
HtrA-Dependent E-Cadherin Shedding Impairs the Epithelial Barrier Function in Primary Gastric Epithelial Cells and Gastric Organoids

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
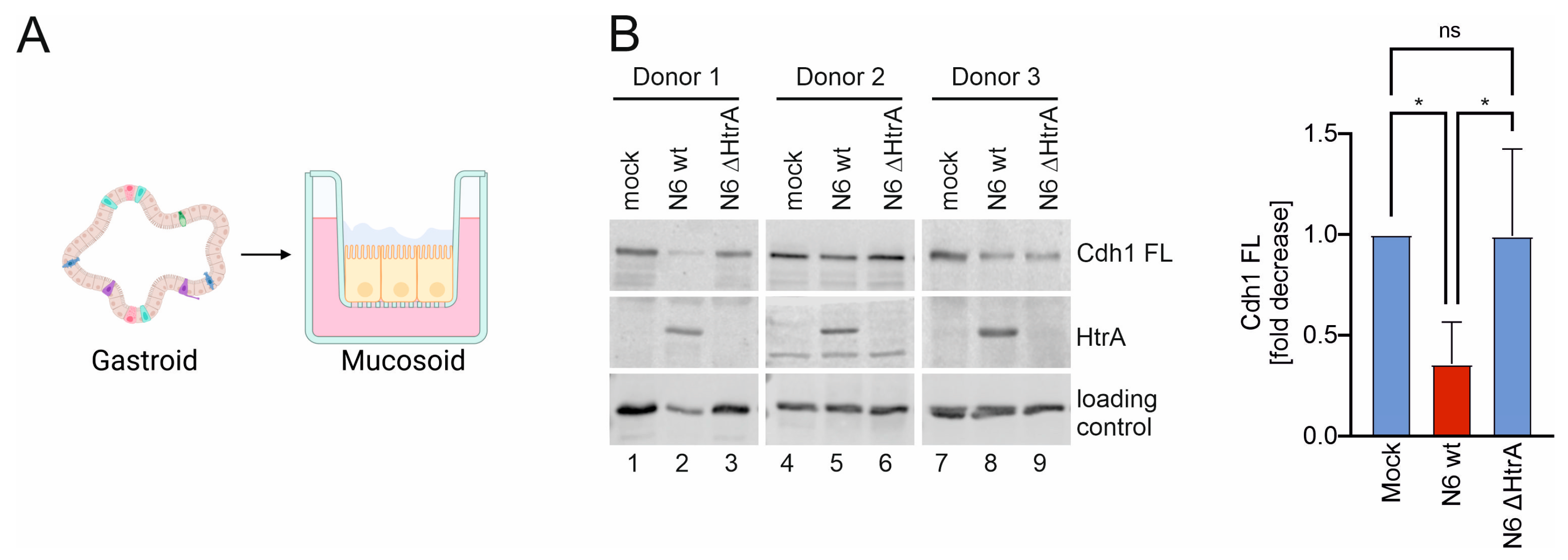
2.1. H. pylori HtrA Cleaves E-Cadherin on Primary Gastric Epithelial Cells
2.2. HtrA-Mediated Cdh1 Shedding Facilitates CagA Translocation into Polarized Mucosoid Cultures
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Gastric Organoids and Mucosoids
5.2. Bacteria and Infection Experiments
5.3. SDS-PAGE and Western Blot
5.4. Real-Time PCR
5.5. Immunofluorescence
5.6. TEER Measurements and Bacterial Transmigration
5.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Engelsberger, V.; Gerhard, M.; Mejías-Luque, R. Effects of Helicobacter pylori infection on intestinal microbiota, immunity and colorectal cancer risk. Front. Cell Infect. Microbiol. 2024, 14, 1339750. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Camargo, M.C.; El-Omar, E.; Liou, J.M.; Peek, R.; Schulz, C.; Smith, S.I.; Suerbaum, S. Helicobacter pylori infection. Nat. Rev. Dis. Primers 2023, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Ferino, L.; Sharafutdinov, I.; Backert, S. Gastric Epithelial Barrier Disruption, Inflammation and Oncogenic Signal Transduction by Helicobacter pylori. Curr. Top. Microbiol. Immunol. 2023, 444, 207–238. [Google Scholar] [PubMed]
- Thompson, C.J.; Vu, V.H.; Leckband, D.E.; Schwartz, D.K. Cadherin cis and trans interactions are mutually cooperative. Proc. Natl. Acad. Sci. USA 2021, 118, e2019845118. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.A.; Reynolds, A.B. Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Dev. Cell 2006, 10, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chu, K.M. E-cadherin and gastric cancer: Cause, consequence, and applications. Biomed. Res. Int. 2014, 2014, 637308. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, P.; Fernandes, M.S.; Figueiredo, J.; Caldeira, J.; Carvalho, J.; Pinheiro, H.; Leite, M.; Melo, S.; Oliveira, P.; Simões-Correia, J.; et al. E-cadherin dysfunction in gastric cancer—Cellular consequences, clinical applications and open questions. FEBS Lett. 2012, 586, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Hu, H.; Chen, B.; Xu, W.; Zhao, J.; Huang, C.; Xing, Y.; Lv, H.; Nie, C.; Wang, J.; et al. Overview on the Role of E-Cadherin in Gastric Cancer: Dysregulation and Clinical Implications. Front. Mol. Biosci. 2021, 8, 689139. [Google Scholar] [CrossRef]
- Costache, S.; de Havilland, R.; Diaz McLynn, S.; Sajin, M.; Baltan, A.; Wedden, S.; D’Arrigo, C. Implementing an On-Slide Molecular Classification of Gastric Cancer: A Tissue Microarray Study. Cancers 2023, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.M.; Reynolds, A.B. The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol. Cell Biol. 1999, 19, 3614–3623. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T.; Sasako, M. Focus on gastric cancer. Cancer Cell 2004, 5, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.N.; Davis, J.L. CDH1 and hereditary diffuse gastric cancer: A narrative review. Chin. Clin. Oncol. 2023, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Hoy, B.; Löwer, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schröder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Bernegger, S.; Vidmar, R.; Fonovic, M.; Posselt, G.; Turk, B.; Wessler, S. Identification of Desmoglein-2 as a novel target of Helicobacter pylori HtrA in epithelial cells. Cell Commun. Signal 2021, 19, 108. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Wessler, S.; Necchi, V.; Rohde, M.; Harrer, A.; Rau, T.T.; Asche, C.I.; Boehm, M.; Loessner, H.; Figueiredo, C.; et al. Helicobacter pylori Employs a Unique Basolateral Type IV Secretion Mechanism for CagA Delivery. Cell Host Microbe 2017, 22, 552–560.e5. [Google Scholar] [CrossRef] [PubMed]
- Poppe, M.; Feller, S.M.; Römer, G.; Wessler, S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 2007, 26, 3462–3472. [Google Scholar] [CrossRef] [PubMed]
- Tammer, I.; Brandt, S.; Hartig, R.; König, W.; Backert, S. Activation of Abl by Helicobacter pylori: A novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 2007, 132, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Boccellato, F.; Woelffling, S.; Imai-Matsushima, A.; Sanchez, G.; Goosmann, C.; Schmid, M.; Berger, H.; Morey, P.; Denecke, C.; Ordemann, J.; et al. Polarised epithelial monolayers of the gastric mucosa reveal insights into mucosal homeostasis and defence against infection. Gut 2019, 68, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Zawilak-Pawlik, A.; Zarzecka, U.; Żyła-Uklejewicz, D.; Lach, J.; Strapagiel, D.; Tegtmeyer, N.; Böhm, M.; Backert, S.; Skorko-Glonek, J. Establishment of serine protease htrA mutants in Helicobacter pylori is associated with secA mutations. Sci. Rep. 2019, 9, 11794. [Google Scholar] [CrossRef]
- Salama, N.R.; Shepherd, B.; Falkow, S. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J. Bacteriol. 2004, 186, 7926–7935. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Moodley, Y.; Yamaoka, Y.; Pernitzsch, S.R.; Schmidt, V.; Traverso, F.R.; Schmidt, T.P.; Rad, R.; Yeoh, K.G.; Bow, H.; et al. Characterisation of worldwide Helicobacter pylori strains reveals genetic conservation and essentiality of serine protease HtrA. Mol. Microbiol. 2016, 99, 925–944. [Google Scholar] [CrossRef] [PubMed]
- Zarzecka, U.; Modrak-Wójcik, A.; Figaj, D.; Apanowicz, M.; Lesner, A.; Bzowska, A.; Lipinska, B.; Zawilak-Pawlik, A.; Backert, S.; Skorko-Glonek, J. Properties of the HtrA Protease From Bacterium Helicobacter pylori Whose Activity Is Indispensable for Growth Under Stress Conditions. Front. Microbiol. 2019, 10, 961. [Google Scholar] [CrossRef] [PubMed]
- Sharafutdinov, I.; Tegtmeyer, N.; Linz, B.; Rohde, M.; Vieth, M.; Tay, A.C.; Lamichhane, B.; Tuan, V.P.; Fauzia, K.A.; Sticht, H.; et al. A single-nucleotide polymorphism in Helicobacter pylori promotes gastric cancer development. Cell Host Microbe 2023, 31, 1345–1358.e6. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Kuo, H.Y.; Chang, W.L.; Yang, H.B.; Lu, C.C.; Cheng, H.C.; Wu, M.S.; Sheu, B.S. H. pylori isolates with amino acid sequence polymorphisms as presence of both HtrA-L171 & CagL-Y58/E59 increase the risk of gastric cancer. J. Biomed. Sci. 2019, 26, 4. [Google Scholar]
- Zarzecka, U.; Tegtmeyer, N.; Sticht, H.; Backert, S. Trimer stability of Helicobacter pylori HtrA is regulated by a natural mutation in the protease domain. Med. Microbiol. Immunol. 2023, 212, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, O.; Naumann, M. Matrix Metalloproteinases in Helicobacter pylori-Associated Gastritis and Gastric Cancer. Int. J. Mol. Sci. 2022, 23, 1883. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.P.; Kuang, J.Y.; Yang, Q.K.; Bian, X.W.; Yu, S.C. Beyond a tumor suppressor: Soluble E-cadherin promotes the progression of cancer. Int. J. Cancer 2016, 138, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- Ivaldo, C.; Passalacqua, M.; Furfaro, A.L.; d’Abramo, C.; Ruiz, S.; Chatterjee, P.K.; Metz, C.N.; Nitti, M.; Marambaud, P. Oxidative stress-induced MMP- and γ-secretase-dependent VE-cadherin processing is modulated by the proteasome and BMP9/10. Sci. Rep. 2023, 13, 597. [Google Scholar] [CrossRef]
- Bernegger, S.; Hutterer, E.; Zarzecka, U.; Schmidt, T.P.; Huemer, M.; Widlroither, I.; Posselt, G.; Skorko-Glonek, J.; Wessler, S. E-Cadherin Orthologues as Substrates for the Serine Protease High Temperature Requirement A (HtrA). Biomolecules 2022, 12, 356. [Google Scholar] [CrossRef]
- Necchi, V.; Ricci, V.; Sommi, P.; Solcia, E. CagA Effector Protein in Helicobacter pylori-Infected Human Gastric Epithelium in Vivo: From Bacterial Core and Adhesion/Injection Clusters to Host Cell Proteasome-Rich Cytosol. Toxins 2019, 11, 618. [Google Scholar] [CrossRef] [PubMed]
- Niessen, C.M. Tight junctions/adherens junctions: Basic structure and function. J. Investig. Dermatol. 2007, 127, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Vasioukhin, V.; Bowers, E.; Bauer, C.; Degenstein, L.; Fuchs, E. Desmoplakin is essential in epidermal sheet formation. Nat. Cell Biol. 2001, 3, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Kwok, T.; Zabler, D.; Urman, S.; Rohde, M.; Hartig, R.; Wessler, S.; Misselwitz, R.; Berger, J.; Sewald, N.; König, W.; et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007, 449, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Javaheri, A.; Kruse, T.; Moonens, K.; Mejías-Luque, R.; Debraekeleer, A.; Asche, C.I.; Tegtmeyer, N.; Kalali, B.; Bach, N.C.; Sieber, S.A.; et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2016, 2, 16189. [Google Scholar] [CrossRef] [PubMed]
- Königer, V.; Holsten, L.; Harrison, U.; Busch, B.; Loell, E.; Zhao, Q.; Bonsor, D.A.; Roth, A.; Kengmo-Tchoupa, A.; Smith, S.I.; et al. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat. Microbiol. 2016, 2, 16188. [Google Scholar] [CrossRef]
- Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults—The Evidence Report; National Institutes of Health: Bethesda, MD, USA, 1998; Volume 6, (Suppl. 2), pp. 51s–209s.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Date 1 | Gender | Age 2 | BMI 3 | Obese Classification 4 | Comorbidity AH/DM-II/PAS 5 | Medicaments 6 | HP Eradication 7 | Histological Diagnosis 8 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2021/04 | male | 37.3 | 41.5 | III | no/no/yes | No | No | Unremarkable |
| 2 | 2021/04 | female | 21.2 | 58.6 | III | yes/yes/yes | Yes (Metformin, Trajenta) | No | Unremarkable |
| 3 | 2021/06 | female | 20.8 | 46.6 | III | no/no/yes | No | No | Type C gastritis |
| 4 | 2021/09 | male | 37.0 | 45.4 | III | no/no/no | Yes (Pantoloc) | Yes (5) | Type C gastritis |
| 5 | 2022/07 | female | 33.2 | 38.4 | II | no/no/no | No | No | Type C gastritis |
| 6 | 2023/10 | male | 44.2 | 50.5 | III | yes/yes/no | Yes (Amlodipin, Pantoloc, Ramipril) | No | Unremarkable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canadas-Ortega, M.; Mühlbacher, I.; Posselt, G.; Diechler, S.; Ferner, C.D.; Boccellato, F.; Koch, O.O.; Neureiter, D.; Weitzendorfer, M.; Emmanuel, K.; et al. HtrA-Dependent E-Cadherin Shedding Impairs the Epithelial Barrier Function in Primary Gastric Epithelial Cells and Gastric Organoids. Int. J. Mol. Sci. 2024, 25, 7083. https://doi.org/10.3390/ijms25137083
Canadas-Ortega M, Mühlbacher I, Posselt G, Diechler S, Ferner CD, Boccellato F, Koch OO, Neureiter D, Weitzendorfer M, Emmanuel K, et al. HtrA-Dependent E-Cadherin Shedding Impairs the Epithelial Barrier Function in Primary Gastric Epithelial Cells and Gastric Organoids. International Journal of Molecular Sciences. 2024; 25(13):7083. https://doi.org/10.3390/ijms25137083
Chicago/Turabian StyleCanadas-Ortega, Marina, Iris Mühlbacher, Gernot Posselt, Sebastian Diechler, Christian Daniel Ferner, Francesco Boccellato, Oliver Owen Koch, Daniel Neureiter, Michael Weitzendorfer, Klaus Emmanuel, and et al. 2024. "HtrA-Dependent E-Cadherin Shedding Impairs the Epithelial Barrier Function in Primary Gastric Epithelial Cells and Gastric Organoids" International Journal of Molecular Sciences 25, no. 13: 7083. https://doi.org/10.3390/ijms25137083