
ProPept-MT: A Multi-Task Learning Model for Peptide Feature Prediction



Abstract
:1. Introduction
2. Results
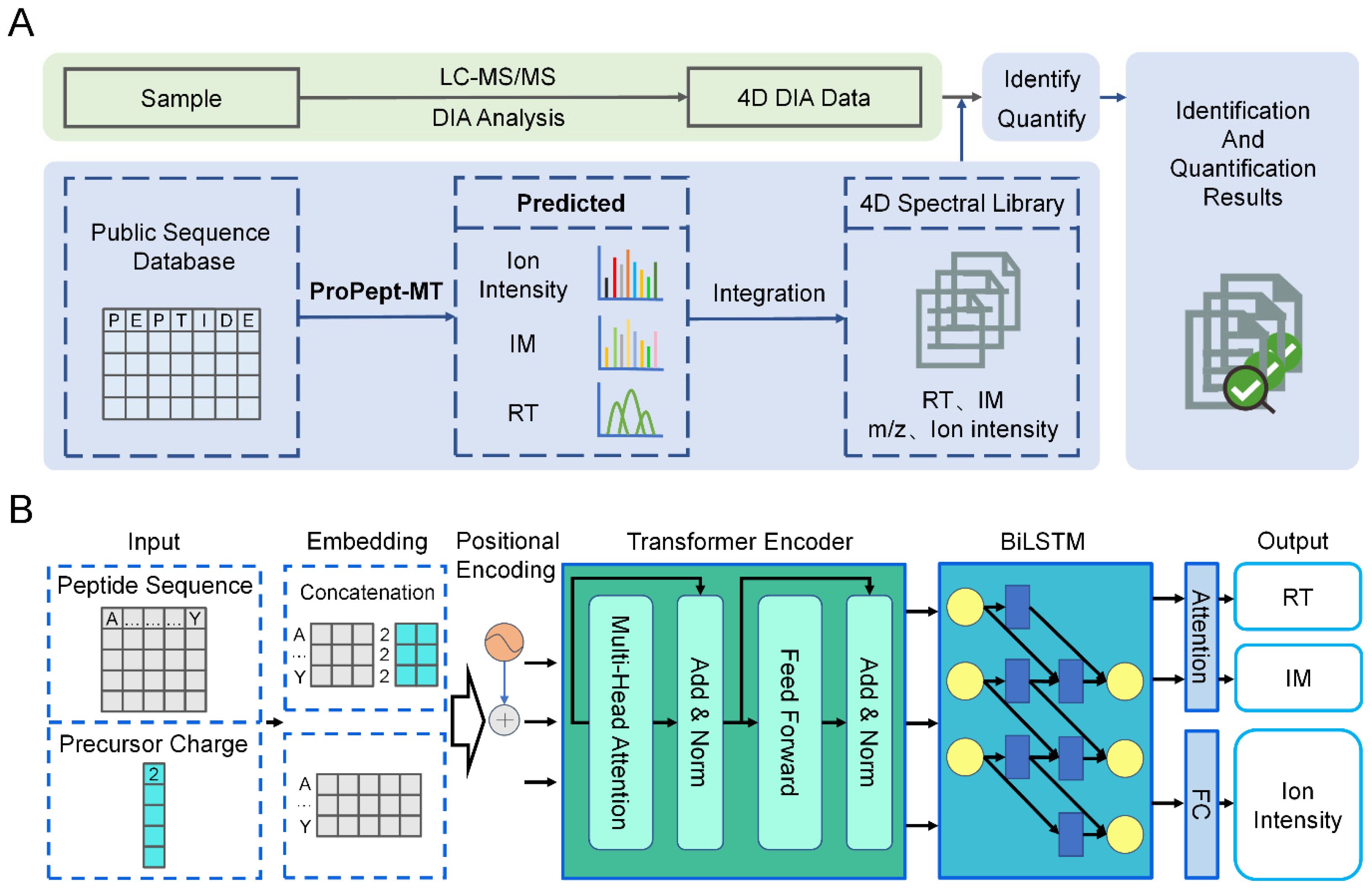
2.1. Development of Model Structure
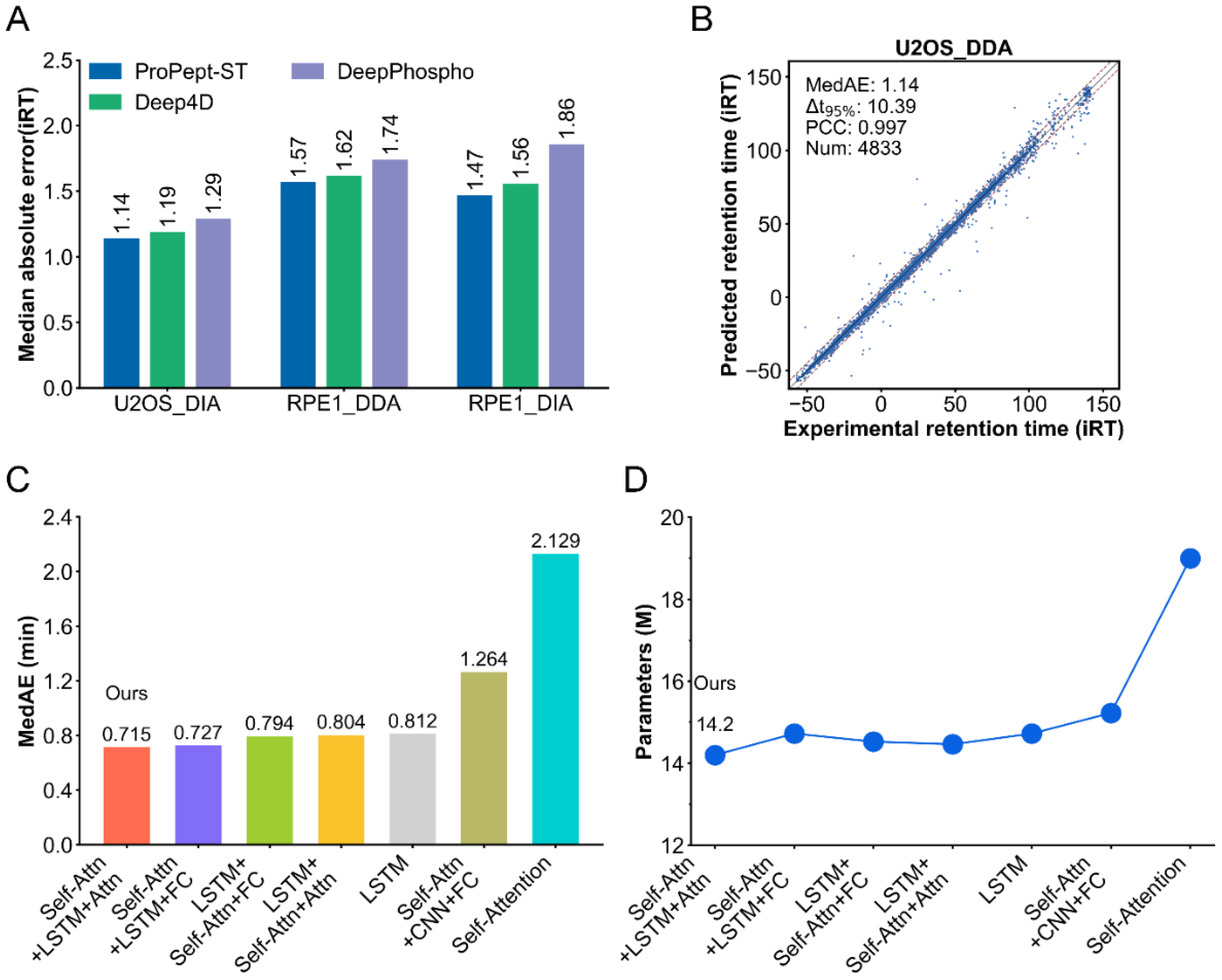
2.2. Performance of ProPept-ST in Predicting Retention Time
2.3. Ablation Studies
2.4. Performance of ProPept-MT on Benchmark Datasets
2.5. Performance Comparison between ProPept-MT and Other Models
3. Materials and Methods
3.1. Dataset Collection and Pre-Processing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Name | Species | Instrument | Peptides | Identifier |
|---|---|---|---|---|
| H1_DDA | Human | timsTOF Pro | 64,358 | PXD041421 [45] |
| H2_DIA | Human | timsTOF Pro | 125,360 | PXD041391 [45] |
| H3_DIAp | Human | timsTOF Pro | 42,351 | PXD034709 [46] |
| H4_DDAp | Human | timsTOF Pro | 31,599 | PXD034709 |
| H5_DDAp | Human | timsTOF Pro | 42,677 | PXD027834 [47] |
| H6_DDAp | Human | timsTOF Pro | 16,784 | PXD042842 [48] |
| H7_DDAp | Human | timsTOF Pro 2 | 9495 | PXD043026 [49] |
| M1_DDAp | Mouse | timsTOF Pro | 12,132 | PXD028051 [50] |
| M2_DDAp | Mouse | timsTOF Pro 2 | 8296 | PXD043026 |
3.2. The Model Architecture of ProPept-MT
3.3. Loss Function
3.4. Model Training
| Algorithm 1. Nash-MTL |
| Input: - initial parameter vector, –differentiable loss functions, –learning rate |
| Output: |
| for t = 1,…, T do |
| Compute task gradients |
| Set the matrix with columns |
| Solve for to obtain |
| Update the parameters |
| end for |
| return |
3.5. Evaluation Metrics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, 016717. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef] [PubMed]
- Ting, Y.S.; Egertson, J.D.; Payne, S.H.; Kim, S.; MacLean, B.; Kall, L.; Aebersold, R.; Smith, R.D.; Noble, W.S.; MacCoss, M.J. Peptide-Centric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data. Mol. Cell. Proteom. 2015, 14, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Searle, B.C.; Swearingen, K.E.; Barnes, C.A.; Schmidt, T.; Gessulat, S.; Kuster, B.; Wilhelm, M. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 2020, 11, 1548. [Google Scholar] [CrossRef]
- Xing, X.; Li, X.; Wei, C.; Zhang, Z.; Liu, O.; Xie, S.; Chen, H.; Quan, S.; Wang, C.; Yang, X.; et al. DP-GAN+B: A lightweight generative adversarial network based on depthwise separable convolutions for generating CT volumes. Comput. Biol. Med. 2024, 174, 108393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, B.; Yang, H.; Zhao, J.; Zhang, Y.; Gao, Y.; Monteiro, O.; Zhang, K.; Liu, B.; Wang, S. Potential rapid intraoperative cancer diagnosis using dynamic full-field optical coherence tomography and deep learning: A prospective cohort study in breast cancer patients. Sci. Bull. 2024, 69, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Z.; Yu, J.; Gao, Y.; Liu, M. Multi-scale nested UNet with transformer for colorectal polyp segmentation. J. Appl. Clin. Med. Phys. 2024, 25, e14351. [Google Scholar] [PubMed]
- Hu, H.; Feng, Z.; Lin, H.; Cheng, J.; Lyu, J.; Zhang, Y.; Zhao, J.; Xu, F.; Lin, T.; Zhao, Q.; et al. Gene function and cell surface protein association analysis based on single-cell multiomics data. Comput. Biol. Med. 2023, 157, 106733. [Google Scholar] [CrossRef]
- Xu, F.; Li, X.; Wu, R.; Qi, H.; Jin, J.; Liu, Z.; Wu, Y.; Lin, H.; Shen, C.; Shuai, J. Incoherent feedforward loop dominates the robustness and tunability of necroptosis biphasic, emergent, and coexistent dynamics. Fundam. Res. 2024. [Google Scholar] [CrossRef]
- Li, X.; Chen, G.; Zhou, X.; Peng, X.; Li, M.; Chen, D.; Yu, H.; Shi, W.; Zhang, C.; Li, Y.; et al. Roles of Akirin1 in early prediction and treatment of graft kidney ischemia–reperfusion injury. Smart Med. 2024, 3, e20230043. [Google Scholar] [CrossRef]
- Zhu, F.; Niu, Q.; Li, X.; Zhao, Q.; Su, H.; Shuai, J. FM-FCN: A Neural Network with Filtering Modules for Accurate Vital Signs Extraction. Research 2024, 7, 0361. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhong, C.Q.; Li, X.; Guo, H.; Li, Y.; Gao, M.; Yu, R.; Liu, X.; Zhang, F.; Guo, D.; et al. Dear-DIA(XMBD): Deep Autoencoder Enables Deconvolution of Data-Independent Acquisition Proteomics. Research 2023, 6, 0179. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, Q.; Guo, H.; Shuai, S.C.; Cheng, J.; Liu, L.; Shuai, J. AttnPep: A Self-Attention-Based Deep Learning Method for Peptide Identification in Shotgun Proteomics. J. Proteome Res. 2024, 23, 834–843. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Guo, H.; Li, Y.; He, G.; Li, X.; Shuai, J. SeFilter-DIA: Squeeze-and-Excitation Network for Filtering High-Confidence Peptides of Data-Independent Acquisition Proteomics. Interdiscip Sci. 2024. [Google Scholar] [CrossRef]
- Jiang, T.T.; Fang, L.; Wang, K. Deciphering “the language of nature”: A transformer-based language model for deleterious mutations in proteins. Innovation 2023, 4, 100487. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zeng, X.; Zhao, Y.; Chen, R. AlphaFold2 and its applications in the fields of biology and medicine. Signal Transduct. Target. Ther. 2023, 8, 115. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.F.; Zhou, X.X.; Zhou, W.J.; Chi, H.; Zhan, J.; He, S.M. MS/MS Spectrum Prediction for Modified Peptides Using pDeep2 Trained by Transfer Learning. Anal. Chem. 2019, 91, 9724–9731. [Google Scholar] [CrossRef]
- Tiwary, S.; Levy, R.; Gutenbrunner, P.; Salinas Soto, F.; Palaniappan, K.K.; Deming, L.; Berndl, M.; Brant, A.; Cimermancic, P.; Cox, J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519–525. [Google Scholar] [CrossRef]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Shen, C.; Lin, Y.; Yang, P.; Qiao, L. In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics. Nat. Commun. 2020, 11, 146. [Google Scholar] [CrossRef]
- Zhou, X.X.; Zeng, W.F.; Chi, H.; Luo, C.; Liu, C.; Zhan, J.; He, S.M.; Zhang, Z. pDeep: Predicting MS/MS Spectra of Peptides with Deep Learning. Anal. Chem. 2017, 89, 12690–12697. [Google Scholar] [CrossRef]
- Lou, R.; Liu, W.; Li, R.; Li, S.; He, X.; Shui, W. DeepPhospho accelerates DIA phosphoproteome profiling through in silico library generation. Nat. Commun. 2021, 12, 6685. [Google Scholar] [CrossRef]
- Guan, S.; Moran, M.F.; Ma, B. Prediction of LC-MS/MS Properties of Peptides from Sequence by Deep Learning. Mol. Cell. Proteom. 2019, 18, 2099–2107. [Google Scholar] [CrossRef]
- Lin, Y.M.; Chen, C.T.; Chang, J.M. MS2CNN: Predicting MS/MS spectrum based on protein sequence using deep convolutional neural networks. BMC Genom. 2019, 20, 906. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Li, S.; Wang, L.; Ye, Y.; Tang, H. Full-Spectrum Prediction of Peptides Tandem Mass Spectra using Deep Neural Network. Anal. Chem. 2020, 92, 4275–4283. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, P.; Wan, Q.; Ruan, X.; Wu, P.; Hao, Y.; Zhang, Z.; Sun, J.; Nie, W.; Chen, S. High-Coverage Four-Dimensional Data-Independent Acquisition Proteomics and Phosphoproteomics Enabled by Deep Learning-Driven Multidimensional Predictions. Anal. Chem. 2023, 95, 7495–7502. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural. Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural. Inf. Process. Syst. 2017, 30. [Google Scholar] [CrossRef]
- Distler, U.; Kuharev, J.; Navarro, P.; Levin, Y.; Schild, H.; Tenzer, S. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 2014, 11, 167–170. [Google Scholar] [CrossRef]
- Demichev, V.; Szyrwiel, L.; Yu, F.; Teo, G.C.; Rosenberger, G.; Niewienda, A.; Ludwig, D.; Decker, J.; Kaspar-Schoenefeld, S.; Lilley, K.S.; et al. dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 2022, 13, 3944. [Google Scholar] [CrossRef] [PubMed]
- Helm, D.; Vissers, J.P.; Hughes, C.J.; Hahne, H.; Ruprecht, B.; Pachl, F.; Grzyb, A.; Richardson, K.; Wildgoose, J.; Maier, S.K.; et al. Ion mobility tandem mass spectrometry enhances performance of bottom-up proteomics. Mol. Cell. Proteom. 2014, 13, 3709–3715. [Google Scholar] [CrossRef] [PubMed]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Navon, A.; Shamsian, A.; Achituve, I.; Maron, H.; Kawaguchi, K.; Chechik, G.; Fetaya, E. Multi-task learning as a bargaining game. arXiv 2022, arXiv:2202.01017. [Google Scholar]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Escher, C.; Reiter, L.; MacLean, B.; Ossola, R.; Herzog, F.; Chilton, J.; MacCoss, M.J.; Rinner, O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 2012, 12, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Wren, S.A. Peak capacity in gradient ultra performance liquid chromatography (UPLC). J. Pharm. Biomed. Anal. 2005, 38, 337–343. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Cote, R.G.; Griss, J.; Dianes, J.A.; Wang, R.; Wright, J.C.; van den Toorn, H.W.; van Breukelen, B.; Heck, A.J.; Hulstaert, N.; Martens, L.; et al. The PRoteomics IDEntification (PRIDE) Converter 2 framework: An improved suite of tools to facilitate data submission to the PRIDE database and the ProteomeXchange consortium. Mol. Cell. Proteom. 2012, 11, 1682–1689. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lim, K.P.; Kong, W.; Gao, H.; Wong, B.J.H.; Phua, S.X.; Guo, T.; Goh, W.W.B. MultiPro: DDA-PASEF and diaPASEF acquired cell line proteomic datasets with deliberate batch effects. Sci. Data 2023, 10, 858. [Google Scholar] [CrossRef] [PubMed]
- Lou, R.; Cao, Y.; Li, S.; Lang, X.; Li, Y.; Zhang, Y.; Shui, W. Benchmarking commonly used software suites and analysis workflows for DIA proteomics and phosphoproteomics. Nat. Commun. 2023, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, H.; Li, C.; Chen, B.; Gong, H.; Zhao, Y.; Qi, R. Water-Soluble Tomato Extract Fruitflow Alters the Phosphoproteomic Profile of Collagen-Stimulated Platelets. Front. Pharmacol. 2021, 12, 746107. [Google Scholar] [CrossRef]
- Tan, D.; Lu, M.; Cai, Y.; Qi, W.; Wu, F.; Bao, H.; Qv, M.; He, Q.; Xu, Y.; Wang, X.; et al. SUMOylation of Rho-associated protein kinase 2 induces goblet cell metaplasia in allergic airways. Nat. Commun. 2023, 14, 3887. [Google Scholar] [CrossRef]
- Teschner, D.; Gomez-Zepeda, D.; Declercq, A.; Lacki, M.K.; Avci, S.; Bob, K.; Distler, U.; Michna, T.; Martens, L.; Tenzer, S.; et al. Ionmob: A Python package for prediction of peptide collisional cross-section values. Bioinformatics 2023, 39, btad486. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.L.; Li, Y.Y.; Meng, T.G.; Ning, Y.; Sun, S.M.; Zhang, C.H.; Gui, Y.; Wang, Z.B.; Qian, W.P.; Sun, Q.Y. Specific deletion of protein phosphatase 6 catalytic subunit in Sertoli cells leads to disruption of spermatogenesis. Cell Death Dis. 2021, 12, 883. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural. Inf. Process. Syst. 2019, 32. [Google Scholar]
- Caruana, R. Multitask learning. Mach. Learn. 1997, 28, 41–75. [Google Scholar] [CrossRef]
- Sun, T.; Shao, Y.; Li, X.; Liu, P.; Yan, H.; Qiu, X.; Huang, X. Learning sparse sharing architectures for multiple tasks. In Proceedings of the AAAI Conference on Artificial Intelligence, New York, NY, USA, 7–12 February 2020; pp. 8936–8943. [Google Scholar]




| d_model | n_head | d_ff_lstm | n_lstm | MedAE |
|---|---|---|---|---|
| 256 | 8 | 512 | 1 | 0.715 |
| 256 | 8 | 512 | 2 | 0.732 |
| 500 | 10 | 512 | 1 | 0.723 |
| 500 | 10 | 512 | 2 | 0.720 |
| 256 | 8 | 256 | 1 | 0.724 |
| 256 | 8 | 256 | 2 | 0.768 |
| 500 | 10 | 256 | 1 | 0.722 |
| 500 | 10 | 256 | 2 | 0.755 |
| Data Name | Metrics/Model | Retention Time | Ion Intensity | Ion Mobility | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MedAE | IQR | PCC | SA | PCC | DP | PCC | ||||||
| H1_DDA | DeepPhospho | 0.975 | 6.588 | 1.157 | 2.315 | 0.990 | 0.852 | 0.958 | 0.973 | - | - | - |
| ProPept-ST | 0.9805 | 5.200 | 0.643 | 1.323 | 0.990 | 0.870 | 0.9657 | 0.979 | 0.977 | 0.9887 | 0.0961 | |
| ProPept-MT | 0.9810 | 4.974 | 0.598 | 1.264 | 0.991 | 0.872 | 0.9663 | 0.980 | 0.978 | 0.9892 | 0.0963 | |
| H2_DIA | DeepPhospho | 0.989 | 5.481 | 1.019 | 2.046 | 0.997 | 0.805 | 0.889 | 0.953 | - | - | - |
| ProPept-ST | 0.998 | 2.333 | 0.319 | 0.638 | 0.999 | 0.817 | 0.900 | 0.959 | 0.986 | 0.9931 | 0.062 | |
| ProPept-MT | 0.997 | 2.807 | 0.395 | 0.772 | 0.9986 | 0.820 | 0.901 | 0.960 | 0.984 | 0.9928 | 0.063 | |
| H3_DIAP | DeepPhospho | 0.986 | 11.428 | 2.103 | 4.111 | 0.997 | 0.786 | 0.872 | 0.944 | - | - | - |
| ProPept-ST | 0.9952 | 7.045 | 0.753 | 1.508 | 0.99764 | 0.797 | 0.882 | 0.9497 | 0.985 | 0.9924 | 0.0614 | |
| ProPept-MT | 0.9951 | 6.973 | 0.870 | 1.713 | 0.99763 | 0.798 | 0.889 | 0.9502 | 0.984 | 0.9922 | 0.0617 | |
| H4_DDAp | DeepPhospho | 0.976 | 10.524 | 1.915 | 3.678 | 0.990 | 0.809 | 0.928 | 0.955 | - | - | - |
| ProPept-ST | 0.9835 | 6.447 | 0.715 | 1.424 | 0.9918 | 0.831 | 0.941 | 0.965 | 0.971 | 0.9856 | 0.102 | |
| ProPept-MT | 0.9839 | 6.203 | 0.730 | 1.422 | 0.9919 | 0.835 | 0.945 | 0.967 | 0.972 | 0.9862 | 0.099 | |
| H5_DDAp | DeepPhospho | 0.980 | 12.658 | 2.408 | 4.822 | 0.993 | 0.819 | 0.935 | 0.960 | - | - | - |
| ProPept-ST | 0.987 | 9.249 | 0.945 | 1.886 | 0.9935 | 0.8324 | 0.940 | 0.966 | 0.961 | 0.981 | 0.100 | |
| ProPept-MT | 0.988 | 8.699 | 1.077 | 2.142 | 0.9939 | 0.8317 | 0.941 | 0.965 | 0.959 | 0.980 | 0.102 | |
| H6_DDAp | DeepPhospho | 0.980 | 12.883 | 2.600 | 4.231 | 0.993 | 0.804 | 0.928 | 0.953 | - | - | - |
| ProPept-ST | 0.991 | 6.519 | 0.820 | 1.660 | 0.996 | 0.814 | 0.934 | 0.958 | 0.963 | 0.9815 | 0.117 | |
| ProPept-MT | 0.990 | 6.187 | 0.802 | 1.588 | 0.995 | 0.824 | 0.940 | 0.962 | 0.960 | 0.9809 | 0.113 | |
| H7_DDAp | DeepPhospho | 0.958 | 5.352 | 0.831 | 1.598 | 0.983 | 0.807 | 0.932 | 0.954 | - | - | - |
| ProPept-ST | 0.977 | 2.853 | 0.318 | 0.650 | 0.988 | 0.823 | 0.941 | 0.961 | 0.982 | 0.991 | 0.080 | |
| ProPept-MT | 0.980 | 2.255 | 0.294 | 0.587 | 0.990 | 0.838 | 0.950 | 0.968 | 0.986 | 0.994 | 0.067 | |
| M1_DDAp | DeepPhospho | 0.976 | 11.716 | 1.809 | 3.535 | 0.991 | 0.815 | 0.938 | 0.958 | - | - | - |
| ProPept-ST | 0.991 | 6.519 | 0.820 | 1.660 | 0.996 | 0.814 | 0.934 | 0.958 | 0.963 | 0.981 | 0.117 | |
| ProPept-MT | 0.989 | 5.498 | 0.702 | 1.423 | 0.995 | 0.834 | 0.949 | 0.966 | 0.982 | 0.992 | 0.077 | |
| M2_DDAp | DeepPhospho | 0.966 | 4.944 | 0.812 | 1.518 | 0.986 | 0.792 | 0.918 | 0.947 | - | - | - |
| ProPept-ST | 0.980 | 3.050 | 0.367 | 0.755 | 0.990 | 0.807 | 0.927 | 0.955 | 0.941 | 0.973 | 0.112 | |
| ProPept-MT | 0.982 | 1.668 | 0.243 | 0.483 | 0.991 | 0.827 | 0.941 | 0.963 | 0.955 | 0.978 | 0.090 | |
| Data Name | Metrics/Model | Retention Time | Ion Intensity(2+) | Ion Intensity(3+) | Ion Mobility | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MedAE | IQR | PCC | SA | PCC | DP | SA | PCC | DP | PCC | ||||||
| H1_DDA | DeepDIA | 0.975 | 7.132 | 0.974 | 1.962 | 0.987 | 0.802 | 0.950 | 0.952 | 0.721 | 0.900 | 0.905 | - | - | - |
| ProPept-ST | 0.980 | 5.422 | 0.670 | 1.399 | 0.9902 | 0.847 | 0.959 | 0.971 | 0.791 | 0.925 | 0.947 | 0.974 | 0.987 | 0.106 | |
| ProPept-MT | 0.981 | 4.823 | 0.662 | 1.340 | 0.9905 | 0.866 | 0.968 | 0.978 | 0.817 | 0.943 | 0.959 | 0.976 | 0.988 | 0.100 | |
| H2_DIA | DeepDIA | 0.994 | 4.214 | 0.590 | 1.178 | 0.997 | 0.738 | 0.913 | 0.917 | 0.708 | 0.893 | 0.897 | - | - | - |
| ProPept-ST | 0.9973 | 2.817 | 0.381 | 0.770 | 0.999 | 0.812 | 0.894 | 0.957 | 0.792 | 0.865 | 0.947 | 0.984 | 0.9921 | 0.066 | |
| ProPept-MT | 0.9970 | 2.993 | 0.426 | 0.852 | 0.998 | 0.821 | 0.907 | 0.961 | 0.794 | 0.875 | 0.948 | 0.983 | 0.9918 | 0.069 | |
| H3_DIAp | DeepDIA | 0.979 | 15.410 | 1.964 | 3.909 | 0.990 | 0.692 | 0.880 | 0.885 | 0.673 | 0.867 | 0.871 | - | - | - |
| ProPept-ST | 0.992 | 9.413 | 1.134 | 2.282 | 0.996 | 0.788 | 0.8507 | 0.945 | 0.7660 | 0.837 | 0.9332 | 0.979 | 0.990 | 0.073 | |
| ProPept-MT | 0.993 | 8.301 | 0.939 | 1.873 | 0.997 | 0.783 | 0.8513 | 0.942 | 0.7664 | 0.852 | 0.9335 | 0.982 | 0.991 | 0.068 | |
| H4_DDAp | DeepDIA | 0.973 | 14.720 | 1.930 | 3.910 | 0.986 | 0.746 | 0.916 | 0.921 | 0.668 | 0.861 | 0.867 | - | - | - |
| ProPept-ST | 0.984 | 8.243 | 0.992 | 1.972 | 0.992 | 0.805 | 0.928 | 0.954 | 0.750 | 0.891 | 0.924 | 0.960 | 0.980 | 0.126 | |
| ProPept-MT | 0.986 | 6.685 | 0.865 | 1.729 | 0.993 | 0.819 | 0.938 | 0.960 | 0.774 | 0.913 | 0.937 | 0.967 | 0.984 | 0.l13 | |
| H5_DDAp | DeepDIA | 0.983 | 12.850 | 2.372 | 3.419 | 0.992 | 0.771 | 0.933 | 0.936 | 0.692 | 0.880 | 0.885 | - | - | - |
| ProPept-ST | 0.989 | 8.484 | 0.942 | 1.888 | 0.9948 | 0.819 | 0.940 | 0.960 | 0.765 | 0.897 | 0.933 | 0.960 | 0.981 | 0.104 | |
| ProPept-MT | 0.991 | 7.225 | 0.850 | 1.705 | 0.9954 | 0.839 | 0.952 | 0.968 | 0.798 | 0.928 | 0.950 | 0.969 | 0.984 | 0.086 | |
| H6_DDAp | DeepDIA | 0.918 | 21.389 | 2.456 | 4.928 | 0.958 | 0.670 | 0.861 | 0.869 | 0.657 | 0.853 | 0.859 | - | - | - |
| ProPept-ST | 0.942 | 16.991 | 1.924 | 3.857 | 0.971 | 0.754 | 0.873 | 0.926 | 0.765 | 0.905 | 0.933 | 0.923 | 0.963 | 0.159 | |
| ProPept-MT | 0.973 | 6.491 | 0.705 | 1.397 | 0.987 | 0.780 | 0.928 | 0.947 | 0.791 | 0.928 | 0.947 | 0.940 | 0.971 | 0.145 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, G.; He, Q.; Cheng, J.; Yu, R.; Shuai, J.; Cao, Y. ProPept-MT: A Multi-Task Learning Model for Peptide Feature Prediction. Int. J. Mol. Sci. 2024, 25, 7237. https://doi.org/10.3390/ijms25137237
He G, He Q, Cheng J, Yu R, Shuai J, Cao Y. ProPept-MT: A Multi-Task Learning Model for Peptide Feature Prediction. International Journal of Molecular Sciences. 2024; 25(13):7237. https://doi.org/10.3390/ijms25137237
Chicago/Turabian StyleHe, Guoqiang, Qingzu He, Jinyan Cheng, Rongwen Yu, Jianwei Shuai, and Yi Cao. 2024. "ProPept-MT: A Multi-Task Learning Model for Peptide Feature Prediction" International Journal of Molecular Sciences 25, no. 13: 7237. https://doi.org/10.3390/ijms25137237
APA StyleHe, G., He, Q., Cheng, J., Yu, R., Shuai, J., & Cao, Y. (2024). ProPept-MT: A Multi-Task Learning Model for Peptide Feature Prediction. International Journal of Molecular Sciences, 25(13), 7237. https://doi.org/10.3390/ijms25137237