
Anti-Idiotypic VHHs and VHH-CAR-T Cells to Tackle Multiple Myeloma: Different Applications Call for Different Antigen-Binding Moieties
 , , , , and
, , , , and 
Abstract

1. Introduction
2. Results
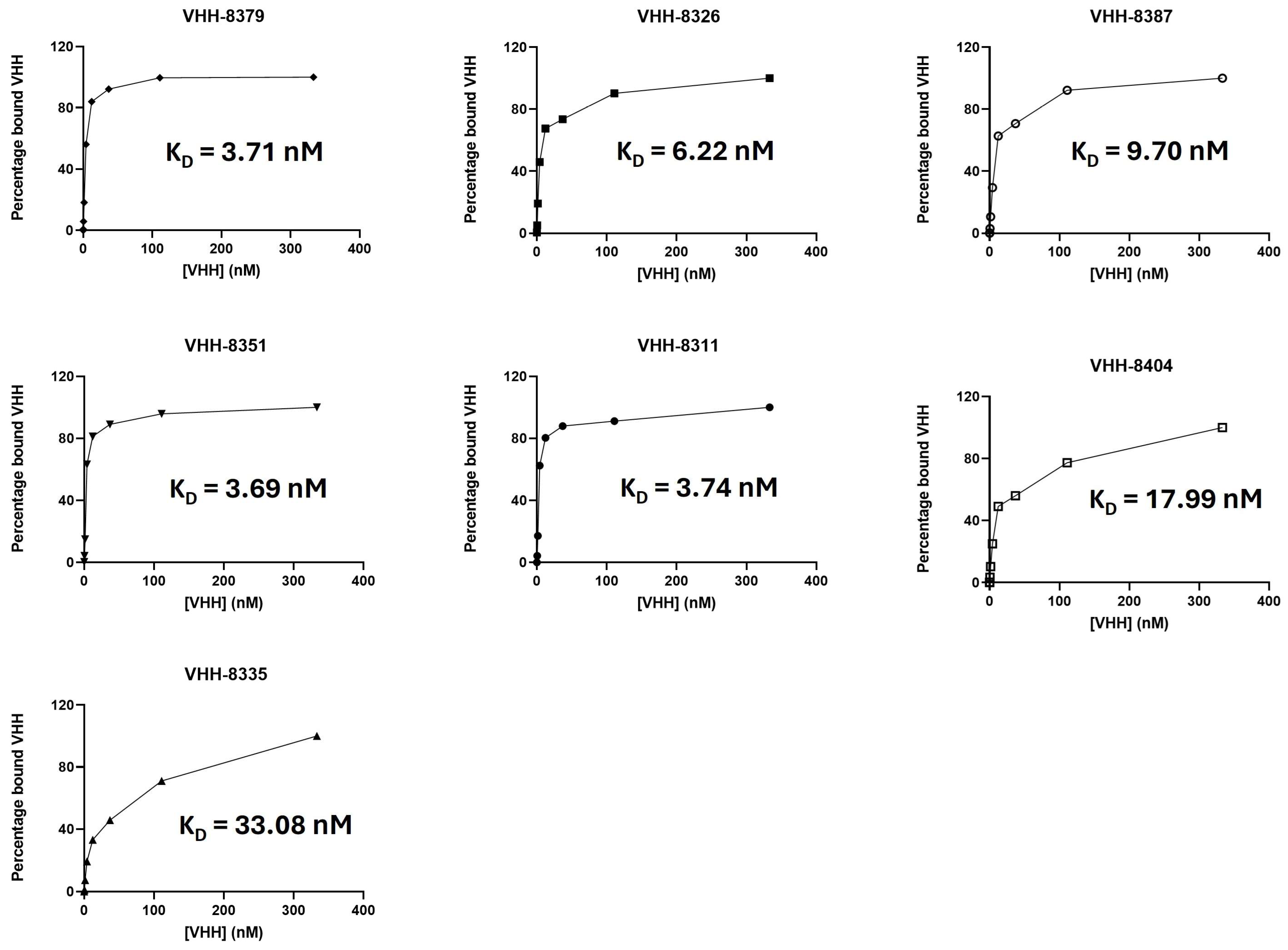
2.1. High-Affinity VHHs Are the Most Capable In Vivo Tracer Moieties
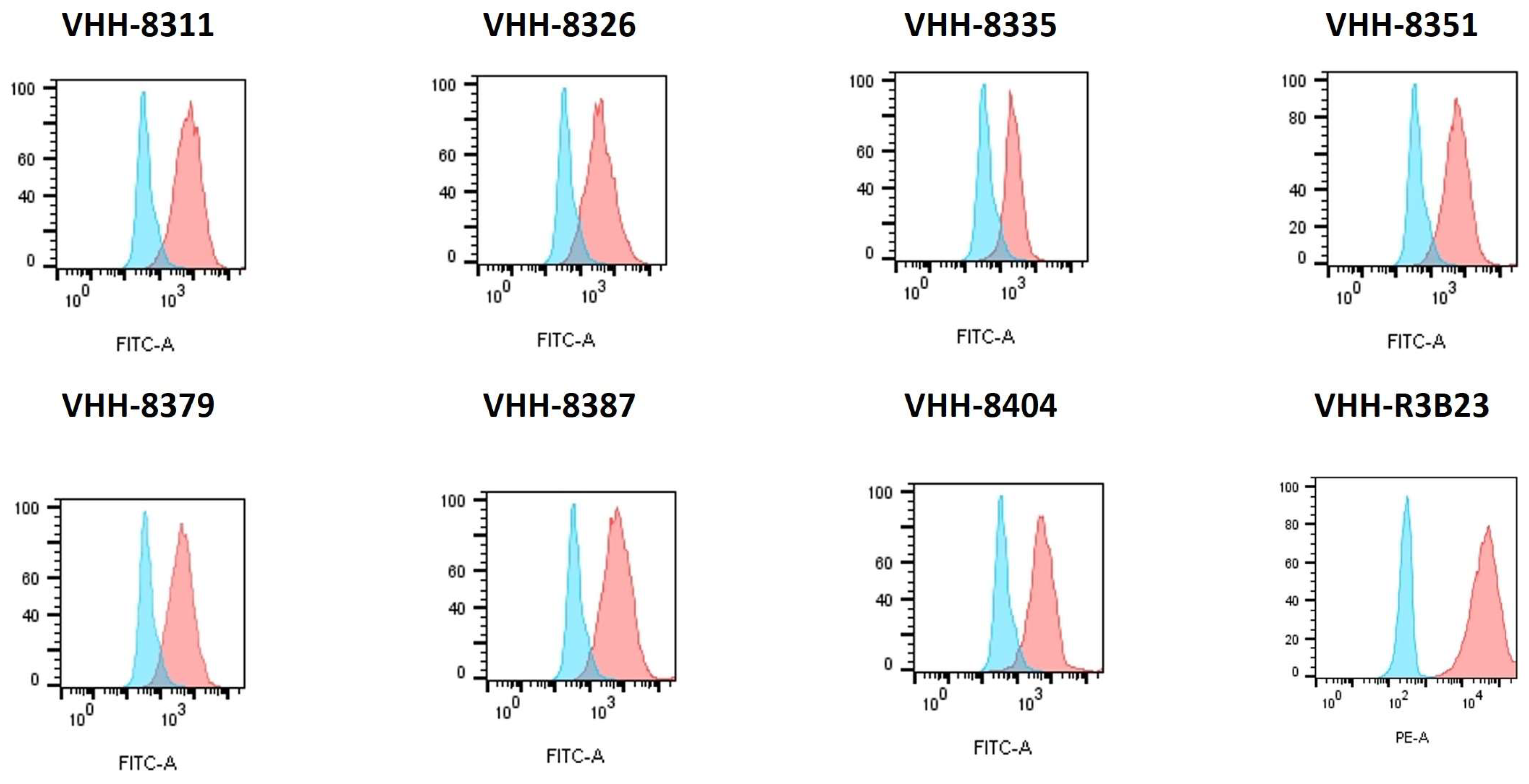
2.2. Reporter T Cells Can Be Stably Transduced with Different Correctly Folded Anti-Idiotypic VHH-CARs
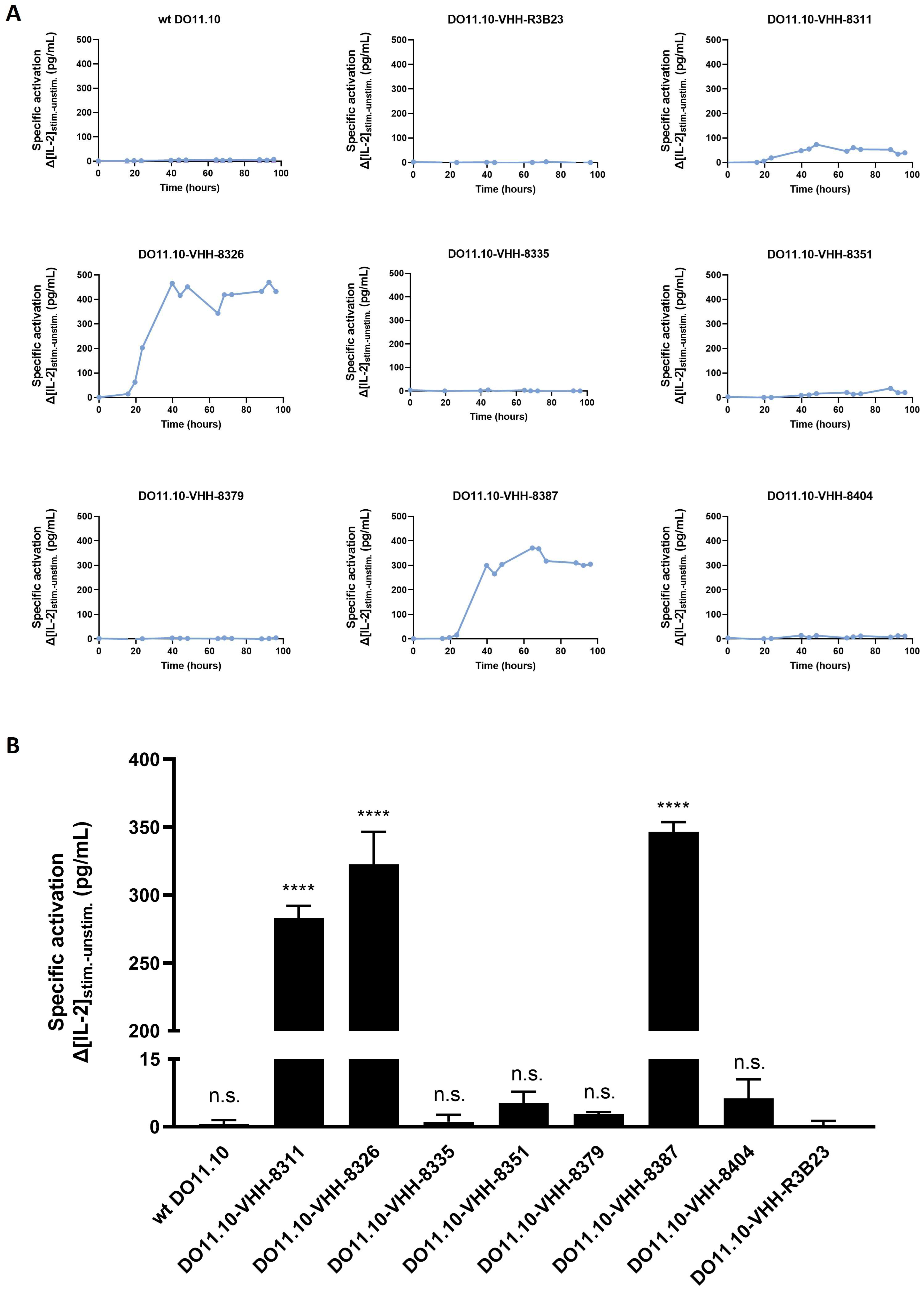
2.3. VHH-Based Anti-Idiotypic CARs Can Induce T Cell Signaling in Reporter T Cells, but the Majority of VHH-CARs Fail
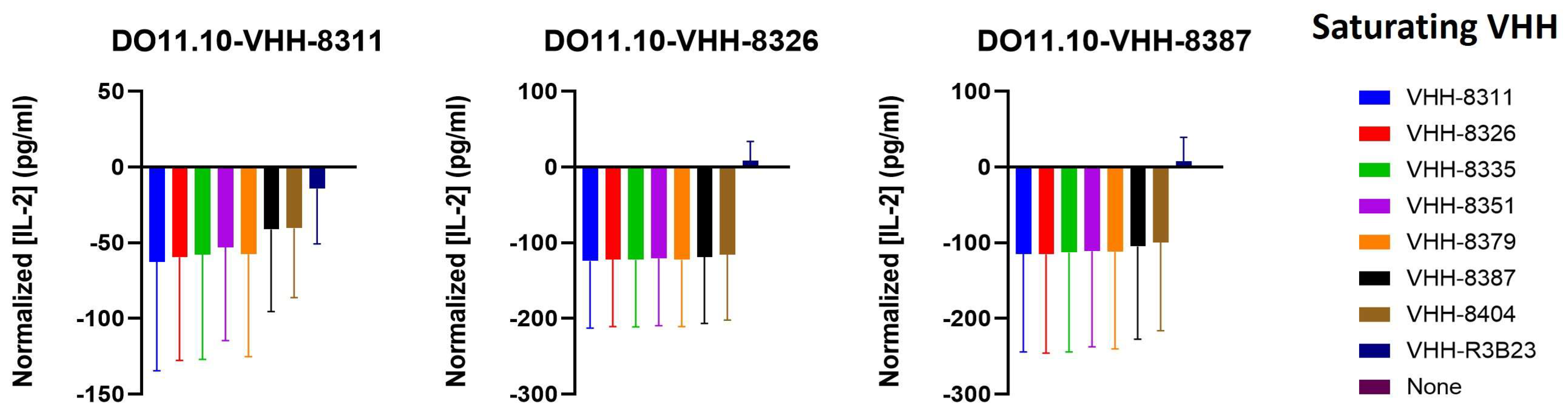
2.4. The VHH-Targeted Epitope Is Non-Determinant for CAR-T Cell Activation
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. VHHs
5.3. Assessment of VHH Binding to Cells and Affinity Determination
5.4. Design and Cloning of a Murine Lentiviral VHH-CAR Transfer Plasmid
5.5. LV Production and T Cell Transduction
5.6. Evaluation of Transduction Efficiency
5.7. Assessment of T Cell Activation by Different VHH-CARs
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. (In English) [Google Scholar] [CrossRef] [PubMed]
- Padala, S.A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, Staging, and Management of Multiple Myeloma. Med. Sci. 2021, 9, 3. (In English) [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Jha, S.B.; Rivera, A.P.; Flores Monar, G.V.; Islam, H.; Puttagunta, S.M.; Islam, R.; Sange, I. Multiple Myeloma and Renal Failure: Mechanisms, Diagnosis, and Management. Cureus 2022, 14, e22585. (In English) [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhuang, J. Pathophysiology and therapeutic advances in myeloma bone disease. Chronic Dis. Transl. Med. 2022, 8, 264–270. (In English) [Google Scholar] [CrossRef] [PubMed]
- Banaszkiewicz, M.; Małyszko, J.; Vesole, D.H.; Woziwodzka, K.; Jurczyszyn, A.; Żórawski, M.; Krzanowski, M.; Batko, K.; Kuźniewski, M.; Krzanowska, K. New Biomarkers of Ferric Management in Multiple Myeloma and Kidney Disease-Associated Anemia. J. Clin. Med. 2019, 8, 1828. (In English) [Google Scholar] [CrossRef] [PubMed]
- Mittelman, M. The implications of anemia in multiple myeloma. Clin. Lymphoma 2003, 4 (Suppl. 1), S23–S29. (In English) [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 1086–1107. (In English) [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 10, 94. (In English) [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.; Grieb, N.; Platzbecker, U.; Vucinic, V.; Merz, M. CAR T cell therapy in multiple myeloma, where are we now and where are we heading for? Eur. J. Haematol. 2024, 112, 19–27. (In English) [Google Scholar] [CrossRef]
- Gazeau, N.; Beauvais, D.; Yakoub-Agha, I.; Mitra, S.; Campbell, T.B.; Facon, T.; Manier, S. Effective anti-BCMA retreatment in multiple myeloma. Blood Adv. 2021, 5, 3016–3020. (In English) [Google Scholar] [CrossRef]
- Rasche, L.; Hudecek, M.; Einsele, H. CAR T-cell therapy in multiple myeloma: Mission accomplished? Blood 2024, 143, 305–310. (In English) [Google Scholar] [CrossRef] [PubMed]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. (In English) [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. (In English) [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. (In English) [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Masutani, M.; Tachibana, M.; Okada, N. Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem. Biophys. Res. Commun. 2020, 527, 350–357. (In English) [Google Scholar] [CrossRef]
- Zhu, X.; Li, Q. Mechanisms of CAR T cell exhaustion and current counteraction strategies. Front. Cell Dev. Biol. 2022, 10, 1034257. (In English) [Google Scholar] [CrossRef] [PubMed]
- Hanssens, H.; Meeus, F.; De Veirman, K.; Breckpot, K.; Devoogdt, N. The antigen-binding moiety in the driver’s seat of CARs. Med. Res. Rev. 2022, 42, 306–342. (In English) [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. (In English) [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Pont, M.J.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Libby, E.N.; Coffey, D.G.; Tuazon, S.A.; Wood, B.; Gooley, T.; et al. γ-Secretase inhibitor in combination with BCMA chimeric antigen receptor T-cell immunotherapy for individuals with relapsed or refractory multiple myeloma: A phase 1, first-in-human trial. Lancet Oncol. 2023, 24, 811–822. (In English) [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. (In English) [Google Scholar] [CrossRef]
- Da Vià, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 2021, 27, 616–619. (In English) [Google Scholar] [CrossRef]
- Nguyen, V.K.; Hamers, R.; Wyns, L.; Muyldermans, S. Camel heavy-chain antibodies: Diverse germline V(H)H and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 2000, 19, 921–930. (In English) [Google Scholar] [CrossRef] [PubMed]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. (In English) [Google Scholar] [CrossRef] [PubMed]
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity Risk Profile of Nanobodies. Front. Immunol. 2021, 12, 632687. (In English) [Google Scholar] [CrossRef] [PubMed]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. (In English) [Google Scholar] [CrossRef] [PubMed]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 868. (In English) [Google Scholar] [CrossRef] [PubMed]
- Parkin, J.; Cohen, B. An overview of the immune system. Lancet 2001, 357, 1777–1789. (In English) [Google Scholar] [CrossRef]
- Bogen, B.; Ruffini, P.A.; Corthay, A.; Fredriksen, A.B.; Frøyland, M.; Lundin, K.; Røsjø, E.; Thompson, K.; Massaia, M. Idiotype-specific immunotherapy in multiple myeloma: Suggestions for future directions of research. Haematologica 2006, 91, 941–948. (In English) [Google Scholar]
- Stevenson, F.K.; Stevenson, G.T. Therapeutic strategies for B cell malignancies involving idiotype-anti-idiotype interactions. Int. Rev. Immunol. 1986, 1, 303–333. (In English) [Google Scholar] [CrossRef]
- Ocqueteau, M.; San Miguel, J.F.; González, M.; Almeida, J.; Orfao, A. Do myelomatous plasma cells really express surface immunoglobulins? Haematologica 1996, 81, 460–463. (In English) [Google Scholar]
- Ghosh, N.; Matsui, W. Cancer stem cells in multiple myeloma. Cancer Lett. 2009, 277, 1–7. (In English) [Google Scholar] [CrossRef]
- Wen, Y.J.; Barlogie, B.; Yi, Q. Idiotype-specific cytotoxic T lymphocytes in multiple myeloma: Evidence for their capacity to lyse autologous primary tumor cells. Blood 2001, 97, 1750–1755. (In English) [Google Scholar] [CrossRef] [PubMed]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019, 134, 30–43. (In English) [Google Scholar] [CrossRef]
- Puttemans, J.; Stijlemans, B.; Keyaerts, M.; Vander Meeren, S.; Renmans, W.; Fostier, K.; Debie, P.; Hanssens, H.; Rodak, M.; Pruszynski, M.; et al. The Road to Personalized Myeloma Medicine: Patient-specific Single-domain Antibodies for Anti-idiotypic Radionuclide Therapy. Mol. Cancer Ther. 2022, 21, 159–169. (In English) [Google Scholar] [CrossRef] [PubMed]
- Minnie, S.A.; Hill, G.R. Immunotherapy of multiple myeloma. J. Clin. Investig. 2020, 130, 1565–1575. (In English) [Google Scholar] [CrossRef]
- Curti, A.; Tosi, P.; Comoli, P.; Terragna, C.; Ferri, E.; Cellini, C.; Massaia, M.; D’Addio, A.; Giudice, V.; Di Bello, C.; et al. Phase I/II clinical trial of sequential subcutaneous and intravenous delivery of dendritic cell vaccination for refractory multiple myeloma using patient-specific tumour idiotype protein or idiotype (VDJ)-derived class I-restricted peptides. Br. J. Haematol. 2007, 139, 415–424. (In English) [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.; Szmania, S.; Freeman, J.; Qian, J.; Rosen, N.A.; Viswamitra, S.; Cottler-Fox, M.; Barlogie, B.; Tricot, G.; van Rhee, F. Optimizing dendritic cell-based immunotherapy in multiple myeloma: Intranodal injections of idiotype-pulsed CD40 ligand-matured vaccines led to induction of type-1 and cytotoxic T-cell immune responses in patients. Br. J. Haematol. 2010, 150, 554–564. (In English) [Google Scholar] [CrossRef] [PubMed]
- Hansson, L.; Abdalla, A.O.; Moshfegh, A.; Choudhury, A.; Rabbani, H.; Nilsson, B.; Osterborg, A.; Mellstedt, H. Long-term idiotype vaccination combined with interleukin-12 (IL-12), or IL-12 and granulocyte macrophage colony-stimulating factor, in early-stage multiple myeloma patients. Clin. Cancer Res. 2007, 13, 1503–1510. (In English) [Google Scholar] [CrossRef]
- Reichardt, V.L.; Milazzo, C.; Brugger, W.; Einsele, H.; Kanz, L.; Brossart, P. Idiotype vaccination of multiple myeloma patients using monocyte-derived dendritic cells. Haematologica 2003, 88, 1139–1149. (In English) [Google Scholar] [PubMed]
- Reichardt, V.L.; Okada, C.Y.; Liso, A.; Benike, C.J.; Stockerl-Goldstein, K.E.; Engleman, E.G.; Blume, K.G.; Levy, R. Idiotype vaccination using dendritic cells after autologous peripheral blood stem cell transplantation for multiple myeloma—A feasibility study. Blood 1999, 93, 2411–2419. (In English) [Google Scholar] [CrossRef]
- Massaia, M.; Borrione, P.; Battaglio, S.; Mariani, S.; Beggiato, E.; Napoli, P.; Voena, C.; Bianchi, A.; Coscia, M.; Besostri, B.; et al. Idiotype vaccination in human myeloma: Generation of tumor-specific immune responses after high-dose chemotherapy. Blood 1999, 94, 673–683. (In English) [Google Scholar] [CrossRef]
- Rhee, F. Idiotype vaccination strategies in myeloma: How to overcome a dysfunctional immune system. Clin. Cancer Res. 2007, 13, 1353–1355. (In English) [Google Scholar] [CrossRef] [PubMed]
- Ruffini, P.A.; Neelapu, S.S.; Kwak, L.W.; Biragyn, A. Idiotypic vaccination for B-cell malignancies as a model for therapeutic cancer vaccines: From prototype protein to second generation vaccines. Haematologica 2002, 87, 989–1001. [Google Scholar] [PubMed]
- Lemaire, M.; D’Huyvetter, M.; Lahoutte, T.; Van Valckenborgh, E.; Menu, E.; De Bruyne, E.; Kronenberger, P.; Wernery, U.; Muyldermans, S.; Devoogdt, N.; et al. Imaging and radioimmunotherapy of multiple myeloma with anti-idiotypic Nanobodies. Leukemia 2014, 28, 444–447. (In English) [Google Scholar] [CrossRef] [PubMed]
- D’Huyvetter, M.; Xavier, C.; Caveliers, V.; Lahoutte, T.; Muyldermans, S.; Devoogdt, N. Radiolabeled nanobodies as theranostic tools in targeted radionuclide therapy of cancer. Expert. Opin. Drug Deliv. 2014, 11, 1939–1954. (In English) [Google Scholar] [CrossRef] [PubMed]
- Asosingh, K.; Radl, J.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. The 5TMM series: A useful in vivo mouse model of human multiple myeloma. Hematol. J. 2000, 1, 351–356. (In English) [Google Scholar] [CrossRef]
- Vanderkerken, K.; De Raeve, H.; Goes, E.; Van Meirvenne, S.; Radl, J.; Van Riet, I.; Thielemans, K.; Van Camp, B. Organ involvement and phenotypic adhesion profile of 5T2 and 5T33 myeloma cells in the C57BL/KaLwRij mouse. Br. J. Cancer 1997, 76, 451–460. (In English) [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. (In English) [Google Scholar] [CrossRef]
- Khan, A.N.; Chowdhury, A.; Karulkar, A.; Jaiswal, A.K.; Banik, A.; Asija, S.; Purwar, R. Immunogenicity of CAR-T Cell Therapeutics: Evidence, Mechanism and Mitigation. Front. Immunol. 2022, 13, 886546. (In English) [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. (In English) [Google Scholar] [CrossRef]
- Mishra, A.K.; Gupta, A.; Dagar, G.; Das, D.; Chakraborty, A.; Haque, S.; Prasad, C.P.; Singh, A.; Bhat, A.A.; Macha, M.A.; et al. CAR-T-Cell Therapy in Multiple Myeloma: B-Cell Maturation Antigen (BCMA) and Beyond. Vaccines 2023, 11, 1721. (In English) [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. (In English) [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.; Xia, J.; Li, P.; Cao, J.; Pan, B.; Tan, X.; Li, H.; Qi, K.; Wang, X.; et al. Humoral immune reconstitution after anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. Blood Adv. 2021, 5, 5290–5299. (In English) [Google Scholar] [CrossRef]
- Kumar, L.; Gundu, N.; Kancharia, H.; Sahoo, R.K.; Malik, P.S.; Sharma, A.; Gupta, R.; Sharma, O.; Biswas, A.; Kumar, R.; et al. Multiple Myeloma-Effect of Induction Therapy on Transplant Outcomes. Clin. Lymphoma Myeloma Leuk. 2021, 21, 80–90.e5. (In English) [Google Scholar] [CrossRef] [PubMed]
- Kaddoura, M.; Binder, M.; Dingli, D.; Buadi, F.K.; Lacy, M.Q.; Gertz, M.A.; Dispenzieri, A.; Kapoor, P.; Hwa, L.; Fonder, A.; et al. Impact of achieving a complete response to initial therapy of multiple myeloma and predictors of subsequent outcome. Am. J. Hematol. 2022, 97, 267–273. (In English) [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. (In English) [Google Scholar] [CrossRef]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. (In English) [Google Scholar] [CrossRef]
- Medina-Herrera, A.; Sarasquete, M.E.; Jiménez, C.; Puig, N.; García-Sanz, R. Minimal Residual Disease in Multiple Myeloma: Past, Present, and Future. Cancers 2023, 15, 3687. (In English) [Google Scholar] [CrossRef] [PubMed]
- Gil, D.; Schrum, A.G. Strategies to stabilize compact folding and minimize aggregation of antibody-based fragments. Adv. Biosci. Biotechnol. 2013, 4, 73–84. (In English) [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yang, S.S.; Sun, Y.; Wu, W.; Liu, Y.F.; Zhuang, Y.; Zhang, W.; Weng, X.Q.; Wu, J.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. (In English) [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. (In English) [Google Scholar] [CrossRef]
- Strassl, I.; Podar, K. The preclinical discovery and clinical development of ciltacabtagene autoleucel (Cilta-cel) for the treatment of multiple myeloma. Expert. Opin. Drug Discov. 2024, 19, 377–391. (In English) [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.C.; Ma, B.; Trinklein, N.D.; Schellenberger, U.; Osborn, M.J.; Ouisse, L.H.; Boudreau, A.; Davison, L.M.; Harris, K.E.; Ugamraj, H.S.; et al. Multispecific Antibody Development Platform Based on Human Heavy Chain Antibodies. Front. Immunol. 2018, 9, 3037. (In English) [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. A guide to: Generation and design of nanobodies. FEBS J. 2021, 288, 2084–2102. (In English) [Google Scholar] [CrossRef]
- Chigoho, D.M.; Bridoux, J.; Hernot, S. Reducing the renal retention of low- to moderate-molecular-weight radiopharmaceuticals. Curr. Opin. Chem. Biol. 2021, 63, 219–228. (In English) [Google Scholar] [CrossRef] [PubMed]
- Lecocq, Q.; De Vlaeminck, Y.; Hanssens, H.; D’Huyvetter, M.; Raes, G.; Goyvaerts, C.; Keyaerts, M.; Devoogdt, N.; Breckpot, K. Theranostics in immuno-oncology using nanobody derivatives. Theranostics 2019, 9, 7772–7791. (In English) [Google Scholar] [CrossRef] [PubMed]
- De Vos, J.; Devoogdt, N.; Lahoutte, T.; Muyldermans, S. Camelid single-domain antibody-fragment engineering for (pre)clinical in vivo molecular imaging applications: Adjusting the bullet to its target. Expert. Opin. Biol. Ther. 2013, 13, 1149–1160. (In English) [Google Scholar] [CrossRef] [PubMed]
- Roy, B.M.; Zhukov, D.V.; Maynard, J.A. Flanking residues are central to DO11.10 T cell hybridoma stimulation by ovalbumin 323–339. PLoS ONE 2012, 7, e47585. (In English) [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Lauwereys, M.; Galleni, M.; Matagne, A.; Frère, J.M.; Kinne, J.; Wyns, L.; Muyldermans, S. Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob. Agents Chemother. 2001, 45, 2807–2812. (In English) [Google Scholar] [CrossRef] [PubMed]
- Vincke, C.; Gutiérrez, C.; Wernery, U.; Devoogdt, N.; Hassanzadeh-Ghassabeh, G.; Muyldermans, S. Generation of single domain antibody fragments derived from camelids and generation of manifold constructs. Methods Mol. Biol. 2012, 907, 145–176. (In English) [Google Scholar] [CrossRef]
- Breckpot, K.; Dullaers, M.; Bonehill, A.; van Meirvenne, S.; Heirman, C.; de Greef, C.; van der Bruggen, P.; Thielemans, K. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J. Gene Med. 2003, 5, 654–667. (In English) [Google Scholar] [CrossRef]
- Lecocq, Q.; Zeven, K.; De Vlaeminck, Y.; Martens, S.; Massa, S.; Goyvaerts, C.; Raes, G.; Keyaerts, M.; Breckpot, K.; Devoogdt, N. Noninvasive Imaging of the Immune Checkpoint LAG-3 Using Nanobodies, from Development to Pre-Clinical Use. Biomolecules 2019, 9, 548. (In English) [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VHH | ka (M−1s−1) | kd (s−1) | KD(M) | Specific In Vivo Uptake |
|---|---|---|---|---|
| 8379 * | 2.83 × 105 | 5.48 × 10−4 | 1.94 × 10−9 | High |
| 8326 | 9.84 × 105 | 10.6 × 10−4 | 1.08 × 10−9 | High |
| 8387 | 3.18 × 105 | 12.8 × 10−4 | 4.03 × 10−9 | Mediocre |
| 8351 | 4.66 × 105 | 5.93 × 10−4 | 1.27 × 10−9 | Mediocre |
| 8311 | 6.27 × 105 | 2.39 × 10−9 | 0.38 × 10−9 | Low |
| 8404 | 2.67 × 105 | 14.6 × 10−4 | 5.47 × 10−9 | Low |
| 8335 | 10.6 × 105 | 23.1 × 10−4 | 2.18 × 10−9 | NA ** |
| R3B23 | Not binding | Not binding | Not binding | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanssens, H.; Meeus, F.; Gesquiere, E.L.; Puttemans, J.; De Vlaeminck, Y.; De Veirman, K.; Breckpot, K.; Devoogdt, N. Anti-Idiotypic VHHs and VHH-CAR-T Cells to Tackle Multiple Myeloma: Different Applications Call for Different Antigen-Binding Moieties. Int. J. Mol. Sci. 2024, 25, 5634. https://doi.org/10.3390/ijms25115634
Hanssens H, Meeus F, Gesquiere EL, Puttemans J, De Vlaeminck Y, De Veirman K, Breckpot K, Devoogdt N. Anti-Idiotypic VHHs and VHH-CAR-T Cells to Tackle Multiple Myeloma: Different Applications Call for Different Antigen-Binding Moieties. International Journal of Molecular Sciences. 2024; 25(11):5634. https://doi.org/10.3390/ijms25115634
Chicago/Turabian StyleHanssens, Heleen, Fien Meeus, Emma L. Gesquiere, Janik Puttemans, Yannick De Vlaeminck, Kim De Veirman, Karine Breckpot, and Nick Devoogdt. 2024. "Anti-Idiotypic VHHs and VHH-CAR-T Cells to Tackle Multiple Myeloma: Different Applications Call for Different Antigen-Binding Moieties" International Journal of Molecular Sciences 25, no. 11: 5634. https://doi.org/10.3390/ijms25115634
APA StyleHanssens, H., Meeus, F., Gesquiere, E. L., Puttemans, J., De Vlaeminck, Y., De Veirman, K., Breckpot, K., & Devoogdt, N. (2024). Anti-Idiotypic VHHs and VHH-CAR-T Cells to Tackle Multiple Myeloma: Different Applications Call for Different Antigen-Binding Moieties. International Journal of Molecular Sciences, 25(11), 5634. https://doi.org/10.3390/ijms25115634