
Artificial Intelligence-Powered Molecular Docking and Steered Molecular Dynamics for Accurate scFv Selection of Anti-CD30 Chimeric Antigen Receptors

 , , ,
, , , 
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Generation and Validation of Novel Anti-CD30 mAbs
2.2. AI-Guided 3D Structure Prediction of Anti-CD30 mAbs/CD30 Antigen-Binding Interaction
2.3. Modeling CD30 Antibody–Antigen Complexes by AI Information-Driven Docking
2.4. Molecular Dynamics Simulation Identify the Most Stable CD30 Antibody–Antigen Complex
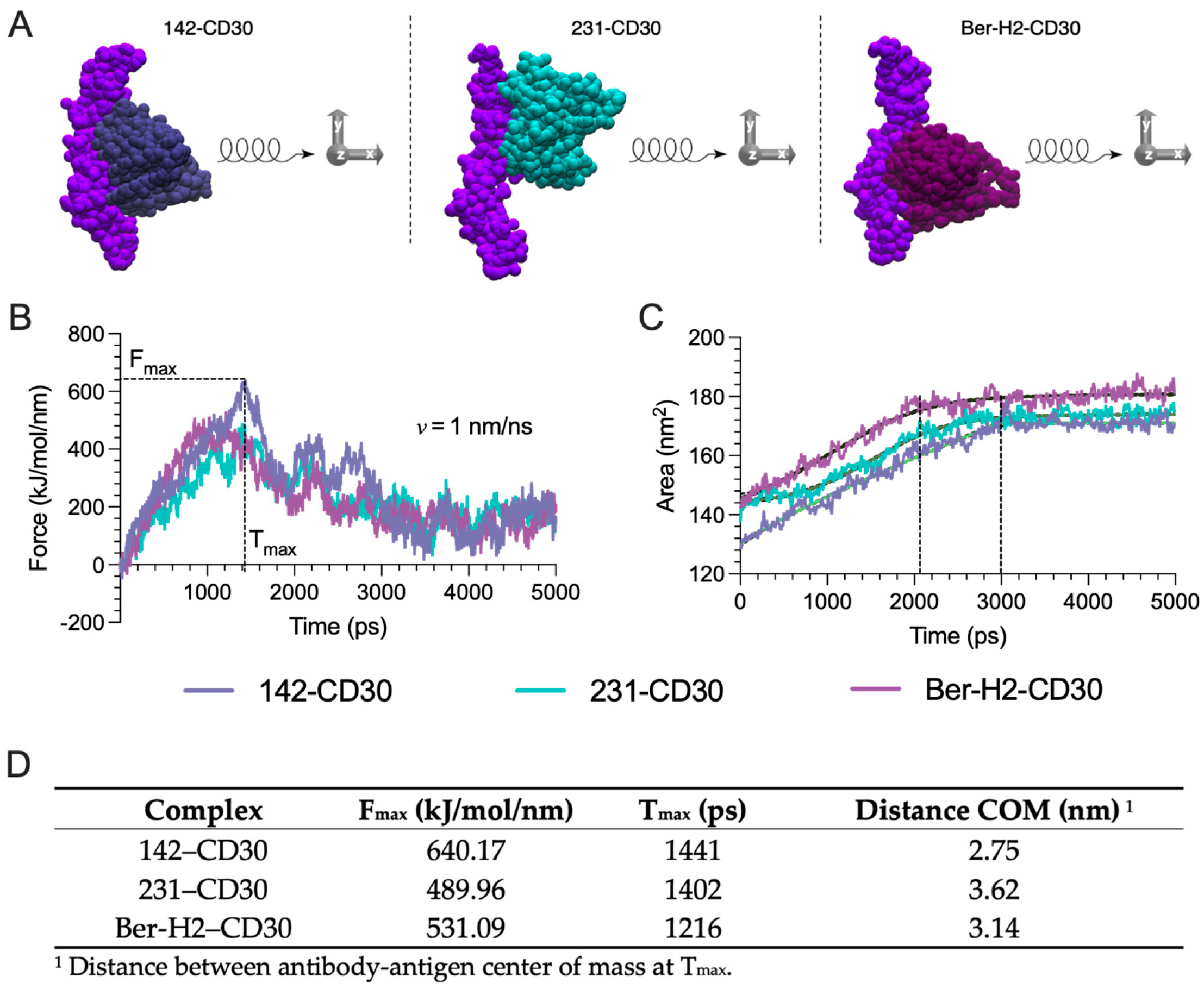
2.5. Coarse-Grained (CG) Steered Molecular Dynamics (SMD) Simulations Provide Insights into the Relative Binding Strengths of CD30 mAb-CD30 Antigen Pairs
2.6. CD30 mAb Binding Affinity Estimation through Umbrella Sampling (US) Simulations Are Comparable to Surface Plasmon Resonance (SPR)
2.7. Anti-Tumor Activity of Newly Generated CD30 CAR-T Cells Confirm AI-Guide In Silico Predictions
3. Discussion
4. Materials and Methods
4.1. Generation of Anti-CD30 Monoclonal Antibodies and scFv Sequencing
4.2. Artificial Intelligence 3D Structure Prediction of the Studied Systems’ Antibody–Antigen
4.3. Artificial Intelligence-Driven Information Molecular Docking
4.4. All-Atom (AA) Molecular Dynamics (MD) Simulations
4.5. Coarse-Grained (CG) Steered Molecular Dynamics (SMD)
4.6. Coarse-Grained (CG) Umbrella Sampling (US) Simulation and Analysis of Binding Free Energy
4.7. Surface Plasmon Resonance (SPR)
4.8. Generation of Anti-CD30 CAR-T Cells
4.9. In Vitro and In Vivo Efficacy Evaluation of Anti-CD30 CAR-T Cells
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-Type Specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Lynn, R.C.; Feng, Y.; Schutsky, K.; Poussin, M.; Kalota, A.; Dimitrov, D.S.; Powell, D.J., Jr. High-Affinity FRβ-Specific CAR T Cells Eradicate AML and Normal Myeloid Lineage without HSC Toxicity. Leukemia 2016, 30, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Lupo-Stanghellini, M.-T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef]
- Leyton-Castro, N.F.; Brigido, M.M.; Maranhão, A.Q. Selection of Antibody Fragments for CAR-T Cell Therapy from Phage Display Libraries. In Chimeric Antigen Receptor T Cells; Humana: New York, NY, USA, 2020; pp. 13–26. [Google Scholar]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T Design: Elements and Their Synergistic Function. eBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Vora, L.K.; Gholap, A.D.; Jetha, K.; Thakur, R.R.S.; Solanki, H.K.; Chavda, V.P. Artificial Intelligence in Pharmaceutical Technology and Drug Delivery Design. Pharmaceutics 2023, 15, 1916. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2020, 9, 71. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Z.; Hong, Q.; Zhou, L.; Ma, Y.; Hu, Y.; Xin, J.; Li, T.; Kong, Z.; Zheng, Q.; et al. A Stepwise Docking Molecular Dynamics Approach for Simulating Antibody Recognition with Substantial Conformational Changes. Comput. Struct. Biotechnol. J. 2022, 20, 710–720. [Google Scholar] [CrossRef]
- Falini, B.; Pileri, S.; Pizzolo, G.; Dürkop, H.; Flenghi, L.; Stirpe, F.; Martelli, M.F.; Stein, H. CD30 (Ki-1) Molecule: A New Cytokine Receptor of the Tumor Necrosis Factor Receptor Superfamily as a Tool for Diagnosis and Immunotherapy. Blood 1995, 85, 1–14. [Google Scholar] [CrossRef]
- Katsin, M.; Dormeshkin, D.; Meleshko, A.; Migas, A.; Dubovik, S.; Konoplya, N. CAR-T Cell Therapy for Classical Hodgkin Lymphoma. HemaSphere 2023, 7, e971. [Google Scholar] [CrossRef]
- Falini, B.; Flenghi, L.; Aversa, F.; Barbabietola, G.; Martelli, M.F.; Comeli, P.; Tazzari, P.L.; Broe, M.K.; Stein, H.; Dürkop, H.; et al. Response of Refractory Hodgkin’s Disease to Monoclonal Anti-CD30 Immunotoxin. Lancet 1992, 339, 1195–1196. [Google Scholar] [CrossRef] [PubMed]
- Arantes, P.R.; Polêto, M.D.; Pedebos, C.; Ligabue-Braun, R. Making It Rain: Cloud-Based Molecular Simulations for Everyone. J. Chem. Inf. Model. 2021, 61, 4852–4856. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Hülsmeyer, M.; Dürkop, H.; Hansen, H.P.; Schneider-Mergener, J.; Ziegler, A.; Uchanska-Ziegler, B. Human CD30: Structural Implications from Epitope Mapping and Modeling Studies. J. Mol. Recognition JMR 2003, 16, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.S.; Ytreberg, F.M. Fast Calculation of Protein–Protein Binding Free Energies Using Umbrella Sampling with a Coarse-Grained Model. J. Chem. Theory Comput. 2018, 14, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Wang, J.; Li, J.; Yuan, H.; Wang, P.; Zhang, M.; Feng, Y.; Cao, X.; Cao, X.; Kang, G.; et al. Design of Nanobody-Based Bispecific Constructs by in Silico Affinity Maturation and Umbrella Sampling Simulations. Comput. Struct. Biotechnol. J. 2023, 21, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Tam, N.M.; Nam, P.C.; Quang, D.T.; Tung, N.T.; Vu, V.V.; Ngo, S.T. Binding of Inhibitors to the Monomeric and Dimeric SARS-CoV-2 Mpro. RSC Adv. 2021, 11, 2926–2934. [Google Scholar] [CrossRef] [PubMed]
- Ramasanoff, R.R.; Sokolov, P.A. The Binding Model of Adenosine-Specific DNA Aptamer: Umbrella Sampling Study. J. Mol. Graph. Model. 2023, 118, 108338. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.Y.; Notman, R. Comparison of Umbrella Sampling and Steered Molecular Dynamics Methods for Computing Free Energy Profiles of Aromatic Substrates through Phospholipid Bilayers. J. Chem. Phys. 2020, 153, 034115. [Google Scholar] [CrossRef] [PubMed]
- Nimrod, G.; Fischman, S.; Austin, M.; Herman, A.; Keyes, F.; Leiderman, O.; Hargreaves, D.; Strajbl, M.; Breed, J.; Klompus, S.; et al. Computational Design of Epitope-Specific Functional Antibodies. Cell Rep. 2018, 25, 2121–2131.e5. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Hernandez, I.; Prasad, V.; Gellad, W.F. Total Costs of Chimeric Antigen Receptor T-Cell Immunotherapy. JAMA Oncol. 2018, 4, 994. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, I.; Hopkins, A.L. Fragment Screening by Surface Plasmon Resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Perriello, V.M.; Rotiroti, M.C.; Pisani, I.; Galimberti, S.; Alberti, G.; Pianigiani, G.; Ciaurro, V.; Marra, A.; Sabino, M.; Tini, V.; et al. IL-3-Zetakine Combined with a CD33 Costimulatory Receptor as a Dual CAR Approach for Safer and Selective Targeting of AML. Blood Adv. 2023, 7, 2855–2871. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Ambrosetti, F.; Jandova, Z.; Bonvin, A.M.J.J. Information-Driven Antibody–Antigen Modelling with HADDOCK. In Computer-Aided Antibody Design; Humana: New York, NY, USA, 2023; pp. 267–282. [Google Scholar]
- Ambrosetti, F.; Olsen, T.H.; Olimpieri, P.P.; Jiménez-García, B.; Milanetti, E.; Marcatilli, P.; Bonvin, A.M.J.J. ProABC-2: PRediction of AntiBody Contacts v2 and Its Application to Information-Driven Docking. Bioinformatics 2020, 36, 5107–5108. [Google Scholar] [CrossRef] [PubMed]
- Høie, M.H.; Gade, F.S.; Johansen, J.M.; Würtzen, C.; Winther, O.; Nielsen, M.; Marcatili, P. DiscoTope-3.0: Improved B-Cell Epitope Prediction Using Inverse Folding Latent Representations. Front. Immunol. 2024, 15, 1322712. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N ⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Darré, L.; Machado, M.R.; Brandner, A.F.; González, H.C.; Ferreira, S.; Pantano, S. SIRAH: A Structurally Unbiased Coarse-Grained Force Field for Proteins with Aqueous Solvation and Long-Range Electrostatics. J. Chem. Theory Comput. 2015, 11, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Darré, L.; Machado, M.R.; Dans, P.D.; Herrera, F.E.; Pantano, S. Another Coarse Grain Model for Aqueous Solvation: WAT FOUR? J. Chem. Theory Comput. 2010, 6, 3793–3807. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella Sampling. WIREs Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. THE Weighted Histogram Analysis Method for Free-energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Souaille, M.; Roux, B. Extension to the Weighted Histogram Analysis Method: Combining Umbrella Sampling with Free Energy Calculations. Comput. Phys. Commun. 2001, 135, 40–57. [Google Scholar] [CrossRef]
- Bravman, T.; Bronner, V.; Lavie, K.; Notcovich, A.; Papalia, G.A.; Myszka, D.G. Exploring “One-Shot” Kinetics and Small Molecule Analysis Using the ProteOn XPR36 Array Biosensor. Anal. Biochem. 2006, 358, 281–288. [Google Scholar] [CrossRef]
- Canziani, G.A.; Klakamp, S.; Myszka, D.G. Kinetic Screening of Antibodies from Crude Hybridoma Samples Using Biacore. Anal. Biochem. 2004, 325, 301–307. [Google Scholar] [CrossRef]
- Beeg, M.; Burti, C.; Allocati, E.; Ciafardini, C.; Banzi, R.; Nobili, A.; Caprioli, F.; Garattini, S.; Gobbi, M. Surface Plasmon Resonance Unveils Important Pitfalls of Enzyme-Linked Immunoassay for the Detection of Anti-Infliximab Antibodies in Patients’ Sera. Sci. Rep. 2021, 11, 14976. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martarelli, N.; Capurro, M.; Mansour, G.; Jahromi, R.V.; Stella, A.; Rossi, R.; Longetti, E.; Bigerna, B.; Gentili, M.; Rosseto, A.; et al. Artificial Intelligence-Powered Molecular Docking and Steered Molecular Dynamics for Accurate scFv Selection of Anti-CD30 Chimeric Antigen Receptors. Int. J. Mol. Sci. 2024, 25, 7231. https://doi.org/10.3390/ijms25137231
Martarelli N, Capurro M, Mansour G, Jahromi RV, Stella A, Rossi R, Longetti E, Bigerna B, Gentili M, Rosseto A, et al. Artificial Intelligence-Powered Molecular Docking and Steered Molecular Dynamics for Accurate scFv Selection of Anti-CD30 Chimeric Antigen Receptors. International Journal of Molecular Sciences. 2024; 25(13):7231. https://doi.org/10.3390/ijms25137231
Chicago/Turabian StyleMartarelli, Nico, Michela Capurro, Gizem Mansour, Ramina Vossoughi Jahromi, Arianna Stella, Roberta Rossi, Emanuele Longetti, Barbara Bigerna, Marco Gentili, Ariele Rosseto, and et al. 2024. "Artificial Intelligence-Powered Molecular Docking and Steered Molecular Dynamics for Accurate scFv Selection of Anti-CD30 Chimeric Antigen Receptors" International Journal of Molecular Sciences 25, no. 13: 7231. https://doi.org/10.3390/ijms25137231
APA StyleMartarelli, N., Capurro, M., Mansour, G., Jahromi, R. V., Stella, A., Rossi, R., Longetti, E., Bigerna, B., Gentili, M., Rosseto, A., Rossi, R., Cencini, C., Emiliani, C., Martino, S., Beeg, M., Gobbi, M., Tiacci, E., Falini, B., Morena, F., & Perriello, V. M. (2024). Artificial Intelligence-Powered Molecular Docking and Steered Molecular Dynamics for Accurate scFv Selection of Anti-CD30 Chimeric Antigen Receptors. International Journal of Molecular Sciences, 25(13), 7231. https://doi.org/10.3390/ijms25137231