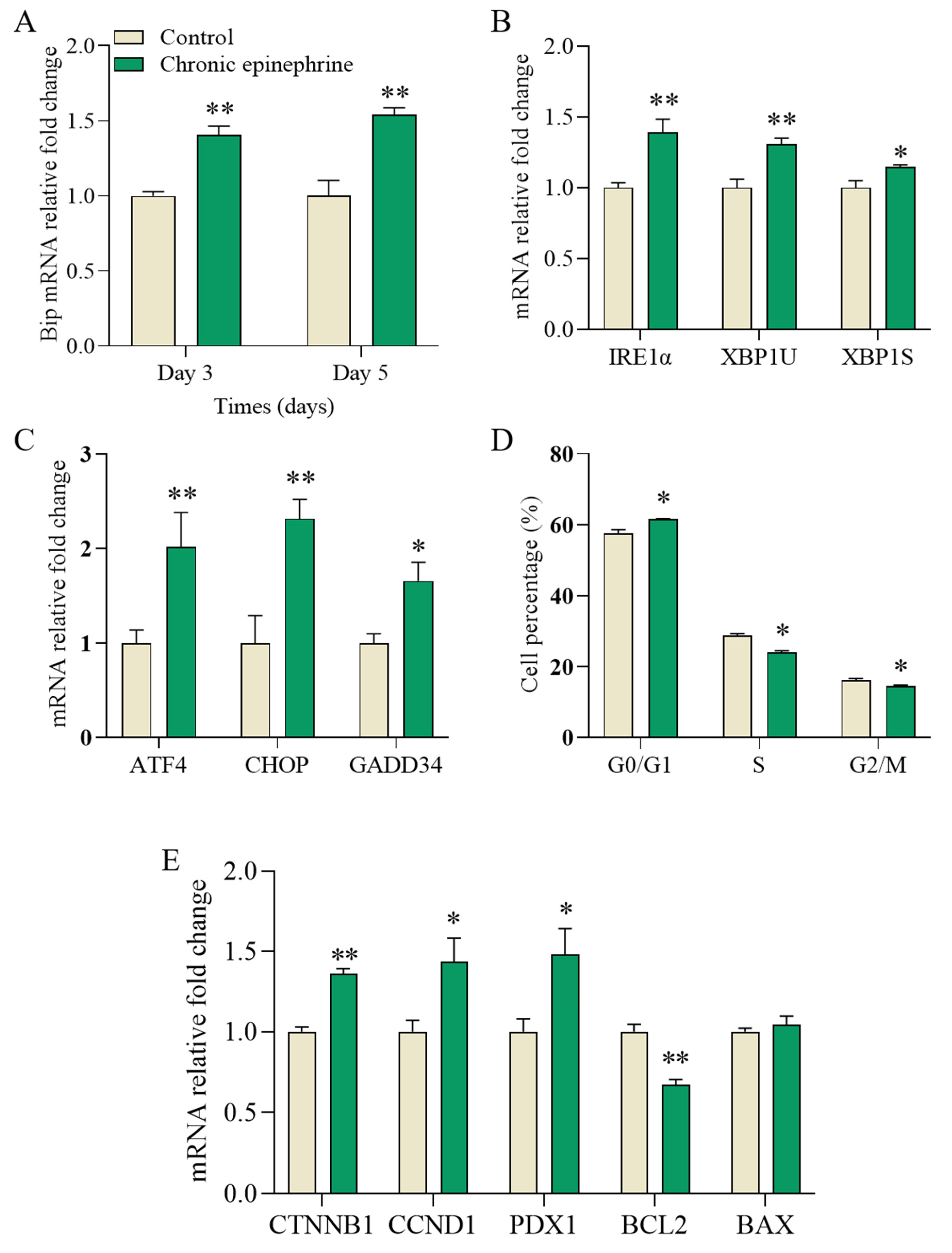
2.6. Cell Fate Signaling Pathway
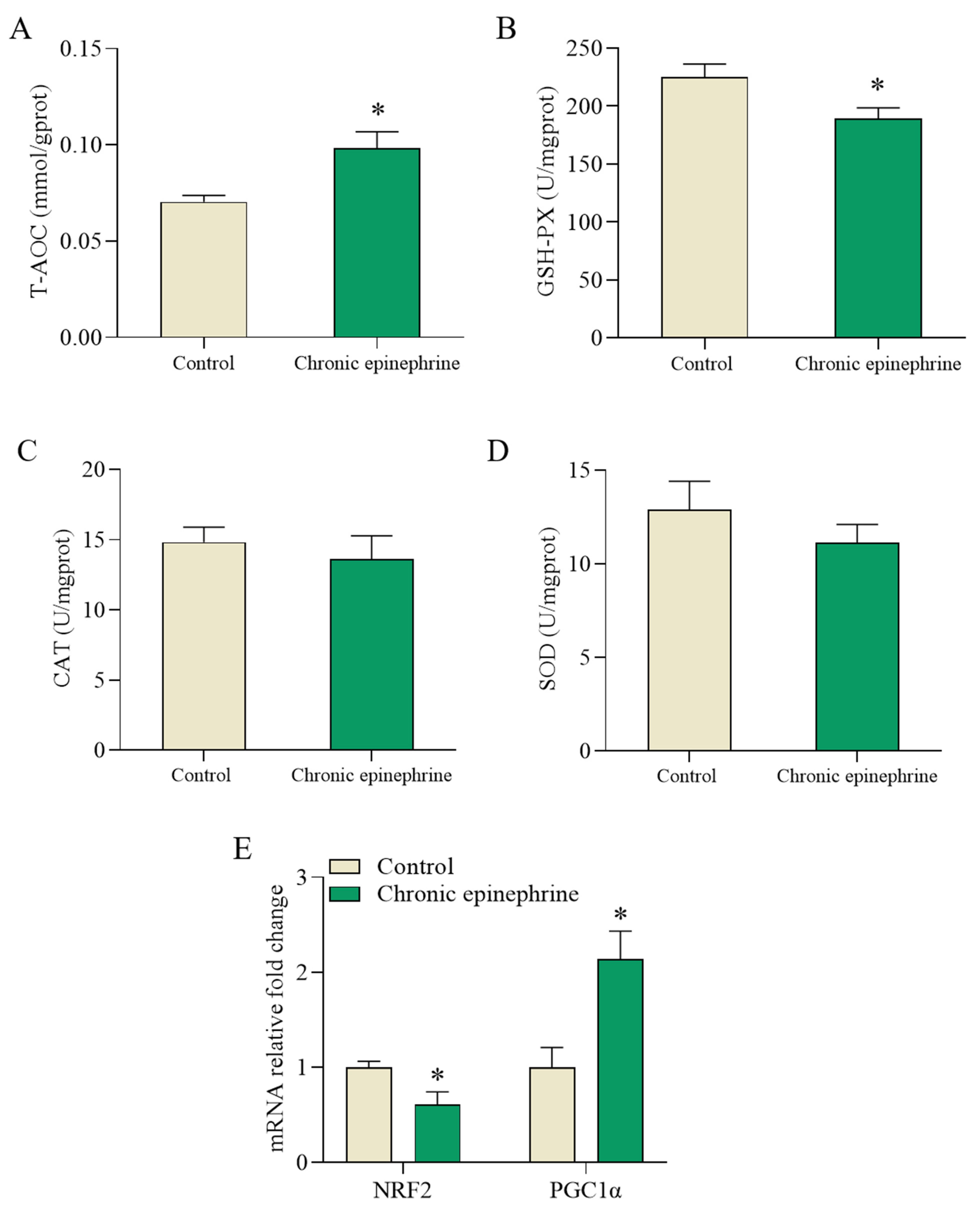
2.7. Impaired ROS Regulation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Number, Cell Viability, and Cycle Assays
4.3. Insulin Secretion Assay
4.4. RNA Sequencing and Analysis
4.5. Biochemical Parameters Assays
4.6. Transmission Electron Microscopy (TEM)
4.7. Quantitative Real Time PCR
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lyssenko, V.; Almgren, P.; Anevski, D.; Perfekt, R.; Lahti, K.; Nissén, M.; Isomaa, B.; Forsen, B.; Homström, N.; Saloranta, C.; et al. Predictors of and Longitudinal Changes in Insulin Sensitivity and Secretion Preceding Onset of Type 2 Diabetes. Diabetes 2005, 54, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Krentz, N.A.J.; Gloyn, A.L. Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat. Rev. Endocrinol. 2020, 16, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, E.; Lim, S.; Lamptey, R.; Webb, D.R.; Davies, M.J. Type 2 diabetes. Lancet 2022, 400, 1803–1820. [Google Scholar] [CrossRef] [PubMed]
- Cannataro, R.; Cione, E.; Cerullo, G.; Rondanelli, M.; Micheletti, P.; Crisafulli, O.; Micheli, M.L.; D’Antona, G. Type 1 diabetes management in a competitive athlete: A five-year case report. Physiol. Rep. 2023, 11, e15740. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.R.; Qiu, X.; Pan, R.; Fu, H.; Zhang, Z.; Wang, Q.; Chen, H.; Wu, Q.Q.; Pan, X.; Zhou, Y.; et al. Dietary intervention preserves β cell function in mice through CTCF-mediated transcriptional reprogramming. J. Exp. Med. 2022, 219, e20211779. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Guan, B.J.; Alzahrani, M.R.; Gao, Z.; Gao, L.; Bracey, S.; Wu, J.; Mbow, C.A.; Jobava, R.; Haataja, L.; et al. Adaptation to chronic ER stress enforces pancreatic β-cell plasticity. Nat. Commun. 2022, 13, 4621. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Krizanova, O.; Babula, P.; Pacak, K. Stress, catecholaminergic system and cancer. Stress 2016, 19, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Lara-Cinisomo, S.; Grewen, K.M.; Girdler, S.S.; Wood, J.; Meltzer-Brody, S. Perinatal Depression, Adverse Life Events, and Hypothalamic-Adrenal-Pituitary Axis Response to Cold Pressor Stress in Latinas: An Exploratory Study. Womens Health Issues 2017, 27, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Huang, H.; Limesand, S.W.; Chen, X. Pancreatic Islets Exhibit Dysregulated Adaptation of Insulin Secretion after Chronic Epinephrine Exposure. Curr. Issues Mol. Biol. 2021, 43, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Green, A.S.; Macko, A.R.; Yates, D.T.; Kelly, A.C.; Limesand, S.W. Enhanced insulin secretion responsiveness and islet adrenergic desensitization after chronic norepinephrine suppression is discontinued in fetal sheep. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E58–E64. [Google Scholar] [CrossRef] [PubMed]
- Yates, D.T.; Macko, A.R.; Chen, X.; Green, A.S.; Kelly, A.C.; Anderson, M.J.; Fowden, A.L.; Limesand, S.W. Hypoxaemia-induced catecholamine secretion from adrenal chromaffin cells inhibits glucose-stimulated hyperinsulinaemia in fetal sheep. J. Physiol. 2012, 590, 5439–5447. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the β cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Hoermann, H.; van Faassen, M.; Roeper, M.; Hagenbeck, C.; Herebian, D.; Muller Kobold, A.C.; Dukart, J.; Kema, I.P.; Mayatepek, E.; Meissner, T.; et al. Association of Fetal Catecholamines with Neonatal Hypoglycemia. JAMA Pediatr. 2024, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.C.; Camacho, L.E.; Pendarvis, K.; Davenport, H.M.; Steffens, N.R.; Smith, K.E.; Weber, C.S.; Lynch, R.M.; Papas, K.K.; Limesand, S.W. Adrenergic receptor stimulation suppresses oxidative metabolism in isolated rat islets and Min6 cells. Mol. Cell Endocrinol. 2018, 473, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.M.; Gannon, M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J. Mol. Endocrinol. 2007, 38, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic β-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 4843. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.B.; Landa-Galván, H.V.; Alonso, L.C. Living Dangerously: Protective and Harmful ER Stress Responses in Pancreatic β-Cells. Diabetes 2021, 70, 2431–2443. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; De Franco, E.; Arvan, P.; Cnop, M. Pathological β-Cell Endoplasmic Reticulum Stress in Type 2 Diabetes: Current Evidence. Front. Endocrinol. 2021, 12, 650158. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.B.; O’Donnell, A.C.; Stamateris, R.E.; Ha, B.; McCloskey, K.M.; Reynolds, P.R.; Arvan, P.; Alonso, L.C. Insulin demand regulates β cell number via the unfolded protein response. J. Clin. Investig. 2015, 125, 3831–3846. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.C.; Bidwell, C.A.; Chen, X.; Macko, A.R.; Anderson, M.J.; Limesand, S.W. Chronic Adrenergic Signaling Causes Abnormal RNA Expression of Proliferative Genes in Fetal Sheep Islets. Endocrinology 2018, 159, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Dezaki, K.; Yoshida, M.; Yamada, H.; Miura, R.; Rita, R.S.; Ookawara, S.; Tabei, K.; Kawakami, M.; Hara, K.; et al. Endogenous α2A-Adrenoceptor-Operated Sympathoadrenergic Tones Attenuate Insulin Secretion via cAMP/TRPM2 Signaling. Diabetes 2017, 66, 699–709. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Huang, Z.; Binns, D.D.; McGlynn, K.; Cobb, M.H. α(2)-Adrenergic Disruption of β Cell BDNF-TrkB Receptor Tyrosine Kinase Signaling. Front. Cell Dev. Biol. 2020, 8, 576396. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; McDermott, A.M.; Gillison, S.L.; Sharp, G.W. Time course of action of pertussis toxin to block the inhibition of stimulated insulin release by norepinephrine. Endocrinology 1995, 136, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes. Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Gaisano, H.Y. Recent new insights into the role of SNARE and associated proteins in insulin granule exocytosis. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kang, F.; Xie, L.; Qin, T.; Miao, Y.; Kang, Y.; Takahashi, T.; Liang, T.; Xie, H.; Gaisano, H.Y. Plasma membrane flipping of Syntaxin-2 regulates its inhibitory action on insulin granule exocytosis. Nat. Commun. 2022, 13, 6512. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2011, 151, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H.; Ichijo, H. Survival and apoptosis signals in ER stress: The role of protein kinases. J. Chem. Neuroanat. 2004, 28, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Scheuner, D.; Han, J.; Song, B.; Ribick, M.; Wang, J.; Gildersleeve, R.D.; Pennathur, S.; Kaufman, R.J. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009, 10, 13–26. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Luo, N.; Cheng, W.; Zhou, Y.; Gu, B.; Zhao, Z.; Zhao, Y. Screening Candidate Genes Regulating Placental Development from Trophoblast Transcriptome at Early Pregnancy in Dazu Black Goats (Capra hircus). Animals 2021, 11, 2132. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Yao, B.; Li, R.; Limesand, S.W.; Zhao, Y.; Chen, X. Chronic Epinephrine-Induced Endoplasmic Reticulum and Oxidative Stress Impairs Pancreatic β-Cells Function and Fate. Int. J. Mol. Sci. 2024, 25, 7029. https://doi.org/10.3390/ijms25137029
Zhang R, Yao B, Li R, Limesand SW, Zhao Y, Chen X. Chronic Epinephrine-Induced Endoplasmic Reticulum and Oxidative Stress Impairs Pancreatic β-Cells Function and Fate. International Journal of Molecular Sciences. 2024; 25(13):7029. https://doi.org/10.3390/ijms25137029
Chicago/Turabian StyleZhang, Ran, Bingpeng Yao, Rui Li, Sean W. Limesand, Yongju Zhao, and Xiaochuan Chen. 2024. "Chronic Epinephrine-Induced Endoplasmic Reticulum and Oxidative Stress Impairs Pancreatic β-Cells Function and Fate" International Journal of Molecular Sciences 25, no. 13: 7029. https://doi.org/10.3390/ijms25137029
APA StyleZhang, R., Yao, B., Li, R., Limesand, S. W., Zhao, Y., & Chen, X. (2024). Chronic Epinephrine-Induced Endoplasmic Reticulum and Oxidative Stress Impairs Pancreatic β-Cells Function and Fate. International Journal of Molecular Sciences, 25(13), 7029. https://doi.org/10.3390/ijms25137029