
Endothelial Protection by Sodium-Glucose Cotransporter 2 Inhibitors: A Literature Review of In Vitro and In Vivo Studies
 , , and
, , and
Abstract
:1. Introduction
2. Molecular Mechanisms Contributing to Endothelial Dysfunction
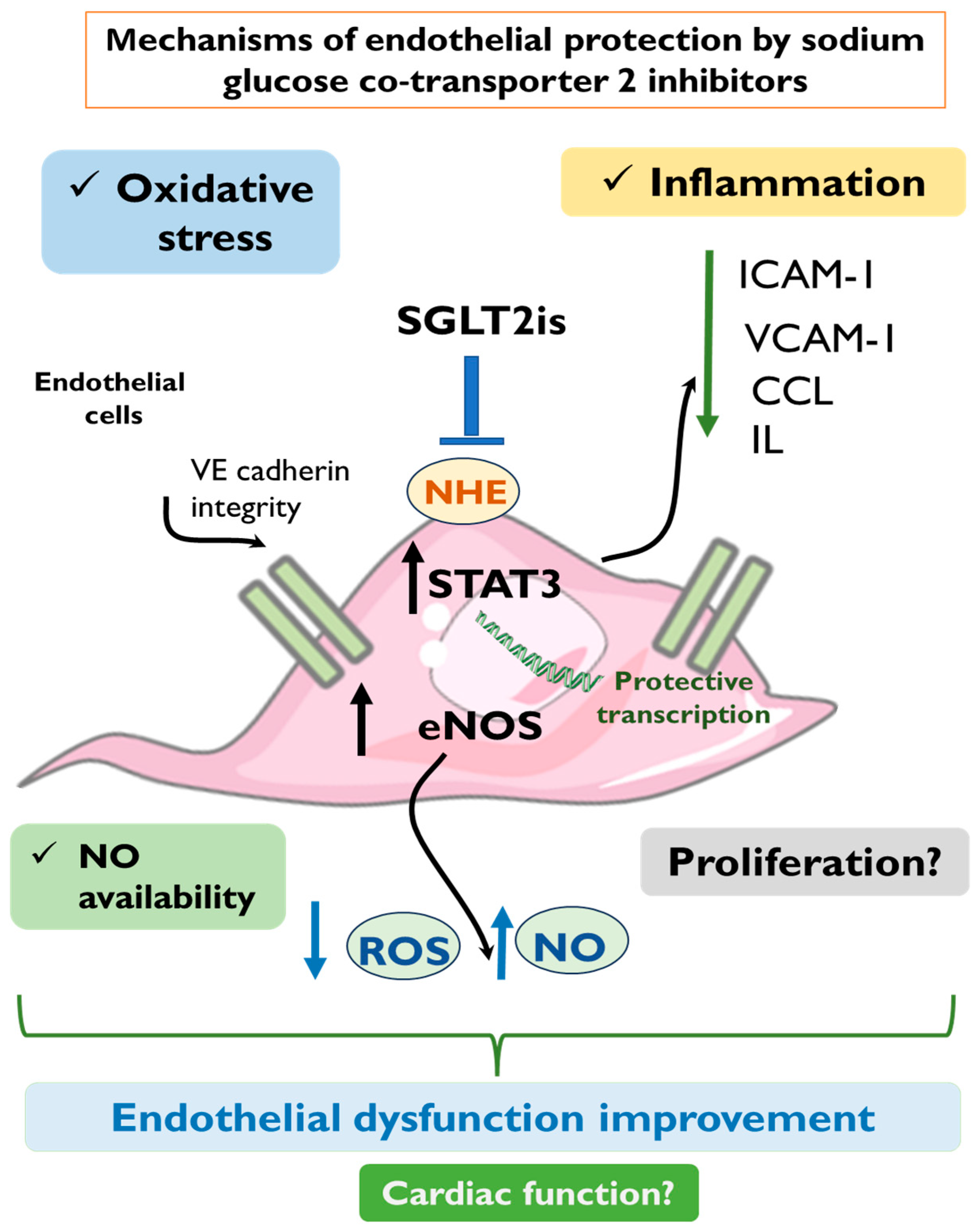
3. Suggested Mechanisms for SGLT2is Endothelial Protection
3.1. Anti-Oxidative Effect of SGLT2is
3.1.1. In Vitro Studies
3.1.2. In Vivo Studies
3.2. Improved NO Production and Vasodilation
3.2.1. In Vitro Studies
3.2.2. In Vivo Studies
3.3. Anti-Inflammatory Effect of SGLT2is
3.3.1. In Vitro Studies
3.3.2. In Vivo Studies
3.4. Impact of SGLT2is on Endothelial Cell Survival and Angiogenesis
3.4.1. In Vitro Studies
3.4.2. In Vivo Studies

{kind=link}
| Drug (Concentration) | Experimental Model | Stimulant | Major Findings | Ref. |
|---|---|---|---|---|
| EMPA (1 μM)/6 h | HCAECs HUVECs | TNF-α (10 ng/mL)/6 h | ↓ NHE activity ↓ ROS | [45] |
| EMPA (1 μM)/6 h | CMECs | Uraemic serum (15%)/6 h | ↓ ROS ↑ NO bioavailability | [71] |
| EMPA (1 μM)/6 h | CMECs | TNF-α (10 ng/mL)/6 h | ↓ ROS ↑ NO | [61] |
| EMPA (10 μM)/12 h | HCAECs | Hypoxia/Reoxygenation | ↓ ROS ↓ Mitochondrial fission ↓ ICAM-1 ↑ VE-cadherin ↑ p-eNOS | [83] |
| EMPA (50 μM)/24 h | HAECs | TNF-α (10 ng/mL)/24 h | ↓ Leukocyte–endothelium adhesion | [70] |
| EMPA (500 nM)/24 h | HMECs | Hypoxia/Reoxygenation | ↓ ROS ↑ p-STAT3 ↑ Cell viability | [79] |
| DAPA (1 μM)/24 h | HUVECs | - | ↓ NHE activity | [44] |
| DAPA (10 μM)/12 h | HCAECs | Hypoxia/Reoxygenation | ↓ Mitochondrial fission ↓ ICAM-1 ↑ p-eNOS ↑ VEGF ↑ Cell Survival | [62] |
| DAPA (10 μM)/72 h | HUVECS | H2O2 (100 µM)/1 h | ↓ β-gal, p21, p53 ↓ Senescence ↑ SIRT-1 ↑ p-eNOS | [63] |
| DAPA (1–5 nM)/24 h | HUVECs | TNF-α (10 ng/mL)/24 h | ↓ ICAM-1 ↓ VCAM-1 | [69] |
| DAPA (0.05–0.5 μM)/24 h | HUVECs | LPS (20 ng/mL)/24 h | ↓ IL-6, IL-8 ↓ NF-κB | [74] |
| CANA (3 or 10 μM)/16 h | HCAECs | LPS (1 μg/mL)/3 h | ↓ IL-6 ↑ p-AMPK | [72] |
| EMPA (1 μM)/ 24 h DAPA (1 μM)/ 24 h CANA (3 μM)/ 24 h | HCAECs | Cyclic stretch (1 Hz, 10%)/24 h | ↓ NHE and NOX activity ↓ ROS ↓ Cell permeability ↑ VE-cadherin | [43] |
| EMPA (30–50 μM)/ 1–3 days DAPA (30–50 μM)/ 1–3 days CANA (10–50 μM)/1–3 days | HUVECs HAECs | - | ↓ Angiogenesis ↓ Cell viability | [80] |
| Drug (Dosage) | Experimental Model | Stimulant/Intervention | Major Findings | Ref. |
|---|---|---|---|---|
| EMPA (10 mg/kg/day)/20 weeks | C57BL/6J mice (Diabetic) | STZ (50 mg/kg/day)/5 days | ↓ ROS ↓ Mitochondrial fission and fusion ↓ Senescence ↑ Angiogenesis | [42] |
| EMPA (10 or 30 mg/kg/day)/ 7 weeks | Wistar rats (Diabetic) | STZ (60 mg/kg) | ↓ ROS, NOX-1, NOX-2 ↓ IL-6, CCL-2 ↓ ICAM-1 ↑ p-eNOS | [48] |
| EMPA (10 or 30 mg/kg/day)/ 6 weeks | ZDF rats (Diabetic) | - | ↓ ROS ↓ ICAM-1 ↓ COX-2, iNOS | [50] |
| EMPA (10 mg/kg/day)/ 6 weeks | C57BL/6J mice (Nondiabetic) | Myocardial ischemia/reperfusion | ↓ Infarct size ↓ MDA ↓ Protein carbonyls ↑ p-STAT3 ↑ SOD-2 | [79] |
| EMPA (10 mg/day)/ 7 days | Yorkshire pigs (Nondiabetic) | Myocardial ischemia/reperfusion | ↓ Infarct size ↓ ROS ↑ Cardiac function | [55] |
| EMPA (14 mg/kg/day)/ 6 weeks | C57BL/6J Aged mice (Nondiabetic) | - | ↓ ROS, MDA ↓ Arterial stiffness ↑ p-eNOS | [82] |
| EMPA (10 mg/day)/ 2 months | Yorkshire pigs (Nondiabetic) | Myocardial ischemia/reperfusion | ↑ Cardiac function ↑ NO ↑ p-eNOS | [65] |
| EMPA (30 mg/kg/day)/ 6 weeks | ZSF1 rats (Diabetic) | - | ↑ NO ↑ p-eNOS | [67] |
| DAPA (60 mg/kg/day)/ 8 weeks | db/db mice (Diabetic) | - | ↓ IL-1β, IL-6, CCL-2 ↑ p-eNOS | [66] |
| DAPA (1 mg/kg/day)/ 8 weeks | db/db mice (Diabetic) | - | ↑ NO ↑ p-eNOS | [63] |
| DAPA (0.1 mg/kg/day)/ 6 weeks | Dahl salt-sensitive rats (Nondiabetic) | 8% NaCl special diet | ↓ NF-κB, IL-6, CCL-2, E-selectin | [66] |
| CANA (20 mg/kg/day)/ 6 weeks | C57BL/6J mice (Diabetic) | STZ (150 mg/kg, single dose) | ↓ ROS ↓ MDA | [53] |
| CANA (10 mg/kg/day)/ 5 weeks | ApoE−/− mice (Diabetic) | High-fat diet | ↓ Atherosclerotic plaques ↓ CCL-2 ↓ VCAM-1 | [77] |
4. Summary and Future Perspectives
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end products |
| CANA | Canagliflozin |
| CCL | Chemokine (C-C motif) ligand |
| CMECs | Cardiac microvascular endothelial cells |
| DAPA | Dapagliflozin |
| COX-2 | Cyclooxygenase-2 |
| EMPA | Empagliflozin |
| eNOS | Endothelial nitric oxide synthase |
| HAECs | Human aortic endothelial cells |
| HCAECs | Human coronary artery endothelial cells |
| HMECs, | Human microvascular endothelial cells |
| HUVECs | Human umbilical vein endothelial cells |
| HF | Heart failure |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| ICAM-1 | Intracellular adhesion molecule-1 |
| IFN-γ | Interferon-γ |
| IL | Interleukins |
| iNOS | Inducible nitric oxide synthase |
| LVEF | Left ventricular ejection fraction |
| NHE1 | Sodium–hydrogen exchanger 1 |
| NCX | Sodium–calcium exchangers |
| NO | Nitric oxide |
| NOXs | Nicotinamide adenine dinucleotide phosphate oxidases |
| RAGE | Receptors for advanced glycation end products |
| ROS | Reactive oxygen species |
| SGLT2is | Sodium-glucose cotransporter 2 inhibitors |
| SIRT1 | Sirtuin 1 |
| SFK | Src family of kinases |
| STAT3 | Signal transducer and activator of transcription 3 |
| T2DM | Type 2 diabetes mellitus |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
| VE-cadherin | Vascular endothelial-cadherin |
References
- Ferrannini, E. Sodium-glucose co-transporters and their inhibition: Clinical physiology. Cell Metab. 2017, 26, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Chao, E.C.; Henry, R.R. SGLT2 inhibition—A novel strategy for diabetes treatment. Nat. Rev. Drug Discov. 2010, 9, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in heart failure with a preserved ejection fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Santulli, G.; Wang, X.; Mone, P. Updated ACC/AHA/HFSA 2022 guidelines on heart failure: What is new? From epidemiology to clinical management. Eur. Heart J.-Cardiovasc. Pharmacother. 2022, 8, e23–e24. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Lam, C.S.; Svedlund, S.; Saraste, A.; Hage, C.; Tan, R.-S.; Beussink-Nelson, L.; Ljung Faxén, U.; Fermer, M.L.; Broberg, M.A.; et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur. Heart J. 2018, 39, 3439–3450. [Google Scholar] [CrossRef]
- Hage, C.; Svedlund, S.; Saraste, A.; Faxén, U.L.; Benson, L.; Fermer, M.L.; Gan, L.-M.; Shah, S.J.; Lam, C.S.; Lund, L.H. Association of coronary microvascular dysfunction with heart failure hospitalizations and mortality in heart failure with preserved ejection fraction: A follow-up in the PROMIS-HFpEF study. J. Card. Fail. 2020, 26, 1016–1021. [Google Scholar] [CrossRef]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G.Á.; Falcão-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Effect of empagliflozin on endothelial function in patients with type 2 diabetes and cardiovascular disease: Results from the multicenter, randomized, placebo-controlled, double-blind EMBLEM trial. Diabetes Care 2019, 42, e159–e161. [Google Scholar] [CrossRef] [PubMed]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, K.; Adamopoulou, E.; Pyrpyris, N.; Sakalidis, A.; Leontsinis, I.; Manta, E.; Mantzouranis, E.; Beneki, E.; Soulaidopoulos, S.; Konstantinidis, D.; et al. The effect of SGLT2 inhibitors on the endothelium and the microcirculation: From bench to bedside and beyond. Eur. Heart J. Cardiovasc. Pharmacother. 2023, 9, 741–757. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.K.; Symons, J.D.; Boudina, S.; Jaishy, B.; Shiu, Y.-T. Role of endothelial cells in myocardial ischemia-reperfusion injury. Vasc. Dis. Prev. 2010, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- De Vriese, A.S.; Verbeuren, T.J.; Van de Voorde, J.; Lameire, N.H.; Vanhoutte, P.M. Endothelial dysfunction in diabetes. Br. J. Pharmacol. 2000, 130, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Mallamaci, F.; Zoccali, C. Endothelial dysfunction in chronic kidney disease, from biology to clinical outcomes: A 2020 update. J. Clin. Med. 2020, 9, 2359. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care 2009, 32, S314. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Obokata, M.; Reddy, Y.N.; Redfield, M.M.; Lerman, A.; Borlaug, B.A. Endothelium-dependent and independent coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2020, 22, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Iliodromitis, E.K.; Rassaf, T.; Schulz, R.; Papapetropoulos, A.; Ferdinandy, P. The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br. J. Pharmacol. 2015, 172, 1587–1606. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Schulz, R.; Papapetropoulos, A.; Turan, B.; Ytrehus, K.; Ferdinandy, P.; Daiber, A.; Di Lisa, F. The role of mitochondrial reactive oxygen species, NO and H2S in ischaemia/reperfusion injury and cardioprotection. J. Cell. Mol. Med. 2020, 24, 6510–6522. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Farmakis, D.; Prokovas, E.; Sigala, F.; Zoga, A.; Spyridaki, K.; Papalois, A.; Papapetropoulos, A.; Anastasiou-Nana, M.; Kremastinos, D.T.; et al. Short-term statin administration in hypercholesterolaemic rabbits resistant to postconditioning: Effects on infarct size, endothelial nitric oxide synthase, and nitro-oxidative stress. Cardiovasc. Res. 2012, 94, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Daiber, A.; Baxter, G.F.; Brizzi, M.F.; Di Lisa, F.; Kaludercic, N.; Lazou, A.; Varga, Z.V.; Zuurbier, C.J.; Schulz, R.; et al. Influence of cardiometabolic comorbidities on myocardial function, infarction, and cardioprotection: Role of cardiac redox signaling. Free Radic. Biol. Med. 2021, 166, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Pavlidis, G.; Tsoumani, M.; Kousathana, F.; Katogiannis, K.; Tsilivarakis, D.; Thymis, J.; Kountouri, A.; Korakas, E.; Pliouta, L.; et al. Endothelial dysfunction is associated with decreased nitric oxide bioavailability in dysglycaemic subjects and first-degree relatives of type 2 diabetic patients. J. Clin. Med. 2022, 11, 3299. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, M.; Mukosera, G.T.; Borchardt, D.; Li, Q.; Tipple, T.E.; Ishtiaq Ahmed, A.S.; Power, G.G.; Blood, A.B. L-NAME releases nitric oxide and potentiates subsequent nitroglycerin-mediated vasodilation. Redox Biol. 2019, 26, 101238. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; Kieda, C.; Grillon, C. Nitric oxide modulates the expression of endothelial cell adhesion molecules involved in angiogenesis and leukocyte recruitment. Exp. Cell Res. 2011, 317, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Srihirun, S.; Sriwantana, T.; Unchern, S.; Kittikool, D.; Noulsri, E.; Pattanapanyasat, K.; Fucharoen, S.; Piknova, B.; Schechter, A.N.; Sibmooh, N. Platelet inhibition by nitrite is dependent on erythrocytes and deoxygenation. PLoS ONE 2012, 7, e30380. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive oxygen species: Drivers of physiological and pathological processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Iliodromitis, E.K.; Lazou, A.; Görbe, A.; Giricz, Z.; Schulz, R.; Ferdinandy, P. Effect of hypercholesterolaemia on myocardial function, ischaemia–reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br. J. Pharmacol. 2017, 174, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative stress and hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef] [PubMed]
- Habas, K.; Shang, L. Alterations in intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in human endothelial cells. Tissue Cell 2018, 54, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Nwariaku, F.E.; Liu, Z.; Zhu, X.; Nahari, D.; Ingle, C.; Wu, R.F.; Gu, Y.; Sarosi, G.; Terada, L.S. NADPH oxidase mediates vascular endothelial cadherin phosphorylation and endothelial dysfunction. Blood 2004, 104, 3214–3220. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A. The role of AGE/RAGE signaling in diabetes-mediated vascular calcification. J. Diabetes Res. 2016, 2016, 6809703. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, N.; Drosatos, K.; Mia, S. The role of glucose in cardiac physiology and pathophysiology. Curr. Opin. Clin. Nutr. Metab. Care 2023, 26, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Klug, N.R.; Chechneva, O.V.; Hung, B.Y.; O’Donnell, M.E. High glucose-induced effects on Na+-K+-2Cl− cotransport and Na+/H+ exchange of blood-brain barrier endothelial cells: Involvement of SGK1, PKCβII, and SPAK/OSR1. Am. J. Physiol.-Cell Physiol. 2021, 320, C619–C634. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front. Cell. Neurosci. 2017, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.D.; Mingorance, C.; Rodríguez-Rodríguez, R.; de Sotomayor, M.A. Endothelial dysfunction and aging: An update. Ageing Res. Rev. 2010, 9, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Romer, G.; Kerindongo, R.P.; Hermanides, J.; Albrecht, M.; Hollmann, M.W.; Zuurbier, C.J.; Preckel, B.; Weber, N.C. Sodium Glucose Co-Transporter 2 Inhibitors Ameliorate Endothelium Barrier Dysfunction Induced by Cyclic Stretch through Inhibition of Reactive Oxygen Species. Int. J. Mol. Sci. 2021, 22, 6044. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Ciuffreda, L.P.; Coppini, R.; Cozzolino, A.; Miccichè, A.; Dell’Aversana, C.; D’Amario, D.; Cianflone, E.; Scavone, C.; et al. Amelioration of diastolic dysfunction by dapagliflozin in a non-diabetic model involves coronary endothelium. Pharmacol. Res. 2020, 157, 104781. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Li, X.; Baartscheer, A.; Schumacher, C.A.; Baumgart, P.; Hermanides, J.; Preckel, B.; Hollmann, M.W.; Coronel, R.; Zuurbier, C.J.; et al. Empagliflozin reduces oxidative stress through inhibition of the novel inflammation/NHE/[Na+] c/ROS-pathway in human endothelial cells. Biomed. Pharmacother. 2022, 146, 112515. [Google Scholar] [CrossRef] [PubMed]
- Juni, R.P.; Al-Shama, R.; Kuster, D.W.; van der Velden, J.; Hamer, H.M.; Vervloet, M.G.; Eringa, E.C.; Koolwijk, P.; van Hinsbergh, V.W. Empagliflozin restores chronic kidney disease–induced impairment of endothelial regulation of cardiomyocyte relaxation and contraction. Kidney Int. 2021, 99, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Borriello, G.; Buonincontri, V.; de Donato, A.; Della Corte, M.; Gravina, I.; Iulianiello, P.; Joshi, R.; Mone, P.; Cacciola, G.; Viggiano, D. The interplay between sodium/glucose cotransporter type 2 and mitochondrial ionic environment. Mitochondrion 2024, 76, 101878. [Google Scholar] [PubMed]
- Oelze, M.; Kröller-Schön, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinßius, E.; Agdauletova, S.; et al. The sodium-glucose co-transporter 2 inhibitor empagliflozin improves diabetes-induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Aroor, A.R.; Das, N.A.; Carpenter, A.J.; Habibi, J.; Jia, G.; Ramirez-Perez, F.I.; Martinez-Lemus, L.; Manrique-Acevedo, C.M.; Hayden, M.R.; Duta, C.; et al. Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury. Cardiovasc. Diabetol. 2018, 17, 108. [Google Scholar] [PubMed]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Ganbaatar, B.; Fukuda, D.; Shinohara, M.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Hirata, K.; Sata, M. Empagliflozin ameliorates endothelial dysfunction and suppresses atherogenesis in diabetic apolipoprotein E-deficient mice. Eur. J. Pharmacol. 2020, 875, 173040. [Google Scholar] [CrossRef] [PubMed]
- Kuno, A.; Kimura, Y.; Mizuno, M.; Oshima, H.; Sato, T.; Moniwa, N.; Tanaka, M.; Yano, T.; Tanno, M.; Miki, T.; et al. Empagliflozin attenuates acute kidney injury after myocardial infarction in diabetic rats. Sci. Rep. 2020, 10, 7238. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Shi, H.; Xiong, L.; Wang, P.; Shi, Y. Canagliflozin mitigates ferroptosis and improves myocardial oxidative stress in mice with diabetic cardiomyopathy. Front. Endocrinol. 2022, 13, 1011669. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, P.E.; Mylonas, N.; Makridakis, M.; Makrecka-Kuka, M.; Iliou, A.; Zerikiotis, S.; Efentakis, P.; Kampoukos, S.; Kostomitsopoulos, N.; Vilskersts, R.; et al. Cardioprotection by selective SGLT-2 inhibitors in a non-diabetic mouse model of myocardial ischemia/reperfusion injury: A class or a drug effect? Basic Res. Cardiol. 2022, 117, 27. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Requena-Ibáñez, J.A.; Picatoste, B.; Fardman, B.; Ishikawa, K.; Mazurek, R.; Pieper, M.; Sartori, S.; Rodriguez-Capitán, J.; Fuster, V.; et al. Cardioprotective effect of empagliflozin and circulating ketone bodies during acute myocardial infarction. Circ. Cardiovasc. Imaging 2023, 16, e015298. [Google Scholar] [CrossRef] [PubMed]
- Chase, D.; Eykyn, T.R.; Shattock, M.J.; Chung, Y.J. Empagliflozin improves cardiac energetics during ischaemia/reperfusion by directly increasing cardiac ketone utilization. Cardiovasc. Res. 2023, 119, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Agnoletti, L.; Curello, S.; Bachetti, T.; Malacarne, F.; Gaia, G.; Comini, L.; Volterrani, M.; Bonetti, P.; Parrinello, G.; Cadei, M.; et al. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: Role of tumor necrosis factor-alpha. Circulation 1999, 100, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.P.; Greer, J.J.; van Haperen, R.; Duncker, D.J.; de Crom, R.; Lefer, D.J. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 4891–4896. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor alpha-Stimulated Human Coronary Arterial Endothelial Cells. Cell Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [PubMed]
- Ma, L.; Zou, R.; Shi, W.; Zhou, N.; Chen, S.; Zhou, H.; Chen, X.; Wu, Y. SGLT2 inhibitor dapagliflozin reduces endothelial dysfunction and microvascular damage during cardiac ischemia/reperfusion injury through normalizing the XO-SERCA2-CaMKII-coffilin pathways. Theranostics 2022, 12, 5034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tai, S.; Zhang, N.; Fu, L.; Wang, Y. Dapagliflozin prevents oxidative stress-induced endothelial dysfunction via sirtuin 1 activation. Biomed. Pharmacother. 2023, 165, 115213. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Haam, C.E.; Byeon, S.; Oh, E.Y.; Choi, S.-K.; Lee, Y.-H. Investigating the Cardiovascular Benefits of Dapagliflozin: Vasodilatory Effect on Isolated Rat Coronary Arteries. Int. J. Mol. Sci. 2023, 24, 16873. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Garcia-Ropero, A.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Vargas-Delgado, A.P.; Flores-Umanzor, E.J.; Sanz, J.; et al. Empagliflozin Ameliorates Diastolic Dysfunction and Left Ventricular Fibrosis/Stiffness in Nondiabetic Heart Failure: A Multimodality Study. JACC Cardiovasc Imaging 2021, 14, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Battson, M.L.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc. Diabetol. 2018, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Farooq, M.A.; Gaertner, S.; Bruckert, C.; Qureshi, A.W.; Lee, H.H.; Benrahla, D.; Pollet, B.; Stephan, D.; Ohlmann, P.; et al. Empagliflozin improved systolic blood pressure, endothelial dysfunction and heart remodeling in the metabolic syndrome ZSF1 rat. Cardiovasc. Diabetol. 2020, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Alsereidi, F.R.; Khashim, Z.; Marzook, H.; Gupta, A.; Shaaban, A.M.; Ramadan, M.M.; Saleh, M.A. Targeting inflammatory signaling pathways with SGLT2 inhibitors: Insights into cardiovascular health and cardiac cell improvement. Curr. Probl. Cardiol. 2024, 49, 102524. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.E.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diabetes Vasc. Dis. Res. 2018, 15, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Teoh, H.; Campeau, M.A.; Verma, S.; Leask, R.L. Empagliflozin restores the integrity of the endothelial glycocalyx in vitro. Mol. Cell. Biochem. 2019, 459, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Juni, R.P.; Kuster, D.W.; Goebel, M.; Helmes, M.; Musters, R.J.; van der Velden, J.; Koolwijk, P.; Paulus, W.J.; van Hinsbergh, V.W. Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. JACC Basic Transl. Sci. 2019, 4, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Kuschma, M.; Römer, G.; Boomsma, M.; Kessler, J.; Hermanides, J.; Hollmann, M.W.; Preckel, B.; Zuurbier, C.J.; Weber, N.C. Novel anti-inflammatory effects of canagliflozin involving hexokinase II in lipopolysaccharide-stimulated human coronary artery endothelial cells. Cardiovasc. Drugs Ther. 2021, 35, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Akoumianakis, I.; Akawi, N.; Kotanidis, C.; Antonopoulos, A.; Carena, M.; Badi, I.; Oikonomou, E.; Reus, E.; Krasopoulos, G.; et al. Direct effects of canagliflozin on human myocardial redox signalling: A novel role for SGLT1 inhibition. Eur. Heart J. 2020, 41, ehaa946.3351. [Google Scholar] [CrossRef]
- Abdollahi, E.; Keyhanfar, F.; Delbandi, A.-A.; Falak, R.; Hajimiresmaiel, S.J.; Shafiei, M. Dapagliflozin exerts anti-inflammatory effects via inhibition of LPS-induced TLR-4 overexpression and NF-κB activation in human endothelial cells and differentiated macrophages. Eur. J. Pharmacol. 2022, 918, 174715. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Bruckert, C.; Matsushita, K.; Mroueh, A.; Amissi, S.; Auger, C.; Houngue, U.; Remila, L.; Chaker, A.B.; Park, S.-H.; Algara-Suarez, P.; et al. Empagliflozin prevents angiotensin II-induced hypertension related micro and macrovascular endothelial cell activation and diastolic dysfunction in rats despite persistent hypertension: Role of endothelial SGLT1 and 2. Vasc. Pharmacol. 2022, 146, 107095. [Google Scholar] [CrossRef] [PubMed]
- Nasiri-Ansari, Ν.; Dimitriadis, G.K.; Agrogiannis, G.; Perrea, D.; Kostakis, I.D.; Kaltsas, G.; Papavassiliou, A.G.; Randeva, H.S.; Kassi, E. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc. Diabetol. 2018, 17, 106. [Google Scholar] [CrossRef]
- He, X.; Yuan, D. A review regarding the article ‘Targeting inflammatory signaling pathways with SGLT2 inhibitors: Insights into cardiovascular health and cardiac cell improvement’. Curr. Probl. Cardiol. 2024, 49, 102563. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, P.E.; Efentakis, P.; Abu Qourah, F.; Femminò, S.; Makridakis, M.; Kanaki, Z.; Varela, A.; Tsoumani, M.; Davos, C.H.; Dimitriou, C.A.; et al. Chronic empagliflozin treatment reduces myocardial infarct size in nondiabetic mice through STAT-3-mediated protection on microvascular endothelial cells and reduction of oxidative stress. Antioxid. Redox Signal. 2021, 34, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Behnammanesh, G.; Durante, Z.; Peyton, K.; Martinez-Lemus, L.; Brown, S.; Bender, S.; Durante, W. Canagliflozin inhibits human endothelial cell proliferation and tube formation. Front. Pharmacol. 2019, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, R.; Wei, L.; Yamada, K.; Hara, T.; Kuriyama, C.; Okuda, S.; Ueta, K.; Shiotani, M.; Nagamori, S.; Kanai, Y. Interaction of the sodium/glucose cotransporter (SGLT) 2 inhibitor canagliflozin with SGLT1 and SGLT2: Inhibition kinetics, sidedness of action, and transporter-associated incorporation accounting for its pharmacodynamic and pharmacokinetic features. J. Pharmacol. Exp. Ther. 2016, 358, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.N.; Ramirez-Perez, F.I.; Cabral-Amador, F.J.; Morales-Quinones, M.; Foote, C.A.; Ghiarone, T.; Sharma, N.; Power, G.; Smith, J.A.; Rector, R.S.; et al. SGLT2 inhibition attenuates arterial dysfunction and decreases vascular F-actin content and expression of proteins associated with oxidative stress in aged mice. Geroscience 2022, 44, 1657–1675. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Shi, W.; Qiu, J.; Zhou, N.; Du, N.; Zhou, H.; Chen, X.; Ma, L. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion injury through improving mitochondrial homeostasis. Cardiovasc Diabetol 2022, 21, 106. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.; Martinez, F.; et al. Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.H.; Rector, T.S.; Bank, A.J.; Williams, R.E.; Heifetz, S.M. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation 1991, 84, 1589–1596. [Google Scholar] [CrossRef]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010, 1, 57–92. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.J.; Boyd, D.; Katwan, O.J.; Strembitska, A.; Almabrouk, T.A.; Kennedy, S.; Palmer, T.M.; Salt, I.P. Canagliflozin inhibits interleukin-1beta-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein kinase-dependent and -independent mechanisms. Sci. Rep. 2018, 8, 5276. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Belcastro, E.; Hasan, H.; Matsushita, K.; Marchandot, B.; Abbas, M.; Toti, F.; Auger, C.; Jesel, L.; Ohlmann, P.; et al. Angiotensin II-induced upregulation of SGLT1 and 2 contributes to human microparticle-stimulated endothelial senescence and dysfunction: Protective effect of gliflozins. Cardiovasc. Diabetol. 2021, 20, 65. [Google Scholar] [CrossRef] [PubMed]
- Khemais-Benkhiat, S.; Belcastro, E.; Idris-Khodja, N.; Park, S.H.; Amoura, L.; Abbas, M.; Auger, C.; Kessler, L.; Mayoux, E.; Toti, F.; et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. J. Cell. Mol. Med. 2020, 24, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, T.W.; Park, G.T.; Kim, J.H.; Lee, H.C.; Han, J.H.; Yoon, A.; Yoon, D.; Kim, S.; Jung, S.M.; et al. Sodium/glucose Co-Transporter 2 Inhibitor, Empagliflozin, Alleviated Transient Expression of SGLT2 after Myocardial Infarction. Korean Circ. J. 2021, 51, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Mroueh, A.; Fakih, W.; Gong, D.S.; Auger, C.; Pieper, M.P.; Morel, O.; Mazzucotelli, J.P.; Schini-Kerth, V. SGLT2 expression in the left ventricle of cardiac patients is correlated with low-grade inflammation involving the pro-oxidant AT1R/NADPH oxidases/SGLT2 crosstalk: Potential role in heart failure. Eur. Heart J. 2023, 44, ehad655.3150. [Google Scholar] [CrossRef]
- Scisciola, L.; Paolisso, P.; Belmonte, M.; Gallinoro, E.; Delrue, L.; Taktaz, F.; Fontanella, R.A.; Degrieck, I.; Pesapane, A.; Casselman, F.; et al. Myocardial sodium–glucose cotransporter 2 expression and cardiac remodelling in patients with severe aortic stenosis: The BIO-AS study. Eur. J. Heart Fail. 2024, 26, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Q.; Christodoulou, A.; Mylonas, N.; Bakker, D.; Nederlof, R.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; Wakker, V.; et al. Sodium Glucose Cotransporter-2 Inhibitor Empagliflozin Reduces Infarct Size Independently of Sodium Glucose Cotransporter-2. Circulation 2023, 147, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yao, Q.; Hu, T.; Yu, J.; Jiang, K.; Wan, Y.; Tang, Q. Dapagliflozin protects against chronic heart failure in mice by inhibiting macrophage-mediated inflammation, independent of SGLT2. Cell Rep. Med. 2023, 4, 101334. [Google Scholar] [CrossRef] [PubMed]
- Trum, M.; Riechel, J.; Lebek, S.; Pabel, S.; Sossalla, S.T.; Hirt, S.; Arzt, M.; Maier, L.S.; Wagner, S. Empagliflozin inhibits Na+/H+ exchanger activity in human atrial cardiomyocytes. ESC Heart Fail. 2020, 7, 4429–4437. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.-M.; Lau, Y.-M.; Dhandhania, V.; Cai, Z.-J.; Lee, Y.-K.; Lai, W.-H.; Tse, H.-F.; Siu, C.-W. Empagliflozin ammeliorates high glucose induced-cardiac dysfuntion in human iPSC-derived cardiomyocytes. Sci. Rep. 2018, 8, 14872. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, S.; Citarella, A.; Trocchianesi, S.; Filardi, T.; Morano, S.; Lenzi, A.; Ferretti, E.; Crescioli, C. Cell-target-specific anti-inflammatory effect of empagliflozin: In vitro evidence in human cardiomyocytes. Front. Mol. Biosci. 2022, 9, 879522. [Google Scholar] [CrossRef]
- Paasche, A.; Wiedmann, F.; Kraft, M.; Seibertz, F.; Herlt, V.; Blochberger, P.L.; Jávorszky, N.; Beck, M.; Weirauch, L.; Seeger, T.; et al. Acute antiarrhythmic effects of SGLT2 inhibitors–dapagliflozin lowers the excitability of atrial cardiomyocytes. Basic Res. Cardiol. 2024, 119, 93–112. [Google Scholar] [CrossRef]
- Dasari, D.; Bhat, A.; Mangali, S.; Ghatage, T.; Lahane, G.P.; Sriram, D.; Dhar, A. Canagliflozin and dapagliflozin attenuate glucolipotoxicity-induced oxidative stress and apoptosis in cardiomyocytes via inhibition of sodium-glucose cotransporter-1. ACS Pharmacol. Transl. Sci. 2022, 5, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Paz, L.; Cristóbal, H.; Ortiz-Perez, J.T.; García de Frutos, P.; Mendieta, G.; Sandoval, E.; Rodriguez, J.J.; Ortega, E.; García-Álvarez, A.; Brugaletta, S.; et al. Direct actions of dapagliflozin and interactions with LCZ696 and spironolactone on cardiac fibroblasts of patients with heart failure and reduced ejection fraction. ESC Heart Fail. 2023, 10, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J. Cell. Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-C.; Lin, Y.-K.; Chen, Y.-C.; Kao, Y.-H.; Yeh, Y.-H.; Trang, N.N.; Chen, Y.-J. Empagliflozin suppressed cardiac fibrogenesis through sodium-hydrogen exchanger inhibition and modulation of the calcium homeostasis. Cardiovasc. Diabetol. 2023, 22, 27. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Lu, J.H.; Lu, R.; Lundenberg, L.; Desjardins, E.M.; Green, A.E.; Lally, J.S.; Schertzer, J.D.; Steinberg, G.R. The SGLT2 inhibitor canagliflozin suppresses lipid synthesis and interleukin-1 beta in ApoE deficient mice. Biochem. J. 2020, 477, 2347–2361. [Google Scholar] [CrossRef] [PubMed]
- Koyani, C.N.; Plastira, I.; Sourij, H.; Hallström, S.; Schmidt, A.; Rainer, P.P.; Bugger, H.; Frank, S.; Malle, E.; von Lewinski, D. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol. Res. 2020, 158, 104870. [Google Scholar] [CrossRef] [PubMed]
- Arefin, A.; Gage, M.C. Metformin, Empagliflozin, and Their Combination Modulate Ex-Vivo Macrophage Inflammatory Gene Expression. Int. J. Mol. Sci. 2023, 24, 4785. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, E.; Swistek, M.; Broncel, M.; Bukowska, B.; Gorzelak-Pabis, P. The protective effects of empagliflozin on DNA oxidative changes in a model of vascular endothelial and smooth muscle cells damaged by oxidized cholesterol. Biomed. Pharmacother. 2024, 170, 116065. [Google Scholar] [CrossRef] [PubMed]
- Behnammanesh, G.; Durante, G.L.; Khanna, Y.P.; Peyton, K.J.; Durante, W. Canagliflozin inhibits vascular smooth muscle cell proliferation and migration: Role of heme oxygenase-1. Redox Biol. 2020, 32, 101527. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, C.; Huang, Z.; Huang, C.; Liu, J.; Wu, T.; Xu, S.; Mai, P.; Geng, D.; Zhou, S.; et al. Empagliflozin alleviates atherosclerotic calcification by inhibiting osteogenic differentiation of vascular smooth muscle cells. Front. Pharmacol. 2023, 14, 1295463. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Jones, W.S.; Udell, J.A.; Anker, S.D.; Petrie, M.C.; Harrington, J.; Mattheus, M.; Zwiener, I.; Amir, O.; Bahit, M.C.; et al. Empagliflozin after acute myocardial infarction. N. Engl. J. Med. 2024, 390, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.F.; Udell, J.A.; Jones, W.S.; Anker, S.D.; Petrie, M.C.; Harrington, J.; Mattheus, M.; Seide, S.; Zwiener, I.; Amir, O.; et al. Effect of Empagliflozin on Heart Failure Outcomes after Acute Myocardial Infarction: Insights from the EMPACT-MI Trial. Circulation 2024, 149, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Konijnenberg, L.S.F.; Damman, P.; Duncker, D.J.; Kloner, R.A.; Nijveldt, R.; van Geuns, R.M.; Berry, C.; Riksen, N.P.; Escaned, J.; van Royen, N. Pathophysiology and diagnosis of coronary microvascular dysfunction in ST-elevation myocardial infarction. Cardiovasc. Res. 2020, 116, 787–805. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mylonas, N.; Nikolaou, P.E.; Karakasis, P.; Stachteas, P.; Fragakis, N.; Andreadou, I. Endothelial Protection by Sodium-Glucose Cotransporter 2 Inhibitors: A Literature Review of In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2024, 25, 7274. https://doi.org/10.3390/ijms25137274
Mylonas N, Nikolaou PE, Karakasis P, Stachteas P, Fragakis N, Andreadou I. Endothelial Protection by Sodium-Glucose Cotransporter 2 Inhibitors: A Literature Review of In Vitro and In Vivo Studies. International Journal of Molecular Sciences. 2024; 25(13):7274. https://doi.org/10.3390/ijms25137274
Chicago/Turabian StyleMylonas, Nikolaos, Panagiota Efstathia Nikolaou, Paschalis Karakasis, Panagiotis Stachteas, Nikolaos Fragakis, and Ioanna Andreadou. 2024. "Endothelial Protection by Sodium-Glucose Cotransporter 2 Inhibitors: A Literature Review of In Vitro and In Vivo Studies" International Journal of Molecular Sciences 25, no. 13: 7274. https://doi.org/10.3390/ijms25137274