
Creation of an Isogenic Human iPSC-Based RGC Model of Dominant Optic Atrophy Harboring the Pathogenic Variant c.1861C>T (p.Gln621Ter) in the OPA1 Gene
 , , , ,
, , , ,  , ,
, , 
Abstract
:1. Introduction
2. Results
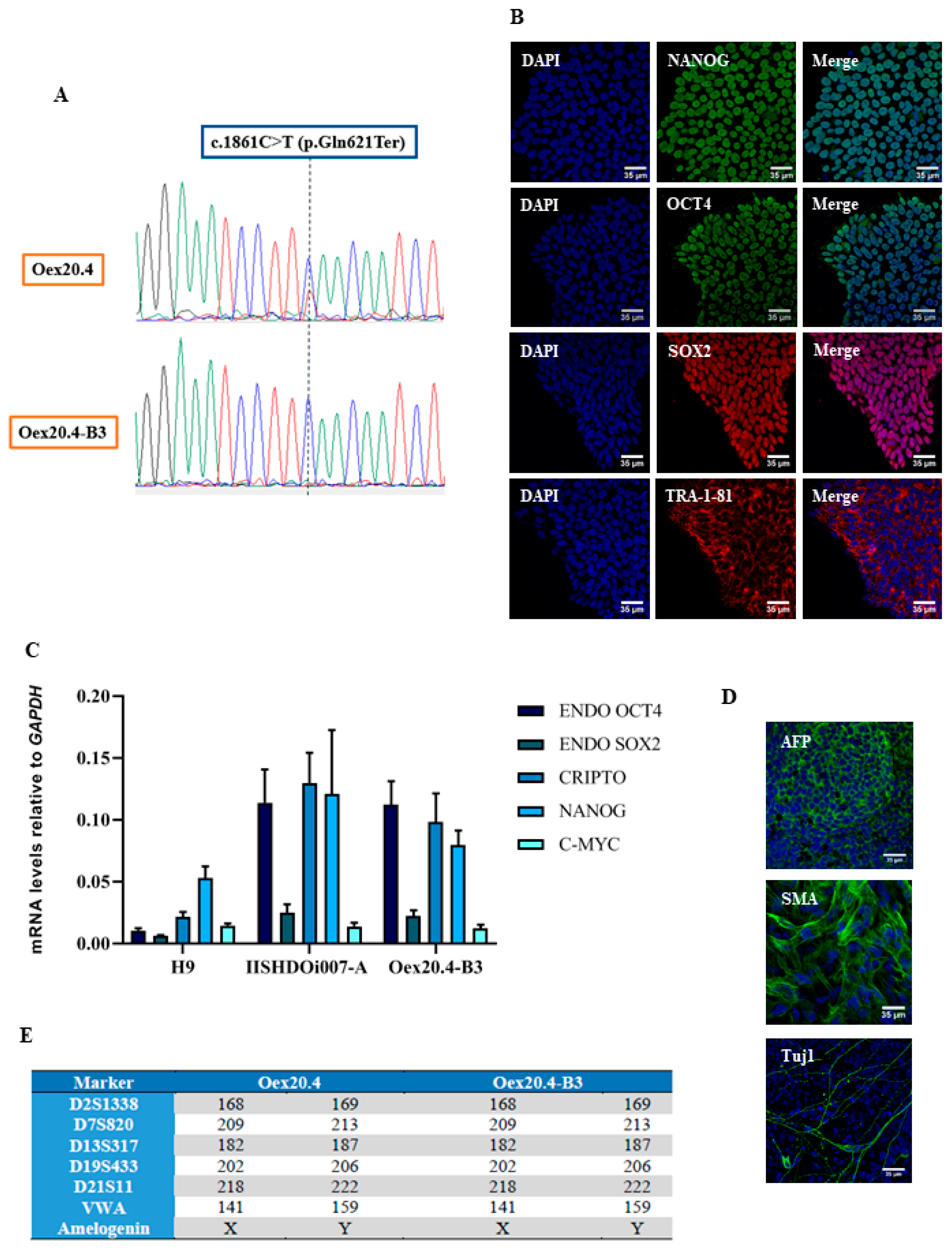
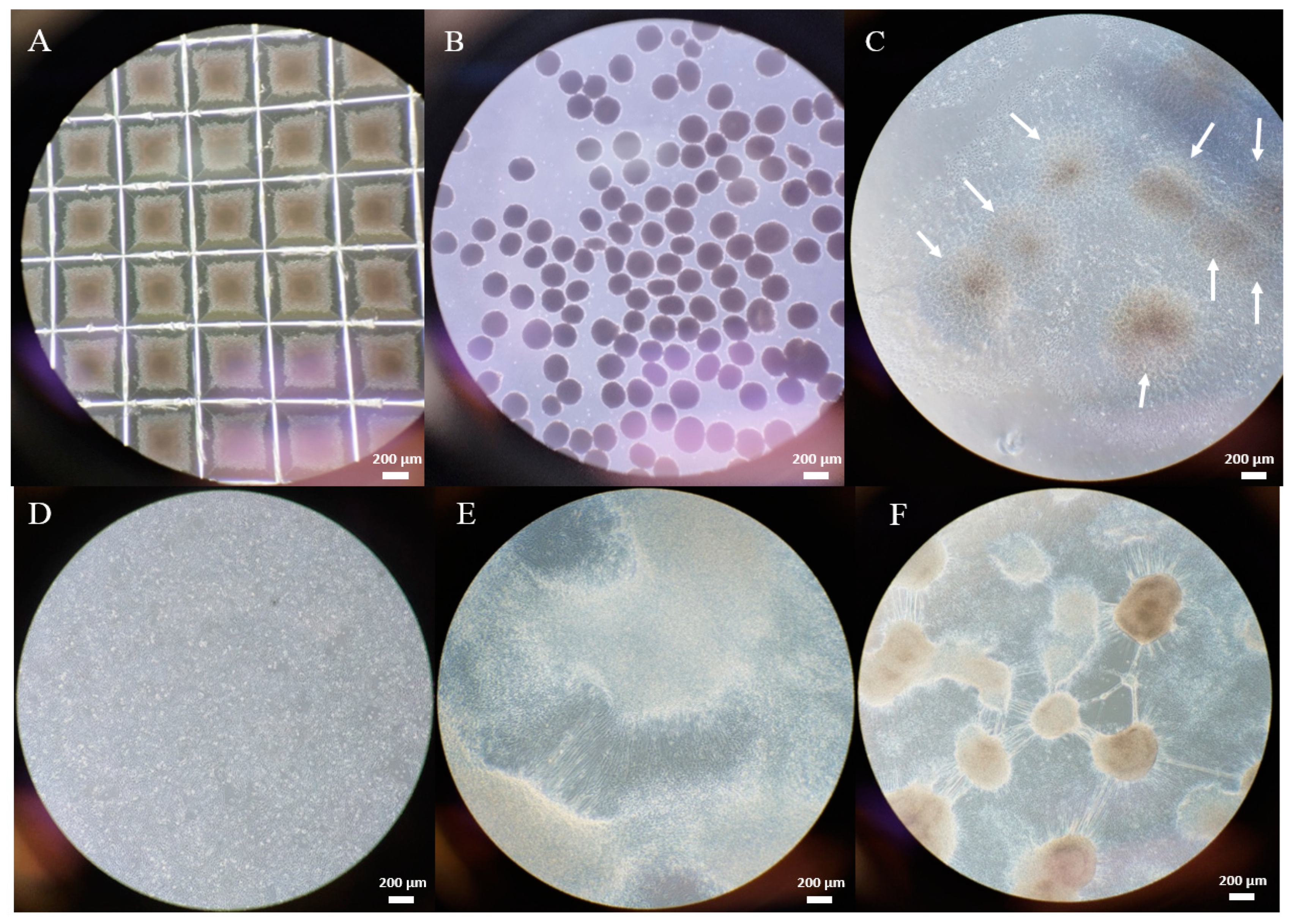
2.1. Gene Editing with CRISPR/Cas9
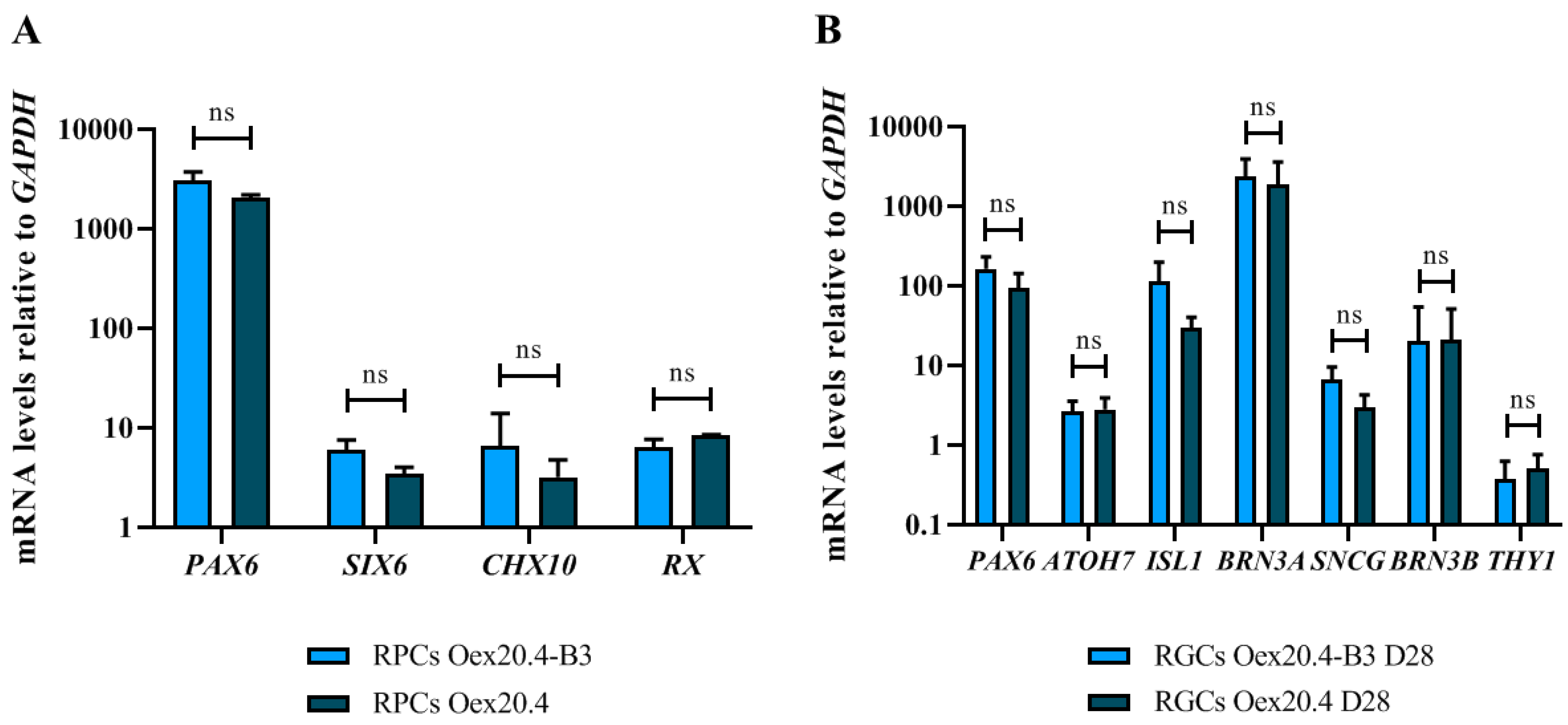
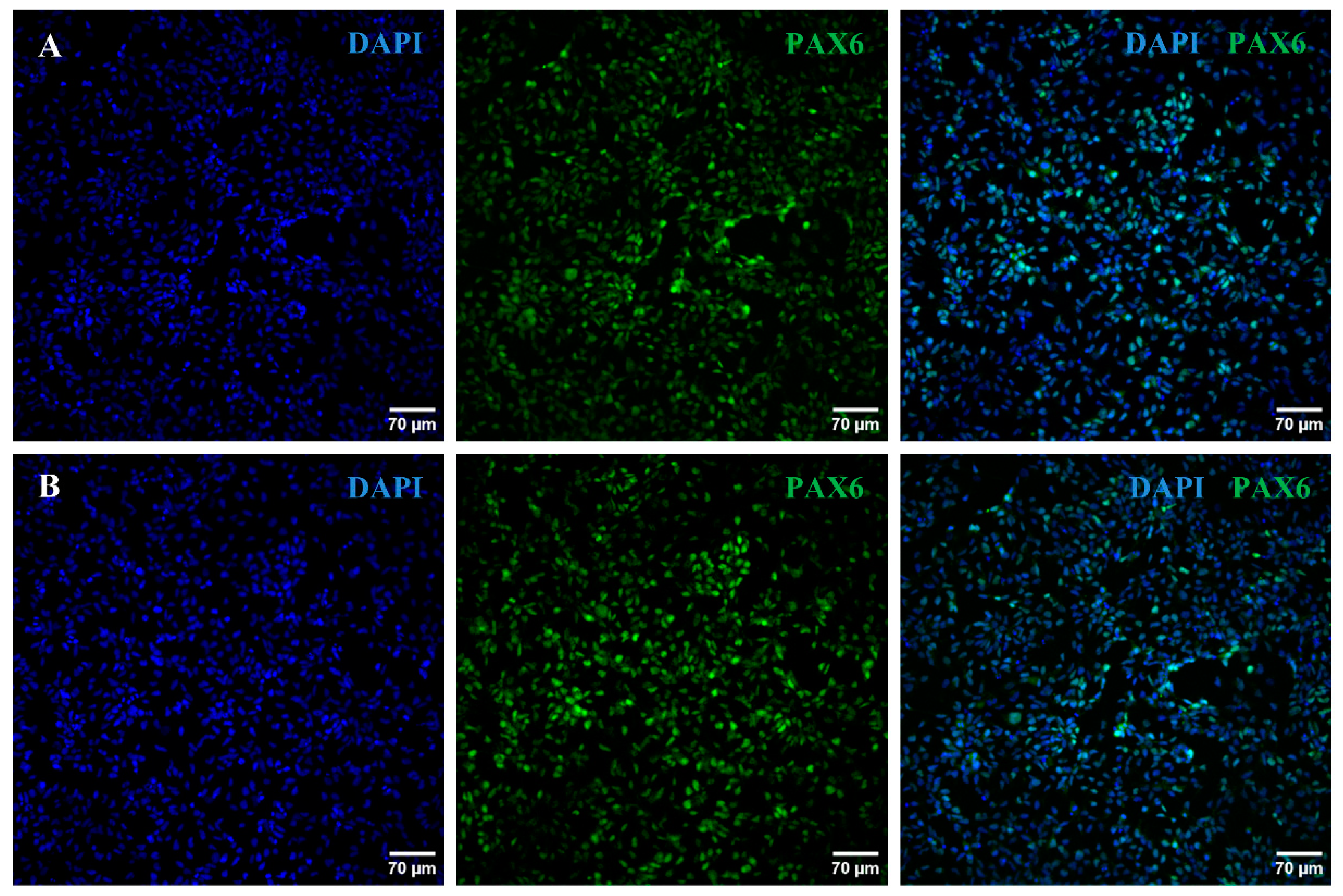
2.2. iPSC Differentiation towards RGCs
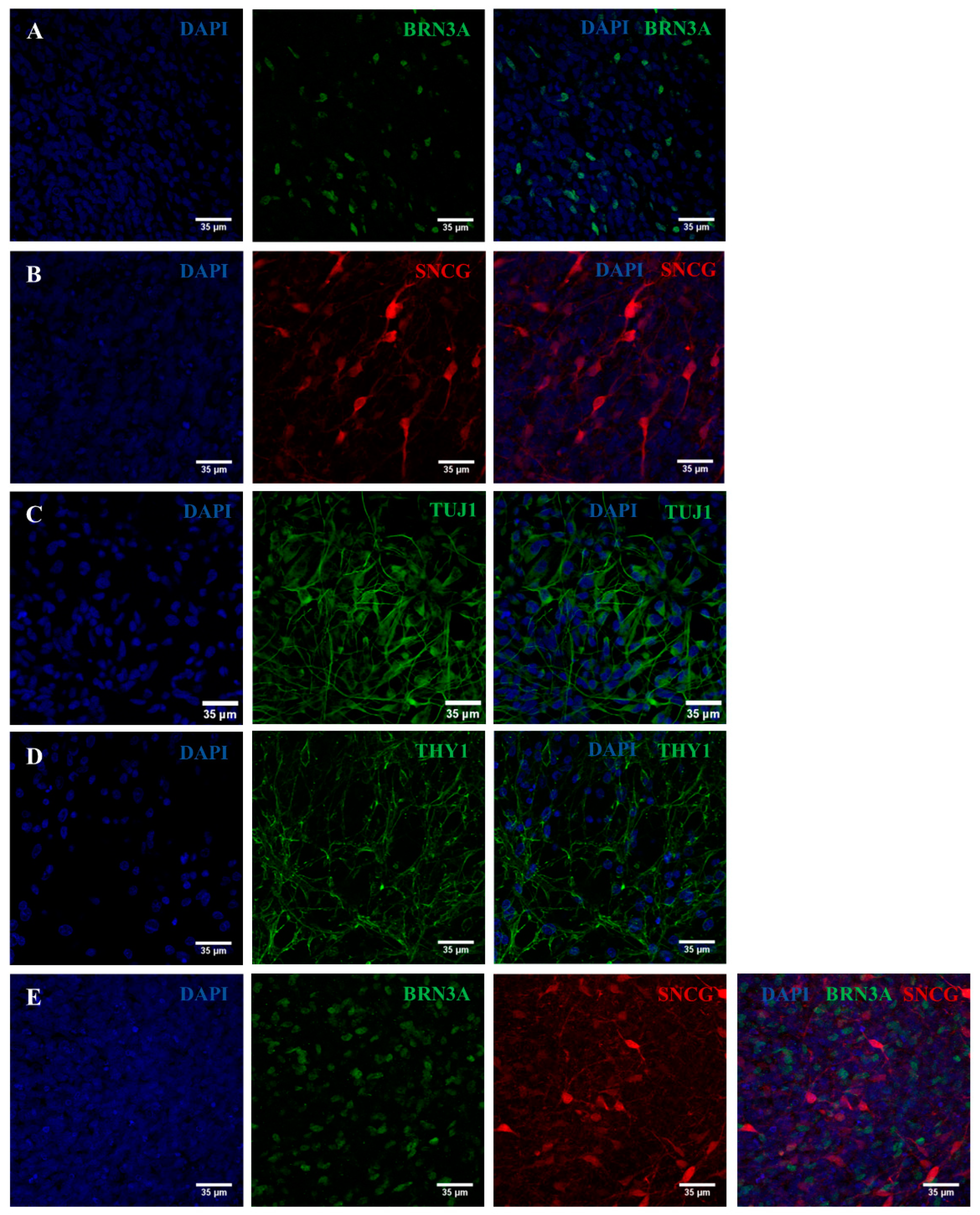
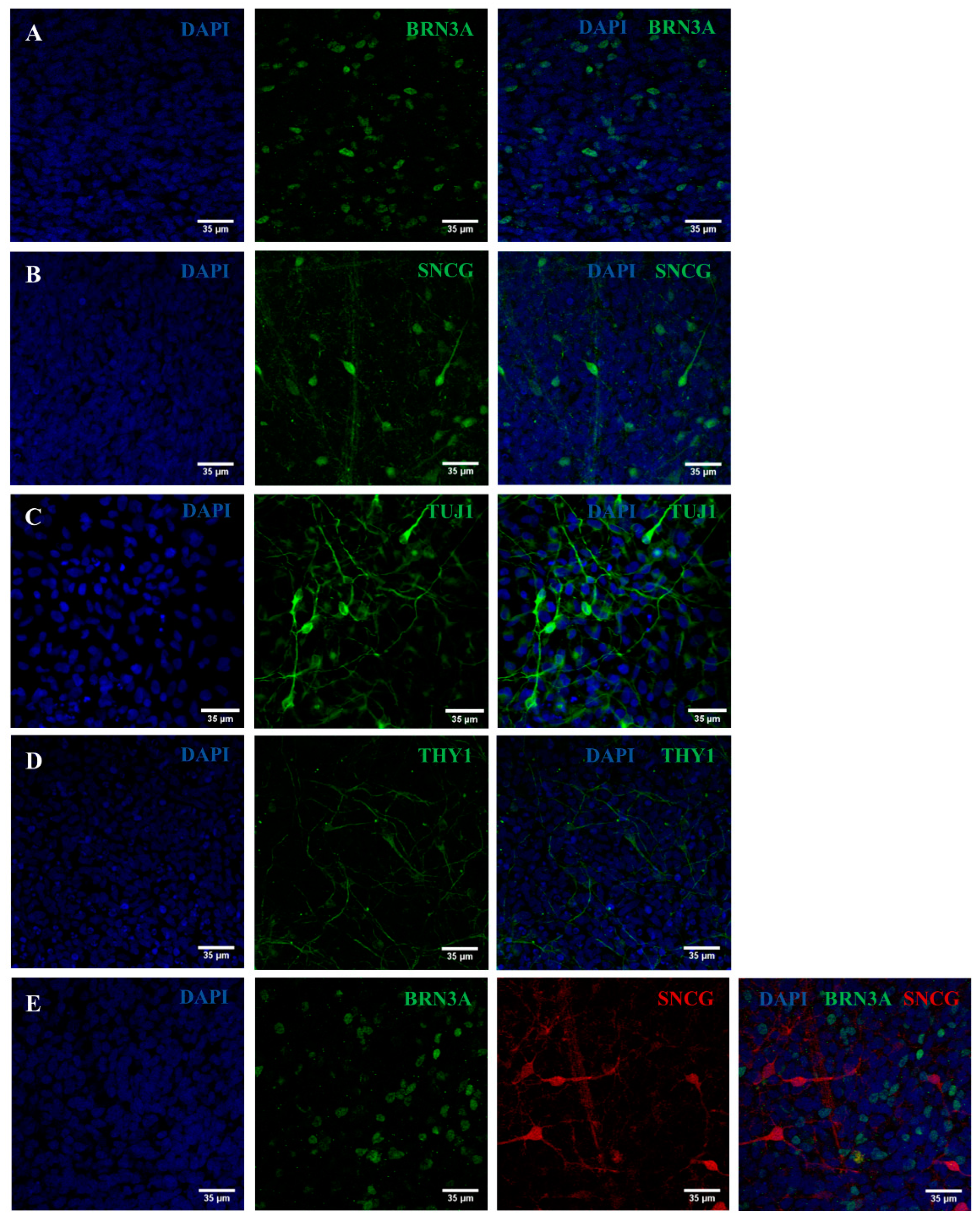
2.3. Validation of the Created RGC-Based Model
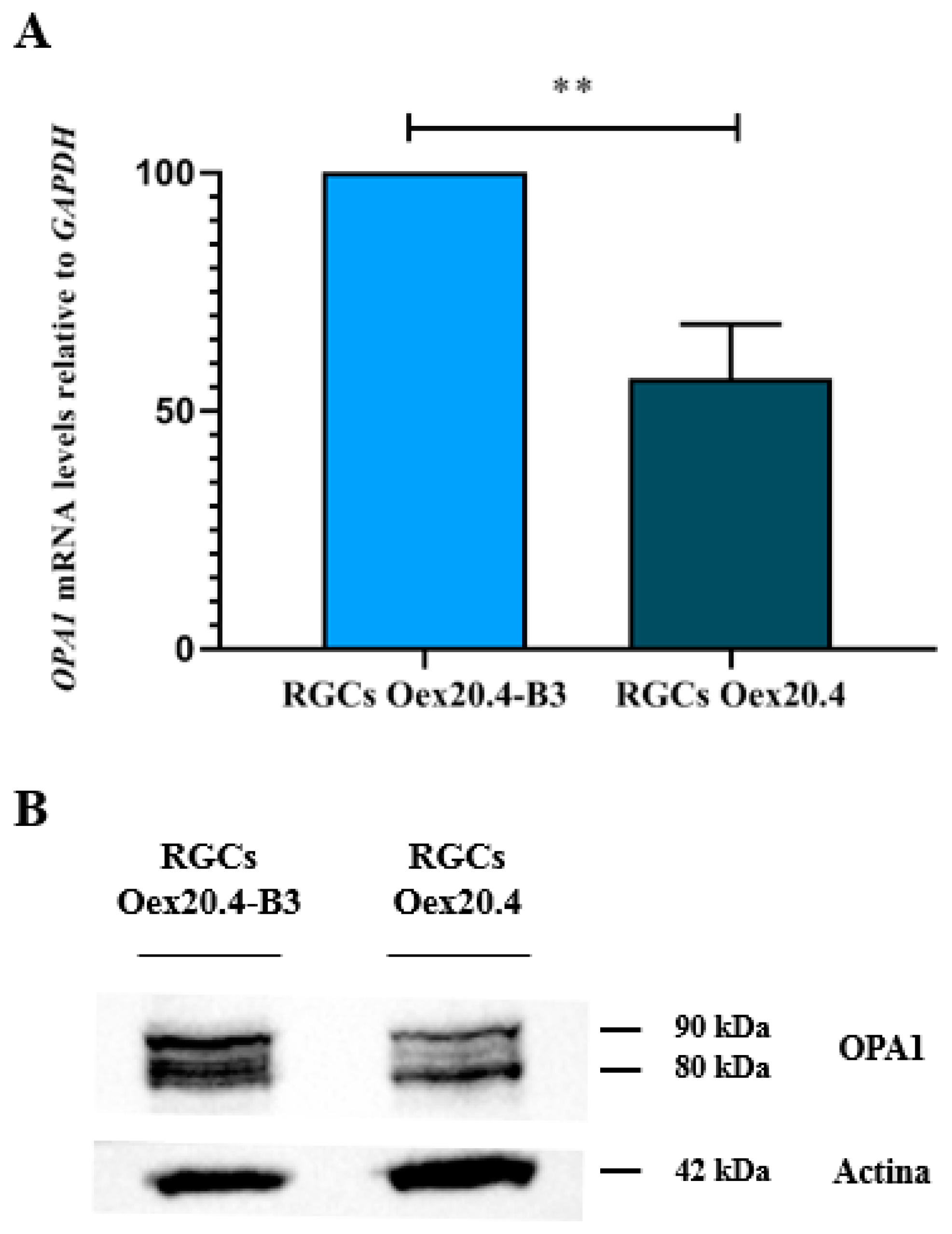
2.3.1. Analysis of OPA1 Expression
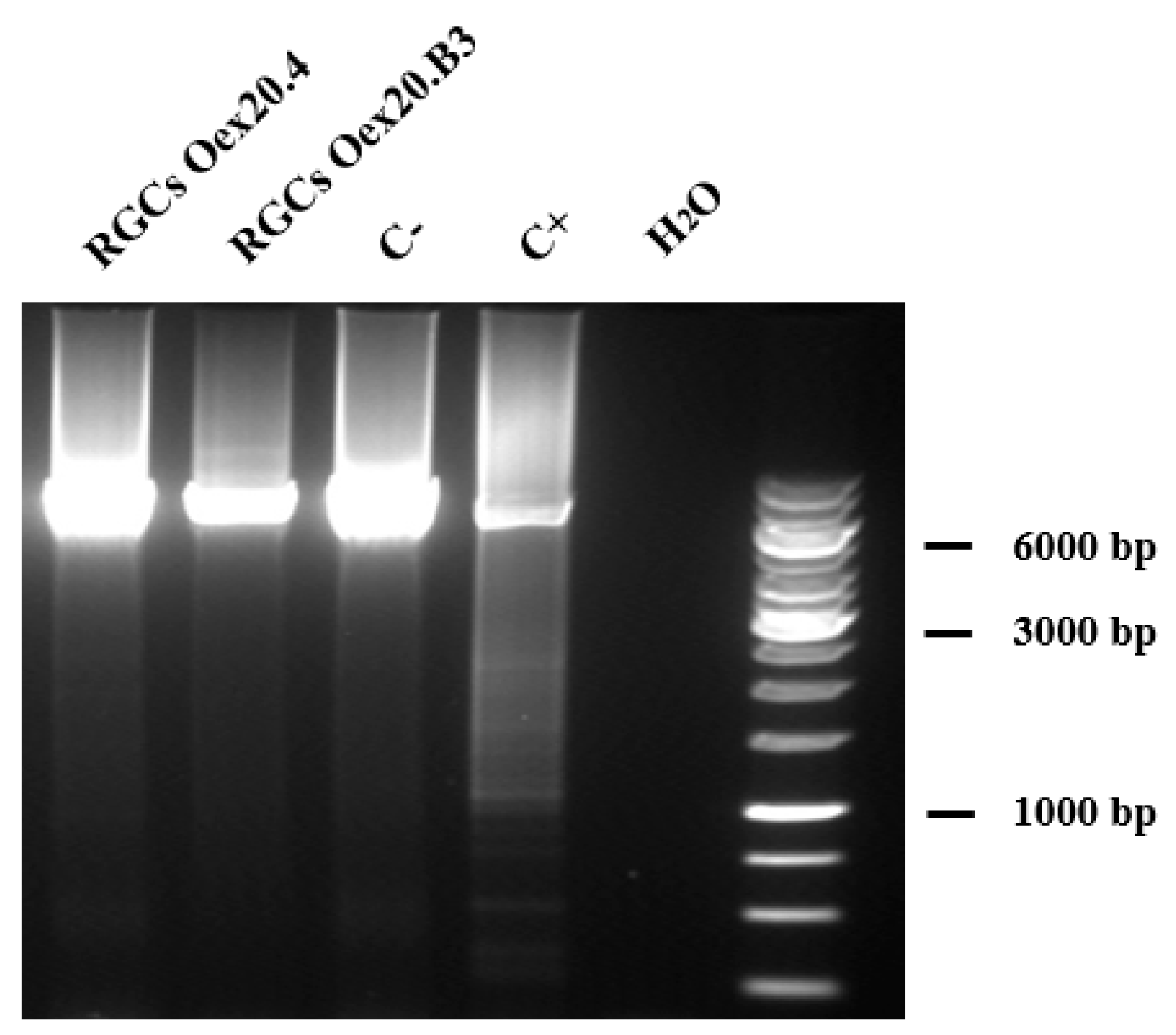
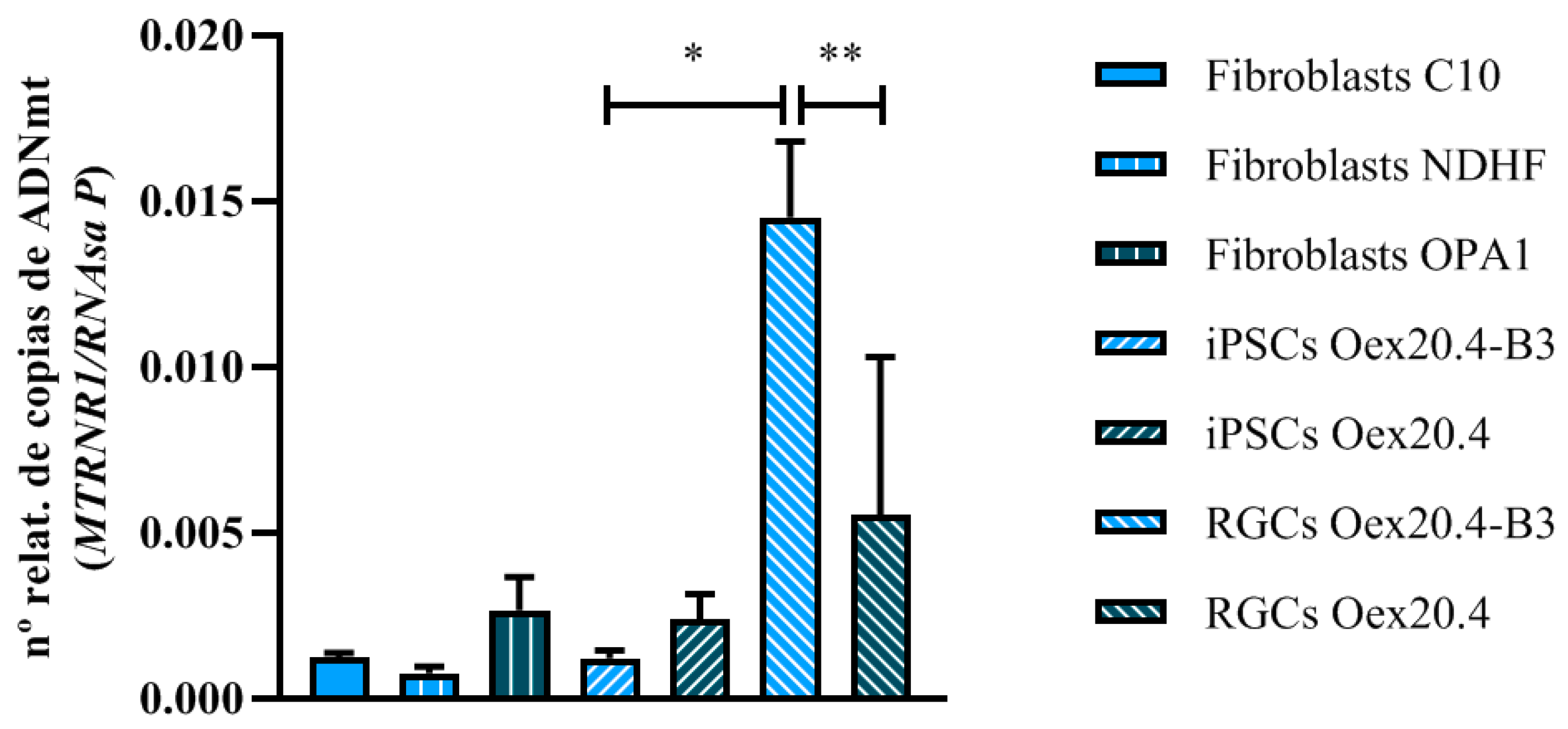
2.3.2. Analyses of mtDNA Multiple Deletions and Depletion
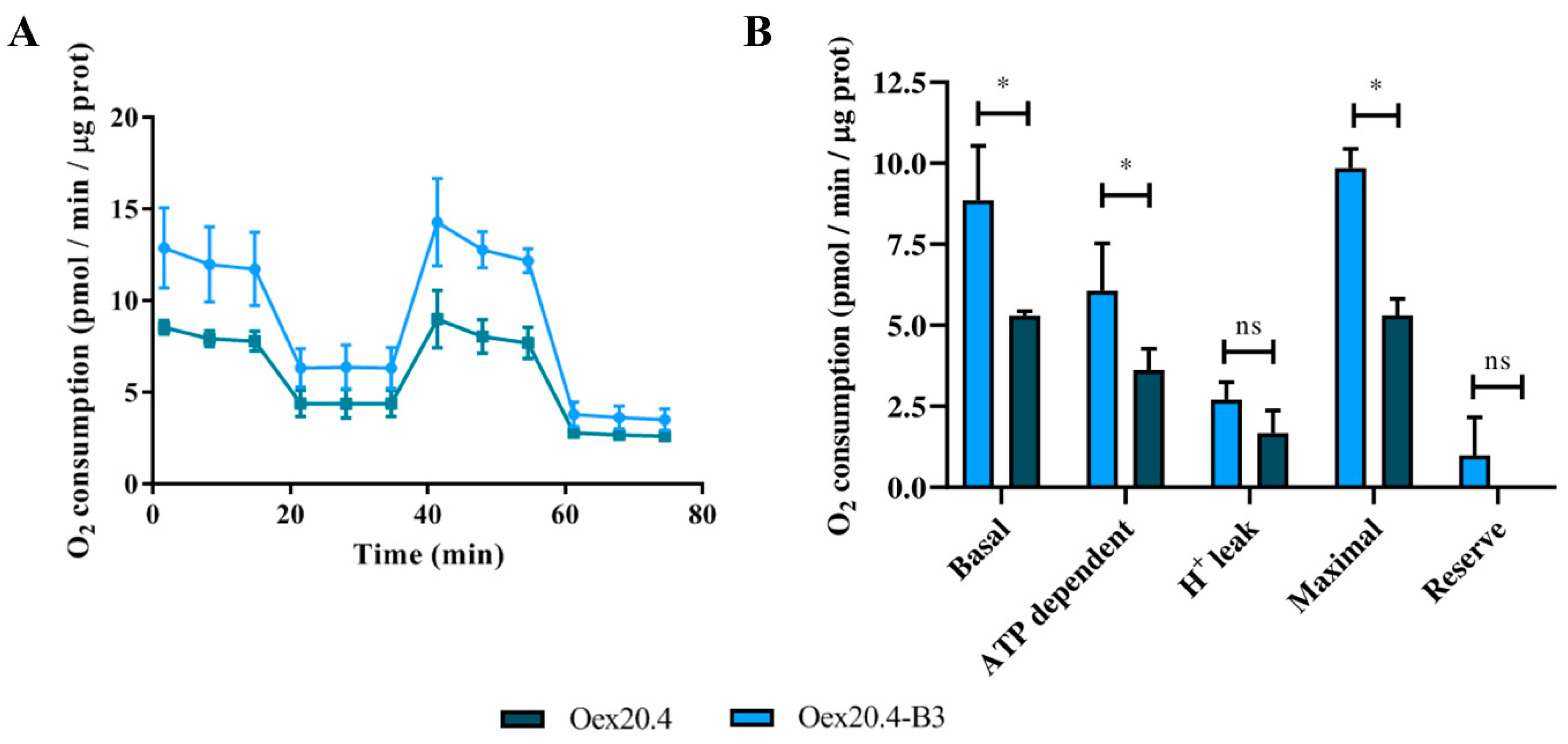
2.3.3. Analysis of the Mitochondrial Respiration
2.3.4. Quantification of Reactive Oxygen Species (ROS) Generation
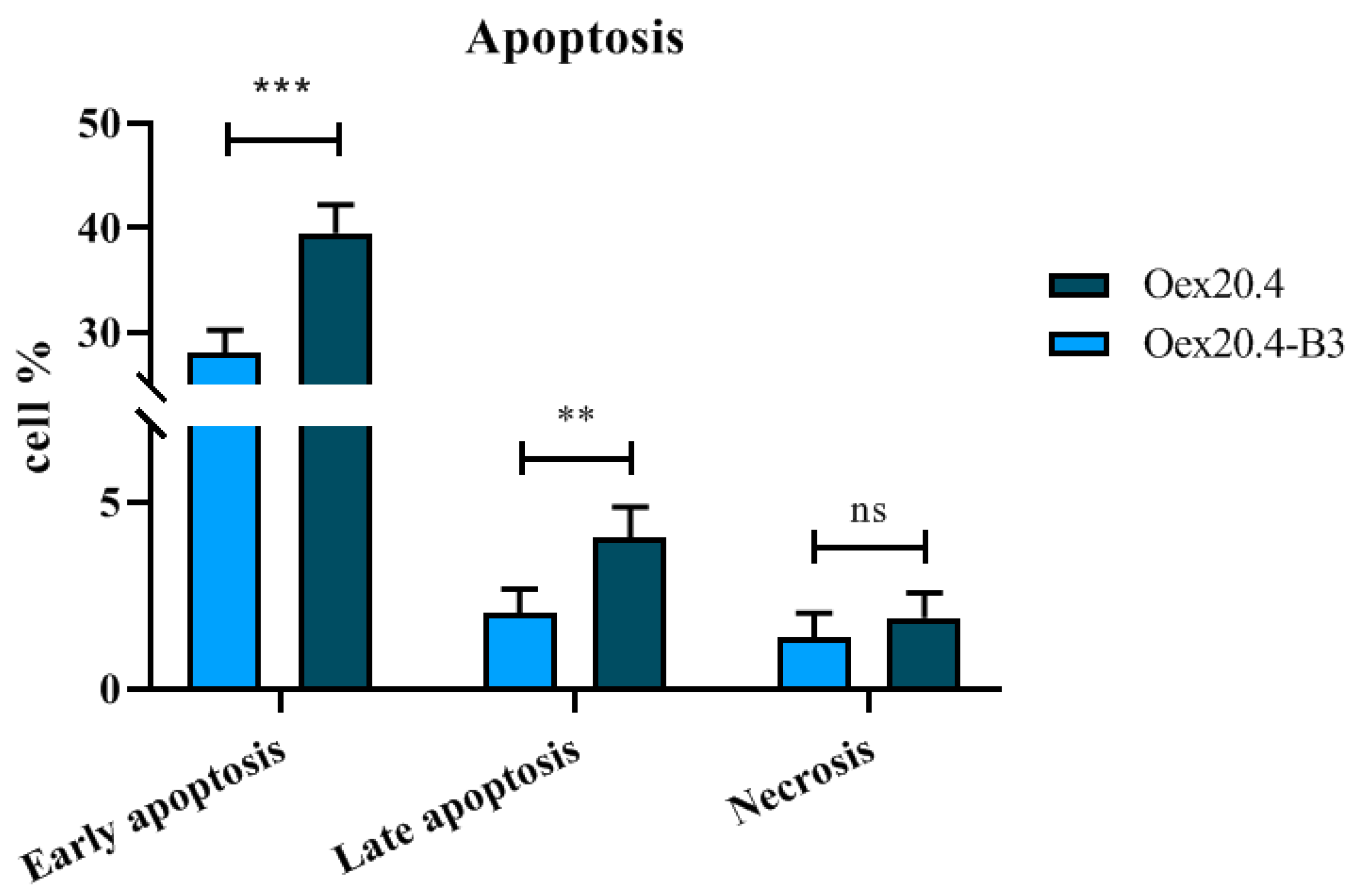
2.3.5. Apoptosis Quantification
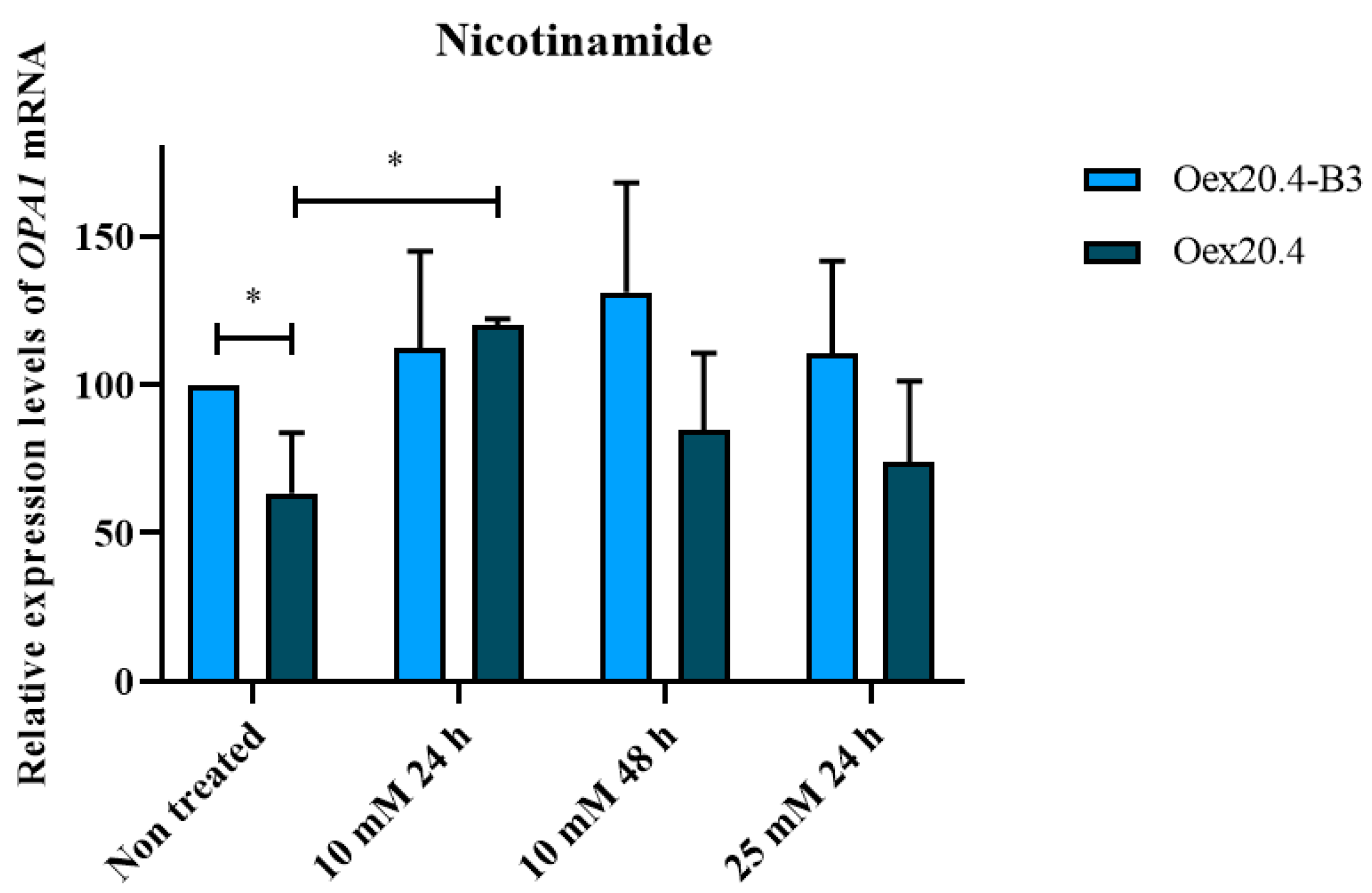
2.4. Nicotinamide Potential for the Rescue of OPA1 Expression
3. Discussion
4. Materials and Methods
4.1. iPSC Culture Conditions
4.2. Gene Editing with CRISPR/Cas9
4.2.1. Design of the sgRNAs
4.2.2. Design of the ssODN
| AATGATACTTCAGTCAAGCTGTTTTTAAAAACAATATTATATTTAGATTTGGTG CTTTTGATACTTTTTTATTTCAGGGAGGAAATCCTTCAACAATCTTTGTGGGA AAGAGTATCAACTCATGTGATTGAAAACATCTACCTTCCAGCTGCGCAGACCA TGAATTCAGGAACTTTTAACA |
4.2.3. Gene Editing
4.2.4. T7E1 Assay to Evaluate Cutting Efficiency
4.2.5. Evaluation of Editing Efficiency with RFLP
4.2.6. Subcloning
4.2.7. Freezing and Analysis of the Clones
4.3. Pluripotency and Integrity Assessment
4.3.1. qPCR
4.3.2. Immunocytochemistry Analyses
4.3.3. In Vitro Differentiation
4.3.4. STR Analysis
4.3.5. Mycoplasma Detection
4.3.6. Karyotype Analysis
4.4. iPSC Differentiation towards RGCs
4.5. Characterization of the Generated RPCs and RGCs
4.5.1. RT-qPCR Analyses
4.5.2. Immunocytochemistry Analyses
4.6. Validation and Functional Analyses of the RGC Model
4.6.1. OPA1 Expression Analysis by RT-PCR
4.6.2. Western Blot for OPA1 Analysis
4.6.3. Identification of mtDNA Deletions by Long-Range PCR
4.6.4. qPCR Relative Quantification of mtDNA
4.6.5. Analysis of Mitochondrial Respiration
4.6.6. Quantification of Reactive Oxygen Species (ROS)
4.6.7. Apoptosis Quantification
4.7. Analysis of the Nicotinamide Potential for the Rescue of OPA1 Expression
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu-Wai-Man, P.; Griffiths, P.G.; Burke, A.; Sellar, P.W.; Clarke, M.P.; Gnanaraj, L.; Ah-kine, D.; Hudson, G.; Czermin, B. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology 2014, 117, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer type) dominant optic atrophy: A novel mitochondrial disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar] [CrossRef]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef]
- Le Roux, B.; Lenaers, G.; Zanlonghi, X.; Amati-Bonneau, P.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J. Rare Dis. 2019, 14, 214. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Griffoin, J.; Kaplan, J.; Dollfus, H.; Lorenz, B.; Faivre, L.; Lenaers, G.; Belenguer, P.; Hamel, C.P. Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 2001, 109, 584–591. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies—Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef]
- Chun, B.Y.; Rizzo, J.F. Dominant Optic Atrophy and Leber’s Hereditary Optic Neuropathy: Update on clinical features and current therapeutic approaches. Semin. Pediatr. Neurol. 2017, 24, 129–134. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, M.; La Morgia, C.; Carbonelli, M.; Di Vito, L.; Zenesini, C.; Cascavilla, M.L.; Barboni, P.; Carelli, V. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann. Clin. Traslational Neurol. 2020, 7, 590–594. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.P.; Zheng, Y.W. Predicting differentiation potential of human pluripotent stem cells: Possibilities and challenges. World J. Stem Cells 2019, 11, 275–382. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, N.; Higuchi, Y.; Harada, S.; Umeda, K.; Isagawa, T.; Aburatani, H.; Kume, K.; Kume, S. Differentiation and characterization of embryonic stem cells into three germ layers. Biochem. Biophys. Res. Commun. 2009, 381, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced pluripotent stem cells: Reprogramming platforms and applications in cell replacement therapy. BioRes. Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Soud, M.A.M.; Alzahrani, A.J.; Mahmoud, A. Induced pluripotent stem cells (iPSCs)—Roles in regenerative therapies, disease modelling and drug screening. Cells 2021, 10, 2319. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Q.; Sun, X.; Shen, W.; Liu, B.; Zhong, X.; Leng, Y.; Li, C.; Zhang, W.; Chai, F.; et al. Generation of retinal ganglion-like cells from reprogrammed mouse fibroblasts. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5970–5978. [Google Scholar] [CrossRef] [PubMed]
- Gill, K.P.; Hewitt, A.W.; Davidson, K.C.; Pébay, A.; Wong, R.C.B. Methods of retinal ganglion cell differentiation from puripotent stem cells. Transl. Vis. Sci. Technol. 2014, 3, 7. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR-Cas9 technology: Applications and human disease modelling. Brief. Funct. Genom. 2017, 16, 4–12. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Díaz, F.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernández, A.F.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with an optic atrophy ‘plus’ phenotype due to a mutation in the OPA1 gene. Stem Cell Res. 2016, 16, 673–676. [Google Scholar] [CrossRef]
- Bruntraeger, M.; Byrne, M.; Long, K.; Bassett, A.R. Editing the genome of human induced pluripotent stem cells using CRISPR/Cas9 ribonucleoprotein complexes. Methods Mol. Biol. 2019, 1961, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Cerrada, V.; García-López, M.; Alvarez-Galeano, S.; Moreno-Izquierdo, A.; Lucia, A.; Rabasa Pérez, M.; Arenas, J.; Gallardo, M.E. Generation of the iPSC line IISHDOi007—A from peripheral blood mononuclear cells from a patient with McArdle disease harbouring the mutation c.2392 T > C; p.Trp798Arg. Stem Cell Res. 2020, 49, 102108. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, S.; Kim, Y.; Jun, I.; Sung, J.J.; Dongjin, R.; Kim, Y.I.; Cho, M.S.; Byeon, S.H.; Kim, D.; et al. Defined conditions for differentiation of functional retinal ganglion cells from human pluripotent stem cells. Retin. Cell Biol. 2018, 59, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Lee, J.S.; Leem, J.W.; Huh, Y.J.; Kim, J.Y.; Kim, H.-S.; Park, I.-J.; Daley, G.Q.; Hwang, D.-Y.; Kim, D.-W. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. 2010, 6, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Maroof, A.M.; Keros, S.; Tyson, J.A.; Ying, S.-W.; Ganat, Y.M.; Merkle, F.T.; Liu, B.; Goulburn, A.; Stanley, E.G.; Elefanty, A.G.; et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 2013, 12, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Pera, E.M.; Wessely, O.; Li, S.Y.; De Robertis, E.M. Neural and head induction by insulin-like growth factor signals. Dev. Cell 2001, 1, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.R.; Gumuscu, B.; Hartman, B.H.; Reh, T.A. Notch activity is downregulated just prior to retinal ganglion cell differentiation. Dev. Neurosci. 2006, 28, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Bhave, S.V.; Ghoda, L.; Hoffman, P.L. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: Signal transduction cascades and site of ethanol action. J. Neurosci. 1999, 19, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, T.A.V.; Corral-Serrano, J.C.; Garanto, A.; Roepman, R.; Cheetham, M.E.; Collin, R.W.J. A look into retinal organoids: Methods, analytical techniques, and applications. Cell. Mol. Life Sci. 2021, 78, 6505–6532. [Google Scholar] [CrossRef]
- Ahmad, I.; Teotia, P.; Erickson, H.; Xia, X. Recapitulating developmental mechanisms for retinal regeneration. Prog. Retin. Eye Res. 2020, 76, 100824. [Google Scholar] [CrossRef]
- Bradley, J.A.; Ramirez, G.; Hagood, J.S. Roles and regulation of Thy-1, a context-dependent modulator of cell phenotype. Nature 2009, 35, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Sladen, P.E.; Jovanovic, K.; Guarascio, R.; Ottaviani, D.; Salsbury, G.; Novoselova, T.; Chapple, J.P.; Yu-wai-man, P.; Cheetham, M.E.; Square, C. Modelling autosomal dominant optic atrophy associated with OPA1 variants in iPSC-derived retinal ganglion cells. Hum. Mol. Genet. 2022, 31, 3478–3493. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Riazifar, H.; Guan, M.X.; Huang, T. Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res. Ther. 2016, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Vu, T.H. Defects of intergenomic communication: Where do we stand? Brain Pathol. 2000, 10, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 2008, 131, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Hwang, J.M.; Ko, H.S.; Seong, M.W.; Park, B.J.; Park, S.S. Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology 2005, 64, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Lodi, R.; Tonon, C.; Valentino, M.L.; Manners, D.; Testa, C.; Malucelli, E.; La Morgia, C.; Barboni, P.; Carbonelli, M.; Schimpf, S.; et al. Defective mitochondrial adenosine triphosphate production in skeletal muscle from patients with dominant optic atrophy due to OPA1 mutations. Arch. Neurol. 2011, 68, 67–73. [Google Scholar] [CrossRef]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley IV, C.A.; Ramalho-Santos, J.; van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef]
- Papa, L.; Djedaini, M.; Hoffman, R. Mitochondrial role in stemness and differentiation of hematopoietic stem cells. Stem Cells Int. 2019, 2019, 4067162. [Google Scholar] [CrossRef]
- Kao, S.H.; Yen, M.Y.; Wang, A.G.; Yeh, Y.L.; Lin, A. Lo Changes in mitochondrial morphology and bioenergetics in human lymphoblastoid cells with four novel opa1 mutations. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2269–2278. [Google Scholar] [CrossRef]
- Nickells, R.W. Apoptosis of Retinal Ganglion Cells in Glaucoma. Surv. Ophthalmol. 1999, 43, S151–S161. [Google Scholar] [CrossRef] [PubMed]
- Bien, A.; Seidenbecher, C.I.; Böckers, T.M.; Sabel, B.A.; Kreutz, M.R. Apoptotic versus necrotic characteristics of retinal ganglion cell death after partial optic nerve injury. J. Neurotrauma 1999, 16, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Wood, J.P.M.; Chidlow, G.; Bae, J.H.; Melena, J.; Nash, M.S. Ganglion cell death in glaucoma: What do we really know? Br. J. Ophthalmol. 1999, 83, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Chevrollier, A.; Guillet, V.; Loiseau, D.; Gueguen, N.; De Crescenzo, M.A.P.; Verny, C.; Ferre, M.; Dollfus, H.; Odent, S.; Milea, D.; et al. Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann. Neurol. 2008, 63, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Karbowski, M.; Youle, R.J.; Schimpf, S.; Wissinger, B.; Pinti, M.; Cossarizza, A.; Vidoni, S.; et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008, 131, 352–367. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P. Therapeutic approaches to inherited optic neuropathies. Semin. Neurol. 2015, 35, 578–586. [Google Scholar] [CrossRef]
- Ferré, M.; Bonneau, D.; Milean, D.; Chevrollier, A.; Verny, C.; Dollfus, H.; Ayuso, C.; Defoort, S.; Vignal, C.; Zanlongui, X.; et al. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum. Mutat. 2009, 30, E692–E705. [Google Scholar] [CrossRef]
- Schimpf, S.; Fuhrmann, N.; Schaich, S.; Wissinger, B. Comprehensive cDNA study and quantitative transcript analysis of mutant OPA1 transcript containing premature termination codons. Hum. Mutat. 2008, 29, 106–112. [Google Scholar] [CrossRef]
- Pereverzev, A.P.; Gurskaya, N.G.; Ermakova, G.V.; Kudryavtseva, E.I.; Markina, N.M.; Kotlobay, A.A.; Lukyanov, S.A.; Zaraisky, A.G.; Lukyanov, K.A. Method for quantitative analysis of nonsense-mediated mRNA decay at the single cell level. Sci. Rep. 2015, 5, 7729. [Google Scholar] [CrossRef]
- Kamat, J.P.; Devasagayam, T.P.A. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox Rep. 1998, 4, 179–784. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin. Exp. Ophthalmol. 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Fuster-garcía, C.; García-garcía, G.; González-romero, E.; Jaijo, T.; Sequedo, M.D.; Ayuso, C.; Vázquez-manrique, R.P.; Millán, J.M.; Aller, E. USH2A gene editing using the CRISPR system. Mol. Ther. Nucleic Acid 2017, 8, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Galera-Monge, T.; Zurita-Díaz, F.; Garesse, R.; Gallardo, M.E. The mutation m.13513G>A impairs cardiac function, favoring a neuroectoderm commitment, in a mutant-load dependent way. J. Cell. Physiol. 2019, 234, 19511–19522. [Google Scholar] [CrossRef] [PubMed]
- Ortuño-Costela, M.d.C.; Rodríguez-Mancera, N.; García-López, M.; Zurita-Díaz, F.; Moreno-Izquierdo, A.; Lucía, A.; Martín, M.Á.; Garesse, R.; Gallardo, M.E. Establishment of a human iPSC line (IISHDOi001-A) from a patient with McArdle disease. Stem Cell Res. 2017, 23, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; De La Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef]
- Cerrada, V.; García-López, M.; Moreno-Izquierdo, A.; Villaverde, C.; Zurita, O.; Martin-Merida, M.I.; Arenas, J.; Ayuso, C.; Gallardo, M.E. Derivation of a human DOA iPSC line, IISHDOi006-A, with a mutation in the ACO2 gene: c.1999G>A; p.Glu667Lys. Stem Cell Res. 2019, 40, 101566. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strand | crRNA Sequence (5′→3′) | PAM | On-Target Score | Off-Target Score | Off-Targets |
|---|---|---|---|---|---|
| + | AATCCTTTAACAATCTTTGT | GGG | 75 | 0 | 1 (3–4 MM) |
| − | TTTCCCACAAAGATTGTTAA | AGG | 79 | 0 | 3 (3–4 MM) |
| Primary Antibodies | ||
| Antibody | Dilution | Reference |
| Mouse Anti-TRA-1-81 | 1:150 | Millipore, Burlington, MA, USA; #MAB4381 |
| Mouse Anti-TRA-1-60 | 1:150 | Millipore, Burlington, MA, USA; #MAB4360 |
| Rabbit Anti-SOX2 | 1:100 | Thermo Fisher Scientific, Waltham, MA, USA; #PA1-16968 |
| Mouse Anti-SSEA4 | 1:10 | Millipore, Burlington, MA, USA; #MAB4304 |
| Rat Anti-SSEA3 | 1:20 | Abcam, Cambridge, UK; #ab16286 |
| Goat Anti-NANOG | 1:25 | R&D Systems, Minneapolis, MN, USA; #sc-5279 |
| Mouse Anti-OCT4 | 1:100 | Santa Cruz Biotechnology, Dallas, TX, USA; #sc-5279 |
| Secondary antibodies | ||
| Antibody | Dilution | Reference |
| CyTM2-conjugated AffiniPure Donkey Anti-Goat IgG (H + L) | 1:50 | Jackson ImmunoResearch Labs, Ely, UK; #705-225-147 |
| CyTM2-conjugated AffiniPure Goat Anti-Mouse IgG, Fcγ Subclass 2b specific | 1:50 | Jackson ImmunoResearch Labs, Ely, UK; #115-225-207 |
| CyTM2-conjugated AffiniPure anti-conejo IgG (H + L) | 1:50 | Jackson ImmunoResearch Labs, Ely, UK; #111-225-144 |
| CyTM3-conjugated AffiniPure Goat Anti-Rat IgM, μ chain specific | 1:250 | Jackson ImmunoResearch Labs, Ely, UK; #112-165-075 |
| CyTM-3conjugated AffiniPure Goat Anti-Mouse IgG, Fcγ Subclass 3 specific | 1:250 | Jackson ImmunoResearch Labs, Ely, UK; #115-165-209 |
| CyTM-3conjugated AffiniPure Donkey Anti-Mouse IgM, μ chain specific | 1:250 | Jackson ImmunoResearch Labs, Ely, UK; #115-165-020 |
| Alexa Fluor 488, Goat Anti-Mouse IgG (H + L) | 1:500 | Thermo Fisher Scientific, Waltham, MA, USA; #A32723 |
| Alexa fluor 568, Goat Anti-Rabbit IgG (H + L) | 1:800 | Thermo Fisher Scientific, Waltham, MA, USA; #A-11011 |
| Primary Antibodies | ||
| Antibody | Dilution | Reference |
| Mouse Anti-AFP (endoderm) | 1:300 | Merck, Darmstadt, Germany; #WH0000174M1 |
| Mouse Anti-SMA (mesoderm) | 1:400 | Merck, Darmstadt, Germany; #A2547 |
| Mouse Anti-Tuj1 (ectoderm) | 1:300 | Merck, Darmstadt, Germany; #T8660 |
| Secondary antibodies | ||
| Antibody | Dilution | Reference |
| Goat Anti-Mouse Alexa Fluor® 488 | 1:500 | Thermo Fisher Scientific, Waltham, MA, USA; #A-11029 |
| N2 Medium | |||
|---|---|---|---|
| Composition | Concentration | Supplier | Reference |
| DMEM/F-12 | - | Gibco | 11320-033 |
| N2 supplement (100X) | 1X | Gibco | 17504044 |
| Insulin solution | 20 mg/mL | Sigma | I9278 |
| XAV939 | 1 µM | StemCell Technologies | 72672 |
| Human FGF-2 | 60 ng/mL | Miltenyi Biotec | 130-093-838 |
| N2B27 Medium | |||
|---|---|---|---|
| Composition | Concentration | Supplier | Reference |
| DMEM/F-12 | - | Gibco | 11320-033 |
| N2 supplement (100X) | 1X | Gibco | 17504044 |
| B27 supplement without vitamin A (50X) | 1X | Gibco | 12587-001 |
| Human FGF-2 | 20 ng/mL | Miltenyi Biotec | 130-093-838 |
| Primer | Sequence (5′-3′) |
|---|---|
| PAX6-Fw | CAGATGTGTTTGCCCGAGAA |
| PAX6-Rv | GCCTGTCTTCTCTGATTCCT |
| RX-Fw | CAAGGTCAACCTACCAGAGGT |
| RX-Rv | CAGCTTCATGGAGGACACTTC |
| CHX10-Fw | CGGAGTATGGGCTCTACGG |
| CHX10-Rv | CTCCATCTTGTCGAGCTTGG |
| SIX6-Fw | TACGCAGGTGGGCAACTGGT |
| SIX6-Rv | GTAGTGCCCGCCCGGAAC |
| BRN3A-Fw | CTCCCTGAGCACAAGTACCC |
| BRN3A-Rv | CCGGCTTGAAAGGATGGCTC |
| BRN3B-Fw | CTCGGAGGCTATGCGGAGA |
| BRN3B-Rv | TGGTAGGTGGCGTCCGGTT |
| ISLET1-Fw | AAGGACAAGAAGCGAAGCAT |
| ISLET1-Rv | TTCCTGTCATCCCCTGGATA |
| SNCG-Fw | TGGTGAGCAGCGTCAACACT |
| SNCG-Rv | CCTCTTTCTCTTTGGATGCC |
| ATOH7-Fw | GTCTCCACTGTGAGCACTTC |
| ATOH7-Rv | GAAGCCGAAGAGTCTCTGGC |
| THY1-Fw | AGGTCCTCTACTTATCCGCC |
| THY1-Rv | ATGCCCTCACACTTGACCAG |
| GAPDH-Fw | GCACCGTCAAGGCTGAGAAC |
| GAPDH-Rv | AGGGATCTCGCTCCTGGAA |
| Primary Antibodies | ||
| Antibody | Dilution | Reference |
| Rabbit anti-PAX6 | 1:500 | Thermo Fisher Scientific, Waltham, MA, USA; #PA5-85374 |
| Mouse anti-BRN3A | 1:50 | Santa Cruz Biotechnology, Dallas, TX, USA; #sc-8429 |
| Rabbit anti-SNCG | 1:500 | Thermo Fisher Scientific, Waltham, MA, USA; #PA5-29142 |
| Mouse anti-THY1 | 1:100 | Thermo Fisher Scientific, Waltham, MA, USA; # MA5-16671 |
| Mouse anti-TUJ1 | 1:300 | Merck, Darmstadt, Germany; #T8660 |
| Secondary antibodies | ||
| Antibody | Dilution | Reference |
| Alexa Fluor 488, Goat Anti-Mouse IgG (H + L) | 1:500 | Thermo Fisher Scientific, Waltham, MA, USA; #A32723 |
| Alexa fluor 568, Goat anti-Rabbit IgG (H + L) | 1:800 | Thermo Fisher Scientific, Waltham, MA, USA; #A-11011 |
| Primer | Sequence (5′-3′) |
|---|---|
| OPA1-Fw | CAGATGTGTTTGCCCGAGAA |
| OPA1-Rv | GCCTGTCTTCTCTGATTCCT |
| GAPDH-Fw | GCACCGTCAAGGCTGAGAAC |
| GAPDH-Rv | AGGGATCTCGCTCCTGGAA |
| Primer | Sequence (5′-3′) |
|---|---|
| F8285-8314 (FW) | CTCTAGAGCCCACTGTAAAGCTAACTTAGC |
| R15600-15574 (RV) | GGGACGGATCGGAGAATTGTGTAGGCG |
| Primer | Sequence (5′-3′) |
|---|---|
| 12S-FW | CCACGGGAAACAGCAGTGAT |
| 12S-RV | CTATTGACTTGGGTTAATCGTGTGA |
| ARNase P-FW | GCTCTCTGAAAGTGACGCC |
| ARNase P-RV | CTCCATGGAGAAGCGCTGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-López, M.; Jiménez-Vicente, L.; González-Jabardo, R.; Dorado, H.; Gómez-Manjón, I.; Martín, M.Á.; Ayuso, C.; Arenas, J.; Gallardo, M.E. Creation of an Isogenic Human iPSC-Based RGC Model of Dominant Optic Atrophy Harboring the Pathogenic Variant c.1861C>T (p.Gln621Ter) in the OPA1 Gene. Int. J. Mol. Sci. 2024, 25, 7240. https://doi.org/10.3390/ijms25137240
García-López M, Jiménez-Vicente L, González-Jabardo R, Dorado H, Gómez-Manjón I, Martín MÁ, Ayuso C, Arenas J, Gallardo ME. Creation of an Isogenic Human iPSC-Based RGC Model of Dominant Optic Atrophy Harboring the Pathogenic Variant c.1861C>T (p.Gln621Ter) in the OPA1 Gene. International Journal of Molecular Sciences. 2024; 25(13):7240. https://doi.org/10.3390/ijms25137240
Chicago/Turabian StyleGarcía-López, Marta, Lydia Jiménez-Vicente, Raquel González-Jabardo, Helena Dorado, Irene Gómez-Manjón, Miguel Ángel Martín, Carmen Ayuso, Joaquín Arenas, and María Esther Gallardo. 2024. "Creation of an Isogenic Human iPSC-Based RGC Model of Dominant Optic Atrophy Harboring the Pathogenic Variant c.1861C>T (p.Gln621Ter) in the OPA1 Gene" International Journal of Molecular Sciences 25, no. 13: 7240. https://doi.org/10.3390/ijms25137240
APA StyleGarcía-López, M., Jiménez-Vicente, L., González-Jabardo, R., Dorado, H., Gómez-Manjón, I., Martín, M. Á., Ayuso, C., Arenas, J., & Gallardo, M. E. (2024). Creation of an Isogenic Human iPSC-Based RGC Model of Dominant Optic Atrophy Harboring the Pathogenic Variant c.1861C>T (p.Gln621Ter) in the OPA1 Gene. International Journal of Molecular Sciences, 25(13), 7240. https://doi.org/10.3390/ijms25137240