
Resistance Exercise Training as a New Trend in Alzheimer’s Disease Research: From Molecular Mechanisms to Prevention

Abstract
1. Introduction
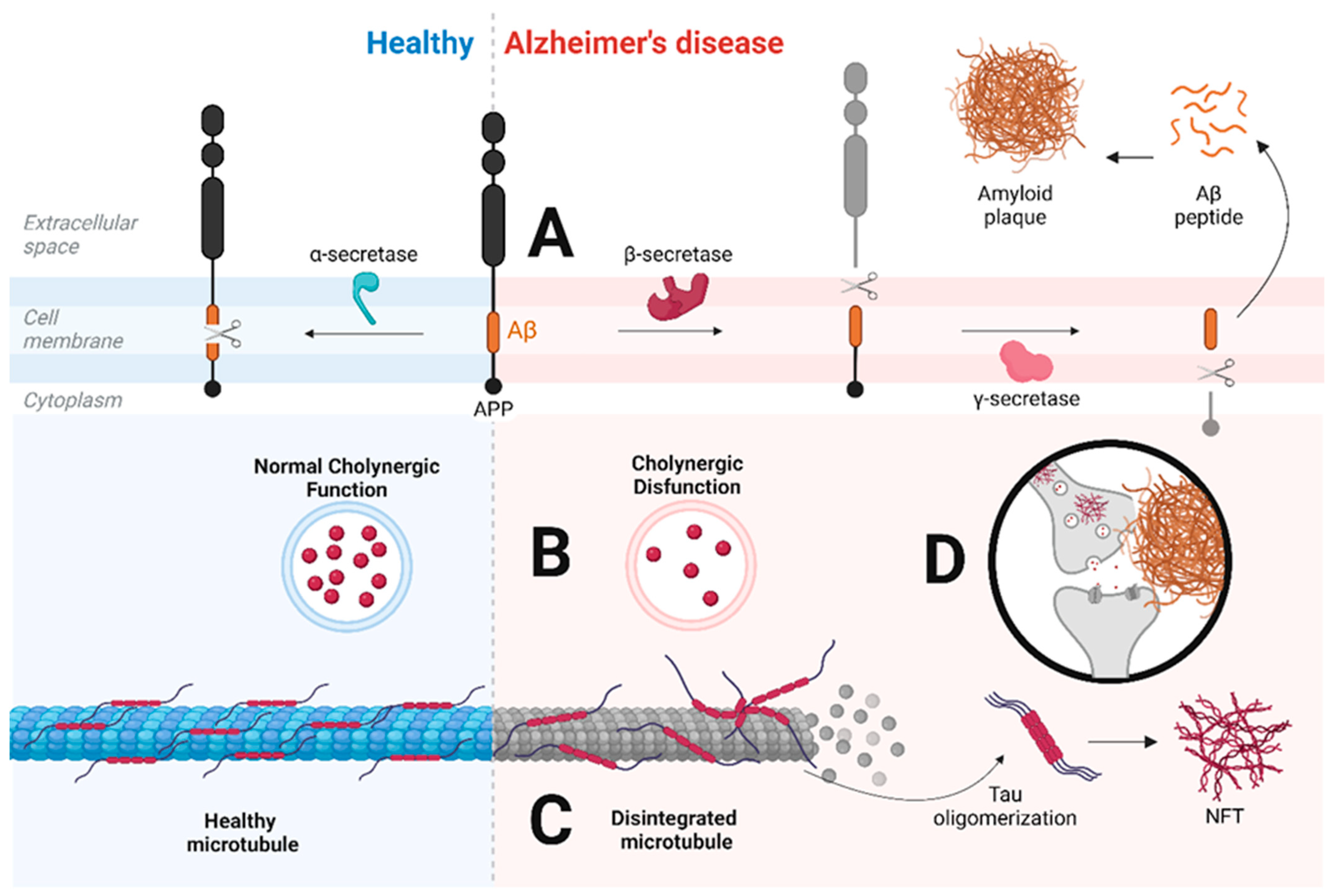
2. Alzheimer’s Disease
2.1. Main Hypotheses for Alzheimer’s Disease
2.2. Other Hypotheses for Alzheimer’s Disease
2.2.1. Vascular Hypothesis
2.2.2. Oxidative Hypothesis
2.2.3. Lipid Hypothesis
2.2.4. Neuroinflammation Hypothesis
3. Resistance Exercise Training
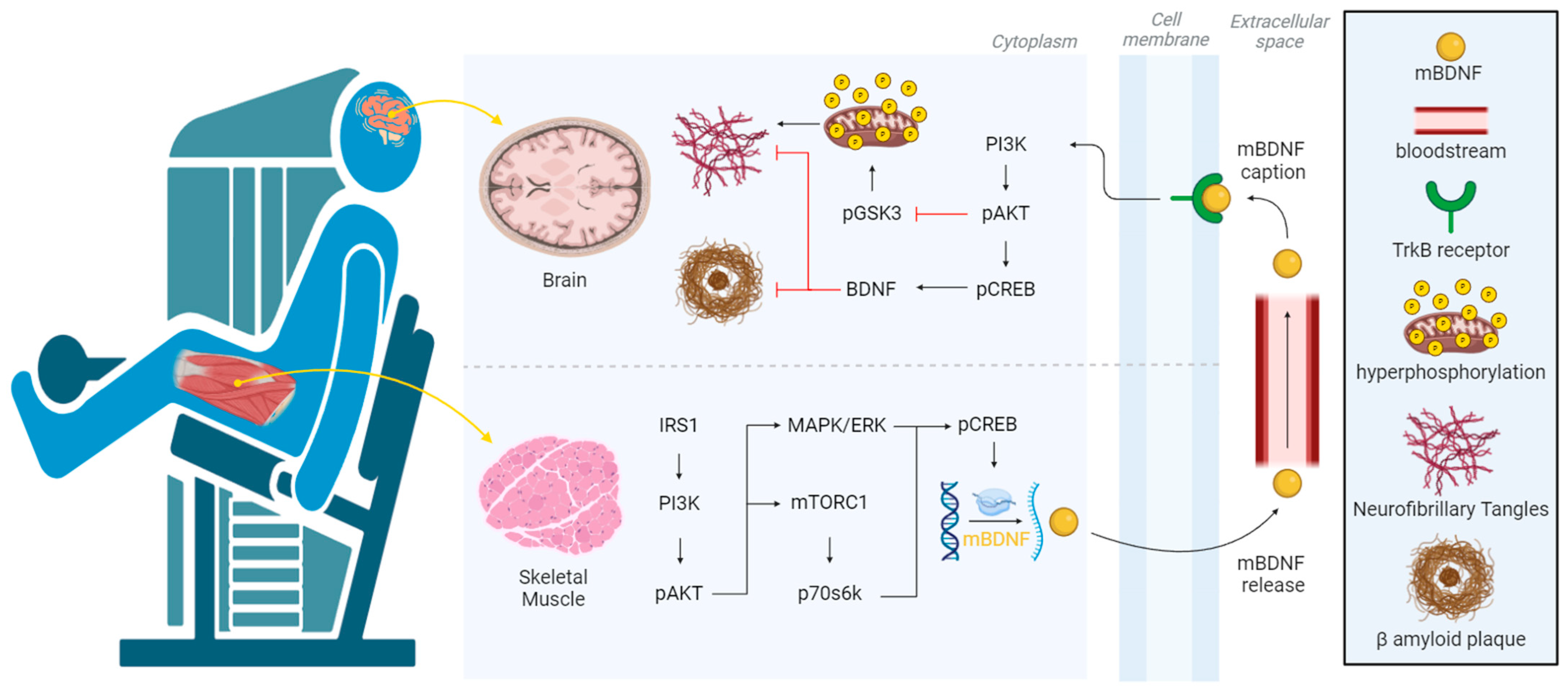
3.1. Molecular Mechanisms
3.2. Considerations
4. Resistance Exercise Training as a Preventive Strategy for Alzheimer’s Disease
4.1. Alzheimer’s Disease in Skeletal Muscle and Brain
4.2. Resistance Exercise Training and Main Hypotheses for Alzheimer’s Disease
4.3. Resistance Exercise Training and Other Hypotheses for Alzheimer’s Disease
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.; Arab-Tehrany, E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int. J. Mol. Sci. 2022, 23, 13954. [Google Scholar] [CrossRef] [PubMed]
- McDade, E.; Llibre-Guerra, J.J.; Holtzman, D.M.; Morris, J.C.; Bateman, R.J. The informed road map to prevention of Alzheimer Disease: A call to arms. Mol. Neurodegener. 2021, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- McKeown, A.; Turner, A.; Angehrn, Z.; Gove, D.; Ly, A.; Nordon, C.; Nelson, M.; Tochel, C.; Mittelstadt, B.; Keenan, A.; et al. Health Outcome Prioritization in Alzheimer’s Disease: Understanding the Ethical Landscape. J. Alzheimer’s Dis. 2020, 77, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.W.; Sano, M. Economic considerations in the management of Alzheimer’s disease. Clin. Interv. Aging 2006, 1, 143–154. [Google Scholar] [CrossRef]
- Kaur, S.; DasGupta, G.; Singh, S. Altered Neurochemistry in Alzheimer’s Disease: Targeting Neurotransmitter Receptor Mechanisms and Therapeutic Strategy. Neurophysiology 2019, 51, 293–309. [Google Scholar] [CrossRef]
- Vasilopoulos, F.; Jeffrey, H.; Wu, Y.; Dumontheil, I. Multi-level meta-analysis of whether fostering creativity during physical activity interventions increases their impact on cognitive and academic outcomes during childhood. Sci. Rep. 2023, 13, 8383. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Shenoy, S. Role of Education and Physical Activity in Executive Function Performance of Adult Population. Curr. Aging Sci. 2023, 16, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Runde, H.A.; Taraldsen, K.; Follestad, T.; Saltvedt, I.; Johnsen, L.G. The impact of cognitive function on physical activity, physical function and quality of life in older adults following a hip fracture. Age Ageing 2023, 52, afad061. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zang, M.; Wang, B.; Guo, W. Does the combination of exercise and cognitive training improve working memory in older adults? A systematic review and meta-analysis. PeerJ 2023, 11, e15108. [Google Scholar] [CrossRef] [PubMed]
- De Sá, C.A.; Saretto, C.B.; Cardoso, A.M.; Remor, A.; Breda, C.O.; da Silva Corralo, V. Effects of a physical exercise or motor activity protocol on cognitive function, lipid profile, and BDNF levels in older adults with mild cognitive impairment. Mol. Cell. Biochem. 2023, 479, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Rosano, C.; Guralnik, J.; Pahor, M.; Glynn, N.W.; Newman, A.B.; Ibrahim, T.S.; Erickson, K.; Cohen, R.; Shaaban, S.E.; MacCloud, R.L.; et al. Hippocampal Response to a 24-Month Physical Activity Intervention in Sedentary Older Adults. Am. J. Geriatr. Psychiatry 2017, 25, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, B.J.; Hsu, F.; Brinkley, T.J.; Church, T.; Goodpaster, B.H.; Kritchevsky, S.B.; Pahor, M. Exercise Training and Plasma C-Reactive Protein and Interleukin-6 in Elderly People. J. Am. Geriatr. Soc. 2008, 56, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, E.J.R.; Ibrahim, H.J.; Chitolina Schetinger, M.R.; de Andrade, C.M.; Cardoso, A.M. Modulation of Inflammatory Mediators and Microglial Activation Through Physical Exercise in Alzheimer’s and Parkinson’s Diseases. Neurochem. Res. 2022, 47, 3221–3240. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chu, J.M.T.; Yan, T.; Zhang, Y.; Chen, Y.; Chang, R.C.C.; Wong, G.T.C. Short-term resistance exercise inhibits neuroinflammation and attenuates neuropathological changes in 3xTg Alzheimer’s disease mice. J. Neuroinflam. 2020, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Özbeyli, D.; Sarı, G.; Özkan, N.; Karademir, B.; Yüksel, M.; Çilingir Kaya, Ö.T.; Kasımay Çakır, Ö. Protective effects of different exercise modalities in an Alzheimer’s disease-like model. Behav. Brain Res. 2017, 328, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.V.; Hashiguchi, D.; Campos, H.C.; Figueiredo, E.V.; Otaviano, S.F.S.D.; Penitente, A.R.; Arida, R.M.; Longo, B.M. The effects of resistance exercise on cognitive function, amyloidogenesis, and neuroinflammation in Alzheimer’s disease. Front. Neurosci. 2023, 17, 1131214. [Google Scholar] [CrossRef]
- Pena, G.S.; Paez, H.G.; Johnson, T.K.; Halle, J.L.; Carzoli, J.P.; Visavadiya, N.P.; Zourdos, M.C.; Whitehurst, M.A.; Khamoui, A.V. Hippocampal Growth Factor and Myokine Cathepsin B Expression following Aerobic and Resistance Training in 3xTg-AD Mice. Int. J. Chronic Dis. 2020, 2020, 5919501. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, D.; Campos, H.C.; Wuo-Silva, R.; Faber, J.; Gomes da Silva, S.; Coppi, A.A.; Arida, R.M.; Longo, B.M. Resistance Exercise Decreases Amyloid Load and Modulates Inflammatory Responses in the APP/PS1 Mouse Model for Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Arida, R.M.; Gomez-Pinilla, F. Physical exercise as an epigenetic modulator of brain plasticity and cognition. Neurosci. Biobehav. Rev. 2017, 80, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Xie, S.X.; Baer, D.R.; Suh, E.; Van Deerlin, V.M.; Loh, N.J.; Irwin, D.J.; McMillan, C.T.; Wolk, D.A.; Chen-Plotkin, A.; et al. Pathological combinations in neurodegenerative disease are heterogeneous and disease-associated. Brain 2023, 146, 2557–2569. [Google Scholar] [CrossRef] [PubMed]
- Chatila, Z.K.; Bradshaw, E.M. Alzheimer’s Disease Genetics: A Dampened Microglial Response? Neuroscientist 2023, 29, 245–263. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Sintini, I. Editorial: New insights into atypical Alzheimer’s disease: From clinical phenotype to biomarkers. Front. Neurosci. 2024, 18, 1414443. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Graff-Radford, J.; Machulda, M.M.; Carlos, A.F.; Schwarz, C.G.; Senjem, M.L.; Jack, C.R., Jr.; Lowe, V.J.; Josephs, K.A.; Whitwell, J.L. Atypical Alzheimer’s disease: New insights into an overlapping spectrum between the language and visual variants. J. Neurol. 2024, 271, 3571–3585. [Google Scholar] [CrossRef]
- Onisiforou, A.; Christodoulou, C.C.; Zamba-Papanicolaou, E.; Zanos, P.; Georgiou, P. Transcriptomic analysis reveals sex-specific patterns in the hippocampus in Alzheimer’s disease. Front. Endocrinol. 2024, 15, 1345498. [Google Scholar] [CrossRef] [PubMed]
- Pelak, V.S.; Krishnan, V.; Serva, S.; Pressman, P.; Mahmood, A.; Noteboom, L.; Bettcher, B.M.; Sillau, S.H.; Callen, A.L. Thaker, A.A. Lobar Microbleeds in the Posterior Cortical Atrophy Syndrome: A Comparison to Typical Alzheimer’s Disease. Curr. Neurol. Neurosci. Rep. 2024, 24, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.A.; Stevens, D.; Kundu, S.; Sanghera, R.; Dagher, R.; Yedavalli, V.; Jones, C.; Sair, H.; Luna, L.P. Detecting Alzheimer’s Disease Stages and Frontotemporal Dementia in Time Courses of Resting-State fMRI Data Using a Machine Learning Approach. J. Imaging Inform. Med. 2024. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, Y.; Howe, I.A.; Eckbo, R.; Wong, B.; Quimby, M.; Hochberg, D.; McGinnis, S.M.; Putcha, D.; Wolk, D.; Touroutoglou, A.; et al. Default mode network tau predicts future clinical decline in atypical early Alzheimer’s disease. medRxiv 2024. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The Cholinergic Hypothesis of Age and Alzheimer’s Disease-Related Cognitive Deficits: Recent Challenges and Their Implications for Novel Drug Development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Arrué, L.; Cigna-Méndez, A.; Barbosa, T.; Borrego-Muñoz, P.; Struve-Villalobos, S.; Oviedo, V.; Martínez-García, C.; Sepúlveda-Lara, A.; Millán, N.; Márquez Montesinos, J.C.E.; et al. New Drug Design Avenues Targeting Alzheimer’s Disease by Pharmacoinformatics-Aided Tools. Pharmaceutics 2022, 14, 1914. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, P.B.S.; Ferreira, A.F.F.; Britto, L.R.; Doussoulin, A.P.; Torrão, A.d.S. Association between thyroid function and Alzheimer’s disease: A systematic review. Metab. Brain Dis. 2021, 36, 1523–1543. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A. Streptozotocin-Induced Cytotoxicity, Oxidative Stress and Mitochondrial Dysfunction in Human Hepatoma HepG2 Cells. Int. J. Mol. Sci. 2012, 13, 5751–5767. [Google Scholar] [CrossRef] [PubMed]
- Fortes, M.A.S.; Scervino, M.V.M.; Marzuca-Nassr, G.N.; Vitzel, K.F.; da Justa Pinheiro, C.H.; Curi, R. Hypertrophy Stimulation at the Onset of Type I Diabetes Maintains the Soleus but Not the EDL Muscle Mass in Wistar Rats. Front. Physiol. 2017, 8, 830. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Jiang, K.; Lin, F.; Zhu, T.; Khan, N.H.; Jiang, E. Pathophysiological Association of Alzheimer’s Disease and Hypertension: A Clinical Concern for Elderly Population. Clin. Interv. Aging 2023, 18, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Kadhim, H.J.; Al-Mumen, H.; Nahi, H.H.; Hamidi, S.M. Streptozotocin-induced Alzheimer’s disease investigation by one-dimensional plasmonic grating chip. Sci. Rep. 2022, 12, 21878. [Google Scholar] [CrossRef] [PubMed]
- Hussey, S.E.; McGee, S.L.; Garnham, A.; McConell, G.K.; Hargreaves, M. Exercise increases skeletal muscle GLUT4 gene expression in patients with type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, A.L.; Cambronero, F.E.; Liu, D.; Moore, E.E.; Neal, J.E.; Terry, J.G.; Nair, S.; Pechman, K.R.; Rane, S.; Davis, L.T.; et al. Higher Aortic Stiffness Is Related to Lower Cerebral Blood Flow and Preserved Cerebrovascular Reactivity in Older Adults. Circulation 2018, 138, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Kaess, B.M.; Rong, J.; Larson, M.G.; Hamburg, N.M.; Vita, J.A.; Levy, D.; Benjamin, E.J.; Vasan, R.S.; Mitchell, G.F. Aortic Stiffness, Blood Pressure Progression, and Incident Hypertension. JAMA 2012, 308, 875. [Google Scholar] [CrossRef] [PubMed]
- Watase, H.; Sun, J.; Hippe, D.S.; Balu, N.; Li, F.; Zhao, X.; Mani, V.; Fayad, Z.A.; Fuster, V.; Hatsukami, T.S.; et al. Carotid Artery Remodeling Is Segment Specific. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, E.; Klegeris, A. Neuroinflammation as a mechanism linking hypertension with the increased risk of Alzheimer’s disease. Neural Regen. Res. 2022, 17, 2342. [Google Scholar] [PubMed]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Fan, X. Systemic Metabolism and Mitochondria in the Mechanism of Alzheimer’s Disease: Finding Potential Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 8398. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: A Current Overview. J. Alzheimer’s Dis. 2023, 92, 751–768. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Brian, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Mitochondrial Metabolism in Astrocytes Regulates Brain Bioenergetics, Neurotransmission and Redox Balance. Front. Neurosci. 2020, 14, 536682. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- El-Osta, H.; Circu, M.L. Mitochondrial ROS and Apoptosis. In Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–23. [Google Scholar]
- Han, Y.; Liu, D.; Cheng, Y.; Ji, Q.; Liu, M.; Zhang, B.; Zhou, S. Maintenance of mitochondrial homeostasis for Alzheimer’s disease: Strategies and challenges. Redox Biol. 2023, 63, 102734. [Google Scholar] [CrossRef] [PubMed]
- de Veij Mestdagh, C.F.; Smit, A.B.; Henning, R.H.; van Kesteren, R.E. Mitochondrial Targeting against Alzheimer’s Disease: Lessons from Hibernation. Cells 2023, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Allinquant, B.; Clamagirand, C.; Potier, M.C. Role of cholesterol metabolism in the pathogenesis of Alzheimer’s disease. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Capitini, C.; Bigi, A.; Parenti, N.; Emanuele, M.; Bianchi, N.; Cascella, R.; Cecchi, C.; Maggi, L.; Annunziato, F.; Pavone, F.S.; et al. APP and Bace1: Differential effect of cholesterol enrichment on processing and plasma membrane mobility. iScience 2023, 26, 106611. [Google Scholar] [CrossRef] [PubMed]
- Windham, I.A.; Cohen, S. The cell biology of APOE in the brain. Trends Cell Biol. 2024, 34, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, J.; Liu, Q. Brain cell type-specific cholesterol metabolism and implications for learning and memory. Trends Neurosci. 2022, 45, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Shi, Q.; Zhang, X.; Gu, L.; Li, J.; Quan, S.; Zhao, X.; Li, Q. ApoE4-mediated blood-brain barrier damage in Alzheimer’s disease: Progress and prospects. Brain Res. Bull. 2023, 199, 110670. [Google Scholar] [CrossRef] [PubMed]
- Akyol, O.; Akyol, S.; Chou, M.C.; Chen, S.; Liu, C.K.; Selek, S.; Soares, J.C.; Chen, C.H. Lipids and lipoproteins may play a role in the neuropathology of Alzheimer’s disease. Front. Neurosci. 2023, 17, 1275932. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Veerhuis, R. The importance of inflammatory mechanisms for the development of Alzheimer’s disease. Exp. Gerontol. 1999, 34, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Wong-Guerra, M.; Pardo-Andreu, G.L.; Nuñez-Figueredo, Y. Modelos animales no transgénicos de demencia. consideraciones metodológicas y relevancia farmacológica. Rev. Cienc. Farm. Aliment. 2023, 1, 1–28. [Google Scholar]
- Morales, I.; Guzmán-MartÃnez, L.; Cerda-Troncoso, C.; FarÃas, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Sepúlveda, P.; Castillo, R.L.; Salazar, L.A. Relationship between Hypoxic and Immune Pathways Activation in the Progression of Neuroinflammation: Role of HIF-1α and Th17 Cells. Int. J. Mol. Sci. 2023, 24, 3073. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Di Benedetto, S.; Müller, L.; Wenger, E.; Düzel, S.; Pawelec, G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev. 2017, 75, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, L.G.G.C.; Heinen, C.; Selivanova, A.; Halff, E.F.; Salomons, F.A.; Dantuma, N.P. Minimal length requirement for proteasomal degradation of ubiquitin-dependent substrates. FASEB J. 2009, 23, 123–133. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Action Plan on Physical Activity 2018–2030: More Active People for a Healthier World: At-A-Glance. 2018. Available online: https://iris.who.int/handle/10665/272721 (accessed on 1 March 2024).
- Caspersen, C.J.; Powell, K.E.; Christenson, G.M. Physical activity, exercise, and physical fitness: Definitions and distinctions for health-related research. Public Health Rep. 1985, 100, 126–131. [Google Scholar] [PubMed]
- Phillips, S.M.; Winett, R.A. Uncomplicated Resistance Training and Health-Related Outcomes. Curr. Sports Med. Rep. 2010, 9, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Baar, K.; Esser, K. Phosphorylation of p70 S6k correlates with increased skeletal muscle mass following resistance exercise. Am. J. Physiol. Cell Physiol. 1999, 276, C120–C127. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.; Kubica, N.; Crozier, S.; Williamson, D.; Farrell, P.; Kimball, S.; Jefferson, L.S. Understanding skeletal muscle hypertrophy: Integration of cell signalling. Physiol. News Mag. 2004, 18–19. [Google Scholar] [CrossRef]
- Glass, D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell. Biol. 2005, 37, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef]
- Zanou, N.; Gailly, P. Skeletal muscle hypertrophy and regeneration: Interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell. Mol. Life Sci. 2013, 70, 4117–4130. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.; Soci, Ú.P.; Melo, S.F.; Alves, C.R.; Oliveira, E.M. Signaling Pathways that Mediate Skeletal Muscle Hypertrophy: Effects of Exercise Training. In Skeletal Muscle—From Myogenesis to Clinical Relations; InTech: London, UK, 2012. [Google Scholar]
- Stone, M.H.; Collins, D.; Plisk, S.; Haff, G.; Stone, M.E. Training Principles: Evaluation of Modes and Methods of Resistance Training. Strength Cond. J. 2000, 22, 65–76. [Google Scholar] [CrossRef]
- Kasper, K. Sports Training Principles. Curr. Sports Med. Rep. 2019, 18, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Tohda, C. Skeletal muscle atrophy-induced hemopexin accelerates onset of cognitive impairment in Alzheimer’s disease. J. Cachexia Sarcopenia Muscle 2021, 12, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Crawford, F.; Mullan, M.; Kokjohn, T.A.; Emmerling, M.R.; Weller, R.O.; Roher, A.E. Elevated Aβ and Apolipoprotein E in AβPP Transgenic Mice and Its Relationship to Amyloid Accumulation in Alzheimer’s Disease. Mol. Med. 2000, 6, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Waite, S.J.; Maitland, S.; Thomas, A.; Yarnall, A.J. Sarcopenia and frailty in individuals with dementia: A systematic review. Arch. Gerontol. Geriatr. 2021, 92, 104268. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease. Neurology 1984, 34, 939. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; Engel, W.K. Inclusion-body myositis: Muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer’s and Parkinson’s disease brains. Acta Neuropathol. 2008, 116, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Uruha, A.; Nishino, I. Pathogenesis of inclusion body myositis: Autoimmune or degenerative disease? Brain Nerve 2013, 65, 1291–1298. [Google Scholar] [PubMed]
- Burns, J.M.; Johnson, D.K.; Watts, A.; Swerdlow, R.H.; Brooks, W.M. Reduced Lean Mass in Early Alzheimer Disease and Its Association With Brain Atrophy. Arch. Neurol. 2010, 67, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Cardoso, V.; Castro, M.; Oliveira, M.M.; Moreira, P.; Peixoto, F.; Videira, R. Age-Dependent Biochemical Dysfunction in Skeletal Muscle of Triple- Transgenic Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.J.d.O.; Oliveira LMd Bittencourt, A.M.V.; Lourenço, L.G.d.C.; Oliveira, G.C.M.d. Brain insulin resistance and Alzheimer’s disease: A systematic review. Dement Neuropsychol. 2024, 18, e20230032. [Google Scholar] [CrossRef] [PubMed]
- Shafi, O. Inverse relationship between Alzheimer’s disease and cancer, and other factors contributing to Alzheimer’s disease: A systematic review. BMC Neurol. 2016, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Culibrk, R.A.; Ebbert, K.A.; Yeisley, D.J.; Chen, R.; Qureshi, F.A.; Hahn, J.; Hahn, M.S. Impact of Suramin on Key Pathological Features of Sporadic Alzheimer’s Disease-Derived Forebrain Neurons. J. Alzheimer’s Dis. 2024, 98, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Kommaddi, R.P.; Gowaikar, R.; PA, H.; Diwakar, L.; Singh, K.; Mondal, A. Akt activation ameliorates deficits in hippocampal-dependent memory and activity-dependent synaptic protein synthesis in an Alzheimer’s disease mouse model. J. Biol. Chem. 2024, 300, 105619. [Google Scholar] [CrossRef]
- Qiao, Y.; Liu, H.; He, C.; Ma, Y. ApoE Mimic Peptide COG1410 Reduces Aβ Deposition and Improves Cognitive Function by Inducing the Transformation of A1/A2 Reactive Astrocytes and Increasing the BDNF Concentration in Brain of APP/PS1 Double Transgenic Mice. Neuroscience 2024, 537, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Kajihara, R. An Interaction between Brain-Derived Neurotrophic Factor and Stress-Related Glucocorticoids in the Pathophysiology of Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 1596. [Google Scholar] [CrossRef] [PubMed]
- Zota, I.; Chanoumidou, K.; Charalampopoulos, I.; Gravanis, A. Dynamics of myelin deficits in the 5xFAD mouse model for Alzheimer’s disease and the protective role of BDNF. Glia 2024, 72, 809–827. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Kojis, D.J.; Ghosh, S.; Beiser, A.S.; Seshadri, S. Association of Neurotrophic Factors at Midlife With In Vivo Measures of β-Amyloid and Tau Burden 15 Years Later in Dementia-Free Adults. Neurology 2024, 102, e209198. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.R.; Gharesouran, J.; Sabaie, H.; Zaboli Mahdiabadi, M.; Mazhari, S.A.; Sharifi-Bonab, M.; Shirvani-Farsani, Z.; Taheri, M.; Sayad, A.; Rezazadeh, M. Neurotrophin growth factors and their receptors as promising blood biomarkers for Alzheimer’s Disease: A gene expression analysis study. Mol. Biol. Rep. 2024, 51, 49. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef]
- Leem, Y.H.; Lim, H.J.; Shim, S.B.; Cho, J.Y.; Kim, B.S.; Han, P.L. Repression of tau hyperphosphorylation by chronic endurance exercise in aged transgenic mouse model of tauopathies. J. Neurosci. Res. 2009, 87, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of brain-derived neurotrophic factor across the blood–brain barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Del Rosso, S.; Baraquet, M.L.; Barale, A.; Defagó, M.D.; Tortosa, F.; Perovic, N.R.; Aoki, M.P. Long-term effects of different exercise training modes on cytokines and adipokines in individuals with overweight/obesity and cardiometabolic diseases: A systematic review, meta-analysis, and meta-regression of randomized controlled trials. Obes. Rev. 2023, 24, e13564. [Google Scholar] [CrossRef] [PubMed]
- Kelley, G.A.; Kelley, K.S.; Stauffer, B.L. Resistance training and inter-interindividual response differences on cardiorespiratory fitness in older adults: An ancillary meta-analysis of randomized controlled trials. Sci. Prog. 2024, 107, 368504241227088. [Google Scholar] [CrossRef] [PubMed]
- Félix-Soriano, E.; Stanford, K.I. Exerkines and redox homeostasis. Redox Biol. 2023, 63, 102748. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.R.; Mendes, L.O.; Veras, A.S.C.; Thorpe, H.H.A.; Fávaro, W.J.; de Almeida Chuffa, L.G.; Pinheiro, P.F.F.; Martinez, F.E. Physical resistance training-induced changes in lipids metabolism pathways and apoptosis in prostate. Lipids Health Dis. 2020, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Morville, T.; Sahl, R.E.; Moritz, T.; Helge, J.W.; Clemmensen, C. Plasma Metabolome Profiling of Resistance Exercise and Endurance Exercise in Humans. Cell. Rep. 2020, 33, 108554. [Google Scholar] [CrossRef] [PubMed]
- de Gregório, E.; Mendes, G.C.; Somensi, L.B.; Freire, C.G.; Lopes, L.F.; Lima, K.R.; Carrazoni, G.S.; Neves, B.S.; Picua, S.S.; da Silva, L.M.; et al. Neuroprotective effects of strength training in a neuroinflammatory animal model. BMC Neurosci. 2022, 23, 22. [Google Scholar] [CrossRef]
- Liu, Y.; Chu, J.M.T.; Ran, Y.; Zhang, Y.; Chang, R.C.C.; Wong, G.T.C. Prehabilitative resistance exercise reduces neuroinflammation and improves mitochondrial health in aged mice with perioperative neurocognitive disorders. J. Neuroinflam. 2022, 19, 150. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.T.K.; Marques, L.S.; Zborowski, V.A.; Silva, G.L.; Nogueira, C.W.; Zeni, G. Resistance Training Modulates Hippocampal Neuroinflammation and Protects Anxiety-Depression-like Dyad Induced by an Emotional Single Prolonged Stress Model. Mol. Neurobiol. 2023, 60, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Vints, W.A.J.; Gökçe, E.; Šeikinaitė, J.; Kušleikienė, S.; Česnaitienė, V.J.; Verbunt, J.; Levin, J.; Masiulis, N. Resistance training’s impact on blood biomarkers and cognitive function in older adults with low and high risk of mild cognitive impairment: A randomized controlled trial. Eur. Rev. Aging Phys. Act. 2024, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.M.; Santagnello, S.B.; de Oliveira Junior, G.N.; de Sousa, J.d.F.R.; Michelin, M.A.; Nomelini, R.S. Lower-Body Resistance Training Reduces Interleukin-1β and Transforming Growth Factor-β1 Levels and Fatigue and Increases Physical Performance in Breast Cancer Survivors. J. Strength Cond. Res. 2023, 37, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Alizaei Yousefabadi, H.; Niyazi, A.; Alaee, S.; Fathi, M.; Mohammad Rahimi, G.R. Anti-Inflammatory Effects of Exercise on Metabolic Syndrome Patients: A Systematic Review and Meta-Analysis. Biol. Res. Nurs. 2021, 23, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.M.P.; Oliveira ECd Becker, L.K.; Costa, G.d.P.; Pinto, K.M.d.C.; Talvani, A.; Carraro, J.C.C.; Coelho, D.B. Resistance Training Associated with Dietetic Advice Reduces Inflammatory Biomarkers in the Elderly. Biomed. Res. Int. 2020, 2020, 7351716. [Google Scholar] [CrossRef] [PubMed]
- Schwappacher, R.; Dieterich, W.; Reljic, D.; Pilarsky, C.; Mukhopadhyay, D.; Chang, D.K.; Biankin, E.V.; Siebler, J.; Herrmann, H.J.; Neurath, M.F.; et al. Muscle-Derived Cytokines Reduce Growth, Viability and Migratory Activity of Pancreatic Cancer Cells. Cancers 2021, 13, 3820. [Google Scholar] [CrossRef] [PubMed]
- Phoemsapthawee, J.; Ammawat, W.; Prasertsri, P.; Sathalalai, P.; Leelayuwat, N. Does Gotu kola supplementation improve cognitive function, inflammation, and oxidative stress more than multicomponent exercise alone?—A randomized controlled study. J. Exerc. Rehabil. 2022, 18, 330–342. [Google Scholar] [CrossRef]
- Jacko, D.; Masur, L.; Schaaf, K.; Zacher, J.; Bersiner, K.; de Marées, M.; Bloch, W.; Gehlert, S. Resistance training does not increase myocellular garbage dumps: A pilot study on lipofuscin in skeletal muscle fibers of resistance trained young men. Physiol. Rep. 2024, 12, e15922. [Google Scholar] [CrossRef] [PubMed]
- Key, M.N.; Szabo-Reed, A.N. Impact of Diet and Exercise Interventions on Cognition and Brain Health in Older Adults: A Narrative Review. Nutrients 2023, 15, 2495. [Google Scholar] [CrossRef] [PubMed]
- Ayari, S.; Abellard, A.; Carayol, M.; Guedj, É.; Gavarry, O. A systematic review of exercise modalities that reduce pro-inflammatory cytokines in humans and animals’ models with mild cognitive impairment or dementia. Exp. Gerontol. 2023, 175, 112141. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, M.A. The Role of Physical Activity in Adjunctive Nursing Management of Neuro-Degenerative Diseases among Older Adults: A Systematic Review of Interventional Studies. Life 2024, 14, 597. [Google Scholar] [CrossRef] [PubMed]
- Zammit, A.R.; Piccinin, A.M.; Duggan, E.C.; Koval, A.; Clouston, S.; Robitaille, A.; Brown, C.L.; Handschuh, P.; Wu, C.; Jarry, V.; et al. A Coordinated Multi-study Analysis of the Longitudinal Association Between Handgrip Strength and Cognitive Function in Older Adults. J. Gerontol. Ser. B 2021, 76, 229–241. [Google Scholar] [CrossRef] [PubMed]
- García-Llorente, A.M.; Casimiro-Andújar, A.J.; Linhares, D.G.; De Souza Vale, R.G.; Marcos-Pardo, P.J. Multidomain interventions for sarcopenia and cognitive flexibility in older adults for promoting healthy aging: A systematic review and meta-analysis of randomized controlled trials. Aging Clin. Exp. Res. 2024, 36, 47. [Google Scholar] [CrossRef] [PubMed]
- Moss, F.P.; Leblond, C.P. Nature of Dividing Nuclei in Skeletal Muscle of Growing Rats. J. Cell. Biol. 1970, 44, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Shariatzadeh Joneydi, M.; Koyanagi, A.; Yang, G.; Ji, B.; Won Lee, S.; Keon Yon, D.; Smith, L.; Shin, J.I.; Yusheng, L. Resistance training restores skeletal muscle atrophy and satellite cell content in an animal model of Alzheimer’s disease. Sci. Rep. 2023, 13, 2535. [Google Scholar] [CrossRef] [PubMed]
- Mcleod, J.C.; Currier, B.S.; Lowisz, C.V.; Phillips, S.M. The influence of resistance exercise training prescription variables on skeletal muscle mass, strength, and physical function in healthy adults: An umbrella review. J. Sport Health Sci. 2024, 13, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Currier, B.S.; Mcleod, J.C.; Banfield, L.; Beyene, J.; Welton, N.J.; D’Souza, A.C.; Keogh, J.A.J.; Lin, L.; Coletta, G.; Yang, A.; et al. Resistance training prescription for muscle strength and hypertrophy in healthy adults: A systematic review and Bayesian network meta-analysis. Br. J. Sports Med. 2023, 57, 1211–1220. [Google Scholar] [CrossRef]
- Foltran, R.B.; Diaz, S.L. BDNF isoforms: A round trip ticket between neurogenesis and serotonin? J. Neurochem. 2016, 138, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Edman, S.; Horwath, O.; Van der Stede, T.; Blackwood, S.J.; Moberg, I.; Strömlind, H.; Nordström, F.; Ekblom, M.; Katz, A.; Apró, W.; et al. Pro-Brain-Derived Neurotrophic Factor (BDNF), but Not Mature BDNF, Is Expressed in Human Skeletal Muscle: Implications for Exercise-Induced Neuroplasticity. Function 2024, 5, zqae005. [Google Scholar] [CrossRef] [PubMed]
- Lomborg, S.D.; Dalgas, U.; Hvid, L.G. The importance of neuromuscular rate of force development for physical function in aging and common neurodegenerative disorders—A systematic review. J. Musculoskelet. Neuronal Interact 2022, 22, 562–586. [Google Scholar] [PubMed]
- Braz de Oliveira, M.P.; Moreira Padovez, R.d.F.C.; Serrão, P.R.M.d.S.; de Noronha, M.A.; Cezar, N.O.d.C.; Andrade, L.P.d. Effectiveness of physical exercise at improving functional capacity in older adults living with Alzheimer’s disease: A systematic review of randomized controlled trials. Disabil. Rehabil. 2023, 45, 391–402. [Google Scholar] [CrossRef]
- Rodrigues Junior, C.F.; Murata, G.M.; Gerlinger-Romero, F.; Nachbar, R.T.; Marzuca-Nassr, G.N.; Gorjão, R.; Vitzel, K.F.; Hirabara, S.M.; Pithon-Curi, T.C.; Curi, R. Changes in Skeletal Muscle Protein Metabolism Signaling Induced by Glutamine Supplementation and Exercise. Nutrients 2023, 15, 4711. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- da Silva Rodrigues, G.; Noronha, N.Y.; Almeida, M.L.; Sobrinho, A.C.d.S.; Watanabe, L.M.; Pinhel, M.A.d.S.; de Lima, J.G.R.; Zhang, R.; Nonino, C.B.; Alves, C.R.R.; et al. Exercise training modifies the whole blood DNA methylation profile in middle-aged and older women. J. Appl. Physiol. 2023, 134, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Dalle Carbonare, L.; Dorelli, G.; Li Vigni, V.; Minoia, A.; Bertacco, J.; Cheri, S.; Deiana, M.; Innamorati, G.; Cominacini, M.; Tarperi, C.; et al. Physical Activity Modulates miRNAs Levels and Enhances MYOD Expression in Myoblasts. Stem Cell Rev. Rep. 2022, 18, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is It Reliable to Use Common Molecular Docking Methods for Comparing the Binding Affinities of Enantiomer Pairs for Their Protein Target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef]
- Coutinho, L.A.; Leão, L.L.; Cassilhas, R.C.; de Paula, A.M.B.; Deslandes, A.C.; Monteiro-Junior, R.S. Alzheimer’s disease genes and proteins associated with resistance and aerobic training: An in silico analysis. Exp. Gerontol. 2022, 168, 111948. [Google Scholar] [CrossRef] [PubMed]
- Nicola, L.; Loo, S.J.Q.; Lyon, G.; Turknett, J.; Wood, T.R. Does resistance training in older adults lead to structural brain changes associated with a lower risk of Alzheimer’s dementia? A narrative review. Ageing Res. Rev. 2024, 98, 102356. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Species | Reference | Aim | Model/ Pathology |
|---|---|---|---|
| Animals | [14] | Determined whether short-term resistance exercise inhibits neuroinflammation and attenuated neuropathological changes in 3xTg-AD mice. | 3xTg-AD mice |
| [15] | Determined the potential protective effects of aerobic, resistance, and combined exercise methods on an AD-like model induced by ovariectomy and D-galactose administration. | AD-like model | |
| [17] | Determined the effects of aerobic training and resistance training on hippocampal BDNF and IGF-1 signaling, Aβ expression, and the myokine cathepsin B in the 3xTg-AD model. | 3xTg-AD mice | |
| [18] | Analyzed hippocampal changes in Aβ load, inflammatory responses, and locomotor activity in a transgenic APP/PS1 mouse model of AD submitted to a resistance exercise program. | APP/PS1 mice | |
| [34] | Determined the mechanisms involved in overload-induced muscle hypertrophy in the context of type I diabetes, specifically examining whether such hypertrophy can counteract the muscle hypotrophy associated with the diabetic state. | Type I diabetes rats | |
| [69] | Determined the acute changes that occur in skeletal muscles following a single bout of high-resistance exercise and assessed the relationship between the acute molecular responses and long-term muscle hypertrophy induced by high-resistance exercise training. | Wistar rats | |
| [78] | Determined the molecular mechanism underlying the accelerated onset of AD induced by skeletal muscle atrophy. | 5XFAD mice | |
| [79] | Determined the relationship between apolipoprotein E levels and Aβ accumulation in the brains of transgenic AβPPswe/tg2576 (tg2576) mice, a commonly used model for AD. | tg2576 mice | |
| [85] | Determined the impact of disease progression in AD on the physiological features of skeletal muscle using a mouse model. | 3xTg-AD mice | |
| [89] | Determined the molecular mechanisms underlying the impairment of new protein synthesis in the synapse in AD pathology, particularly focusing on the Akt1/mTOR signaling pathways. | APP/PS1 mice | |
| [90] | Determined the therapeutic potential of the apolipoprotein E mimetic peptide COG1410 in AD using the transgenic APP/PS1 mouse model. | APP/PS1 mice | |
| [96] | Determined the effect of chronic endurance exercise on tau phosphorylation levels in the brain with an AD-like pathology, using a transgenic mouse model of tauopathies. | AD-like model | |
| [101] | Determined the effect of physical resistance training on lipid metabolism and apoptosis in the adult Wistar rat prostate. | Wistar rats | |
| [103] | Investigated the role of prophylactic muscular strength exercise in diminishing cognitive alterations and modifying antioxidant intracellular scenery in an animal neuroinflammatory model in the CA1 region of the hippocampus. | Wistar rats | |
| [104] | Evaluated the neuroprotective effect of preconditioning resistance training on aged mice undergoing abdominal surgery, and examined the underlying mechanisms related to the neuroinflammatory state and synaptic plasticity in the hippocampus. | C57BL/6N mice | |
| [105] | Evaluated the role of resistance exercise in mitigating anxiety and depression-like behaviors induced by stress in a mouse model. Specifically, the study investigated the effects of resistance exercise on behavioral phenotypes, hippocampal neuroinflammation, and the Akt/mTOR signaling pathway in mice subjected to a single prolonged emotional stress protocol. | Swiss mice | |
| [119] | Evaluated muscle cross-sectional area, myonuclear number, satellite cell content, and myosin heavy chain types in an animal model of AD, and examined the possible role of resistance training in controlling skeletal muscle size in this disease. | Wistar rat with induced AD | |
| [126] | Evaluated the effects of RET and/or glutamine supplementation on signaling protein synthesis in adult rat skeletal muscle. | Rats | |
| Cells | [33] | Determined the molecular mechanisms underlying the cytotoxic effects of streptozotocin on HepG2 hepatoma cells, focusing specifically on the role of oxidative stress, mitochondrial dysfunction, and metabolic alterations. | HepG2 cells |
| [36] | Determined the effect of streptozotocin on neuronal activity using a novel one-dimensional electro-plasmonic nanograting platform. | Neurons | |
| [53] | Determined the influence of cholesterol levels on the proteolytic processing of the amyloid precursor protein by the β-secretase Bace1 in living human neuroblastoma cells. | Neuroblastoma cells | |
| [64] | Determined the mechanism underlying the inefficient degradation of protein UBB (+1), which is generated from an erroneous transcriptional process of the ubiquitin B gene. | HeLa and SH-SY5Y cells | |
| [88] | Determined the impact of forebrain neuron exposure to suramin on the Akt/mTOR signaling pathway, a major regulator of autophagy, in comparison with rapamycin and chloroquine, and investigated the effect of suramin on several AD-related biomarkers in sporadic AD-derived forebrain neurons. | Neurons | |
| [92] | Determined the role of myelin breakdown in AD progression, focusing on its relation to oligodendrocyte progenitor cells and the neurotrophin system. | Oligodendrocytes | |
| Humans | [7] | Examined the influence of age, education, and physical activity on executive function performance and the interaction effects between these factors on two subpopulations of adults, that is, young adults and middle-aged adults. | Healthy individuals |
| [8] | Determined the impact of cognitive function on physical activity, physical function, and health-related quality of life in older adults within the first year after hip fracture surgery. | Healthy individuals | |
| [11] | Determined the hippocampal response to a 24-month physical activity intervention in sedentary older adults. | Healthy individuals | |
| [12] | Determined the effects of a long-term exercise intervention on two prominent biomarkers of inflammation (C-reactive protein and interleukin 6) in elderly men and women. | Healthy individuals | |
| [20] | Determined the incidence and distribution of various pathologies across neurodegenerative diseases and normal aging in a retrospective study of 1647 autopsied individuals. | Autopsied individuals | |
| [23] | Determined how frequently visual function deficits occur in the language variant of AD and how frequently language deficits occur in the visual variant of AD. | Patients with AD | |
| [24] | Determined the sex-specific differences and similarities in the hippocampus and its subfields (CA1 and CA3) in AD. | Autopsied individuals | |
| [26] | Developed an early and accurate diagnostic classification model for neurodegenerative dementia subtypes, specifically AD, frontotemporal dementia, and mild cognitive impairment, using resting-state functional magnetic resonance imaging data and clinical information. | Patients with AD | |
| [27] | Determined the relationship between baseline tau positron-emission tomography and the rate of subsequent clinical decline in individuals with atypical forms of early AD. | Patients with AD | |
| [36] | Determined the effect of a single bout of exercise on GLUT4 gene expression in muscle of patients with type 2 diabetes and control subjects, matched for age and body mass index. | Patients with type 2 diabetes | |
| [38] | Determined the relationship between aortic stiffening, as measured by aortic pulse wave velocity, and brain health indicators, specifically resting cerebral blood flow and cerebrovascular reactivity, in older adults. | Healthy individuals | |
| [39] | Examined temporal relationships between vascular stiffness, central hemodynamics, microvascular function, and blood pressure progression. | Patients with vascular injuries | |
| [84] | Examined body composition in individuals with early AD and without dementia and its relation to cognition and brain volume. | Patients with dementia | |
| [93] | Explored the relationship between BDNF and VEGF serum levels with future brain Aβ and tau pathology in a cohort of cognitively healthy, predominantly middle-aged adults, and tested for possible effect modifications by sex and menopausal status. | Healthy individuals | |
| [94] | Determined the potential of neurotrophin growth factors and their receptors as biomarkers for AD. Specifically, the researchers assessed the messenger RNA expression levels of neurotrophin growth factors (such as BDNF) and their receptors (such as NTRK2, TrkA, and TrkC) in blood samples collected from patients with AD and healthy controls. | Patients with AD | |
| [102] | Investigated and compared the molecular response to two different types of skeletal muscle activity, specifically endurance exercise and resistance exercise, using the untargeted metabolomics profiling of plasma. | Healthy individuals | |
| [106] | Observed the effect of 12 weeks of resistance training on peripheral biomarker levels, cognitive function changes, and their interrelationship in older adults with different risks of mild cognitive impairment. Specifically, the study investigated whether resistance training has differential effects on cognitive function and biomarker levels in older adults with a high risk of mild cognitive impairment compared with those with a low risk of mild cognitive impairment. | Older adults with a low and high risk of mild cognitive impairment | |
| [107] | Determined whether resistance training can improve inflammatory markers, fatigue (both sensations and fatigability), and physical performance in breast cancer survivors. Additionally, the study investigated whether changes in inflammatory markers, fatigue, and physical performance are associated with each other in this population. | Breast cancer survivors | |
| [109] | Evaluated the effects of resistance training combined with dietary advice on chronic inflammation in elderly individuals. Specifically, the study assessed changes in anthropometric parameters and inflammatory biomarkers before and after a long-term progressive resistance training program associated with dietary advice. | Elderly people | |
| [110] | Analyzed the impact of exercise on the serum of patients with advanced-stage pancreatic cancer and its effects on pancreatic cancer cell proliferation, motility, and apoptosis. | Patients with pancreatic cancer | |
| [111] | Determined the synergistic effects of gotu kola supplementation and multicomponent exercise on cognitive function, inflammation, and oxidative stress in older adults with mild cognitive impairment. | Patients with mild cognitive impairment | |
| [123] | Investigated the expression and regulation of BDNF and mature BDNF in human skeletal muscle and plasma under various physiological conditions, including rest, acute exercise, lactate infusion, and fasting. | Healthy individuals | |
| [127] | Characterized the genetic landscape of AD and related dementias through a two-stage genome-wide association study. | Patients with AD | |
| [128] | Investigated whether exercise training modifies the whole blood methylation profile in healthy women. Specifically, the study assessed changes in methylation patterns following a 14-week exercise training protocol, which included aerobic cardiorespiratory and muscle strength exercises. | Healthy individuals | |
| [129] | Investigated the modulation of microRNAs associated with myogenesis following physical performance, particularly in the context of running a half marathon. | Healthy individuals |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sepúlveda-Lara, A.; Sepúlveda, P.; Marzuca-Nassr, G.N. Resistance Exercise Training as a New Trend in Alzheimer’s Disease Research: From Molecular Mechanisms to Prevention. Int. J. Mol. Sci. 2024, 25, 7084. https://doi.org/10.3390/ijms25137084
Sepúlveda-Lara A, Sepúlveda P, Marzuca-Nassr GN. Resistance Exercise Training as a New Trend in Alzheimer’s Disease Research: From Molecular Mechanisms to Prevention. International Journal of Molecular Sciences. 2024; 25(13):7084. https://doi.org/10.3390/ijms25137084
Chicago/Turabian StyleSepúlveda-Lara, Alexis, Paulina Sepúlveda, and Gabriel Nasri Marzuca-Nassr. 2024. "Resistance Exercise Training as a New Trend in Alzheimer’s Disease Research: From Molecular Mechanisms to Prevention" International Journal of Molecular Sciences 25, no. 13: 7084. https://doi.org/10.3390/ijms25137084
APA StyleSepúlveda-Lara, A., Sepúlveda, P., & Marzuca-Nassr, G. N. (2024). Resistance Exercise Training as a New Trend in Alzheimer’s Disease Research: From Molecular Mechanisms to Prevention. International Journal of Molecular Sciences, 25(13), 7084. https://doi.org/10.3390/ijms25137084