
GAL3ST1 Deficiency Reduces Epithelial–Mesenchymal Transition and Tumorigenic Capacity in a Cholangiocarcinoma Cell Line

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
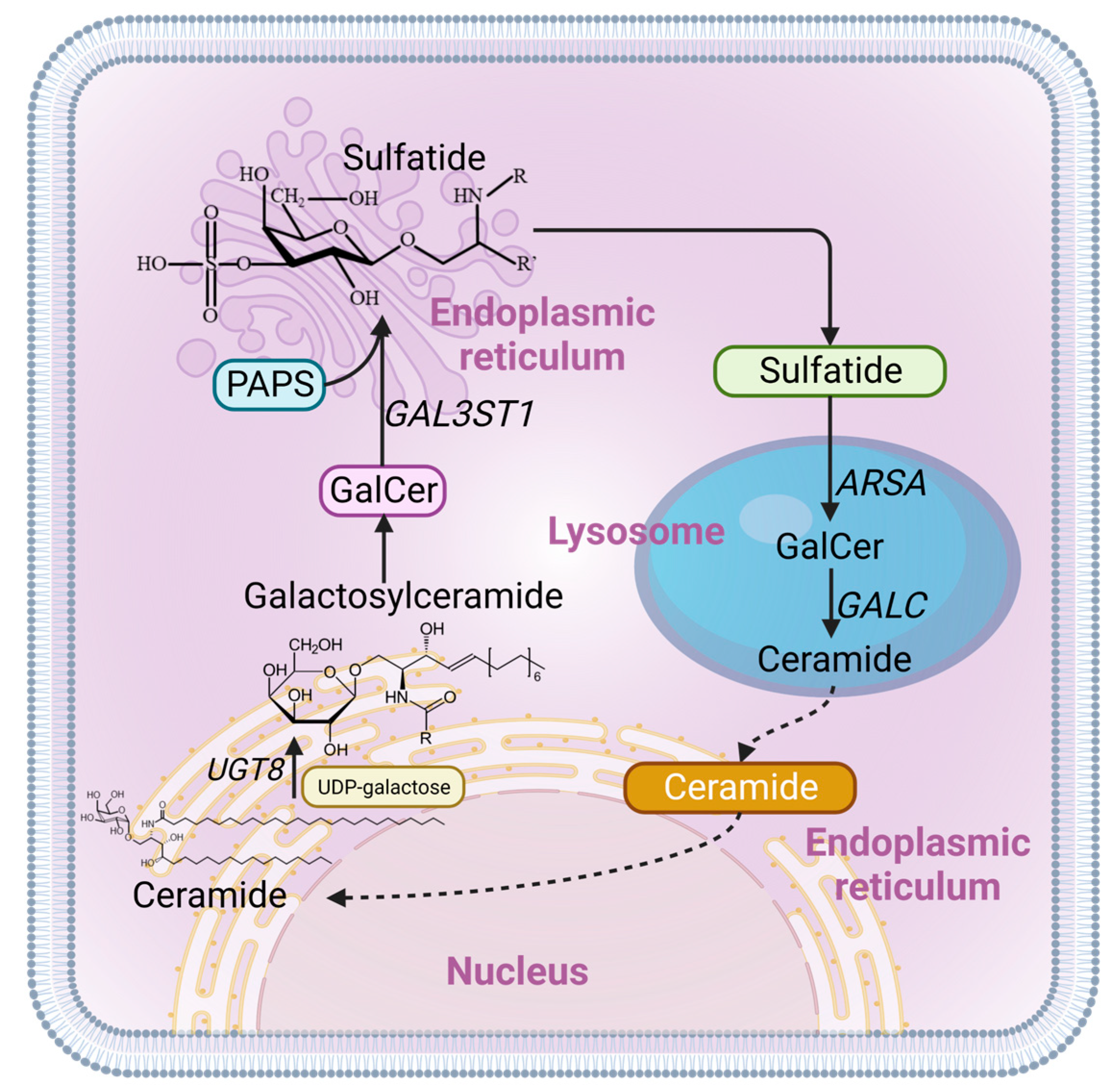
1. Introduction
2. Results
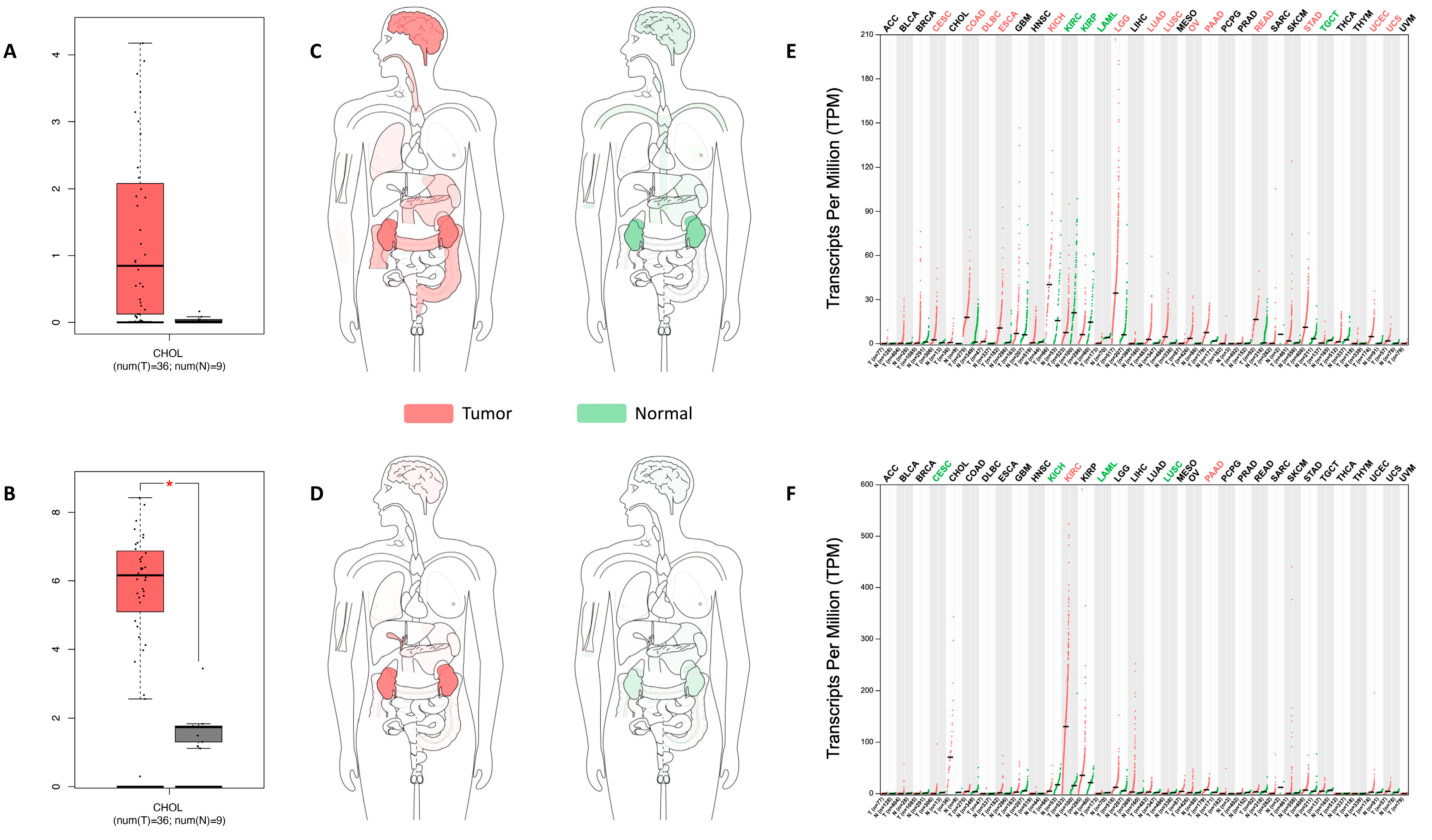
2.1. UGT8 and GAL3ST1 Expression in Human CCA
2.2. GAL3ST1 Deficiency Reduces Cell Viability, Cell Proliferation, and Clonogenic Capacity in TFK1 Cells
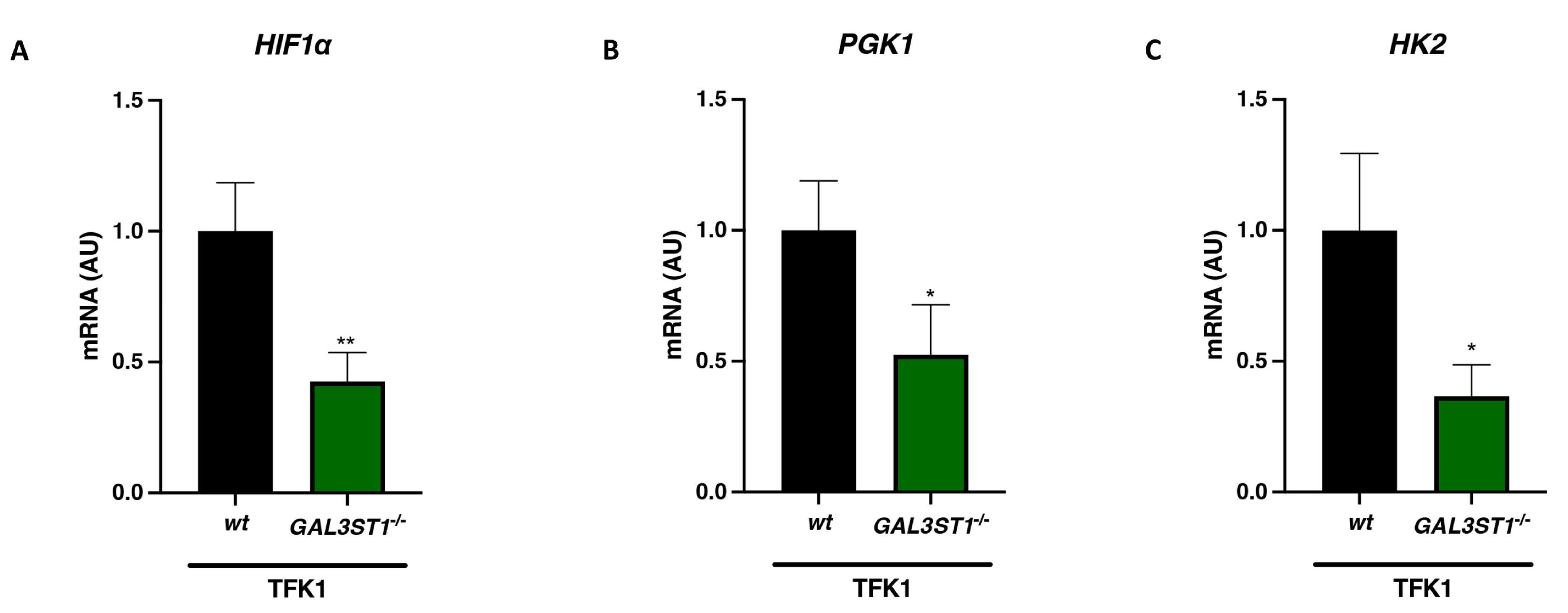
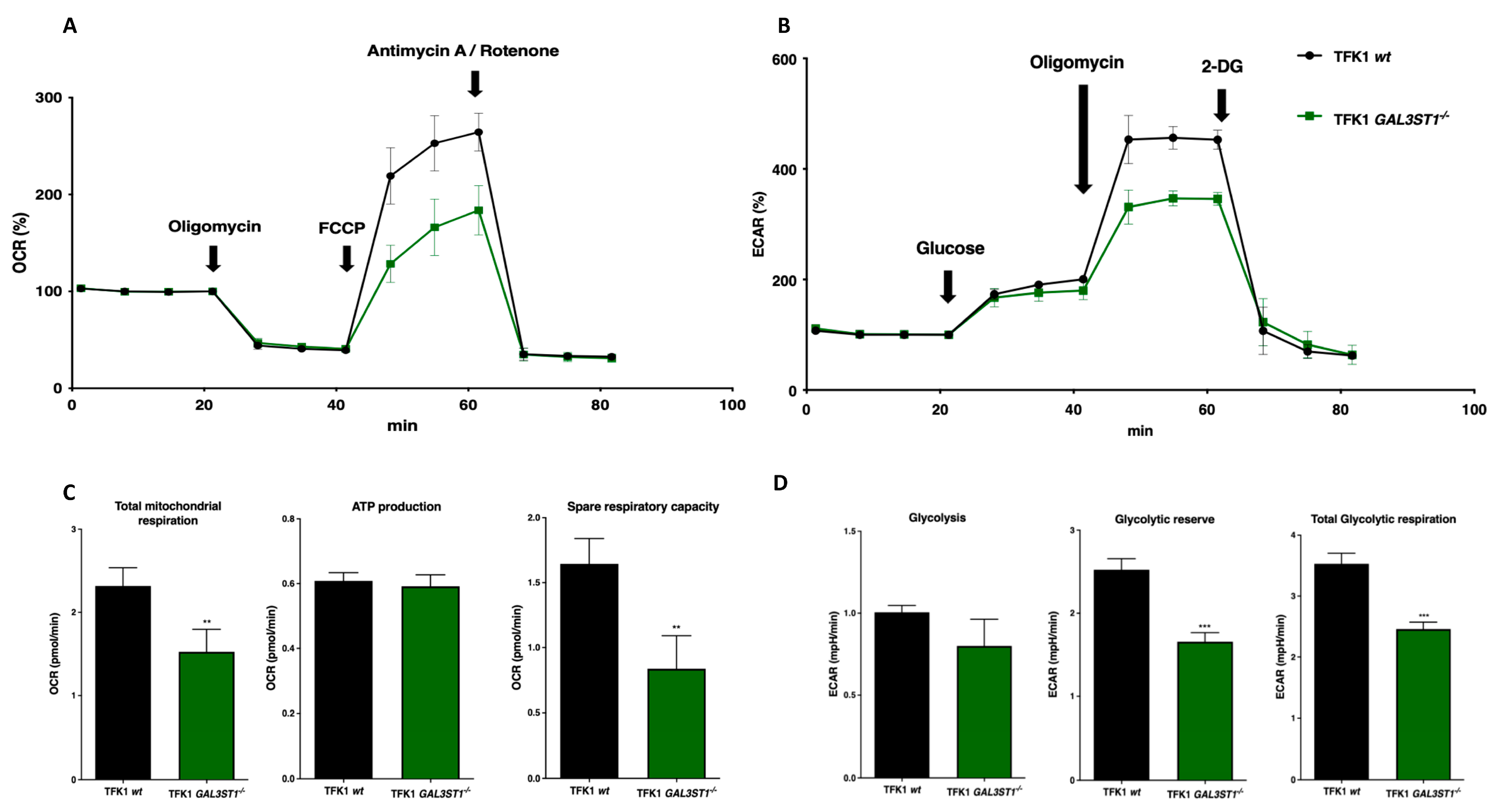
2.3. GAL3ST1 Deficiency Reprograms Glycolysis and Bioenergetic Metabolism in TFK1 Cells
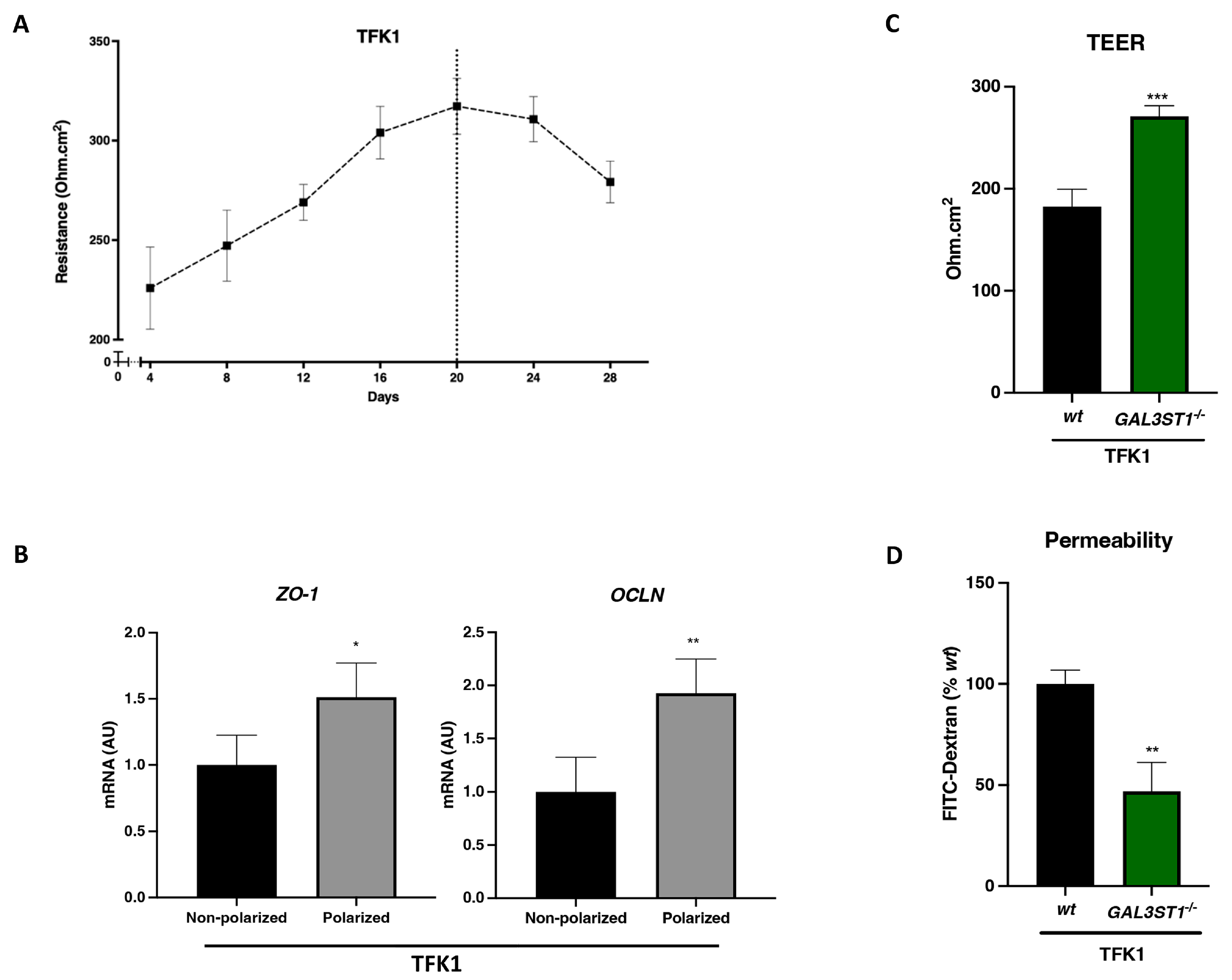
2.4. GAL3ST1 Deficiency Improves the Barrier Function in TFK1 Cells
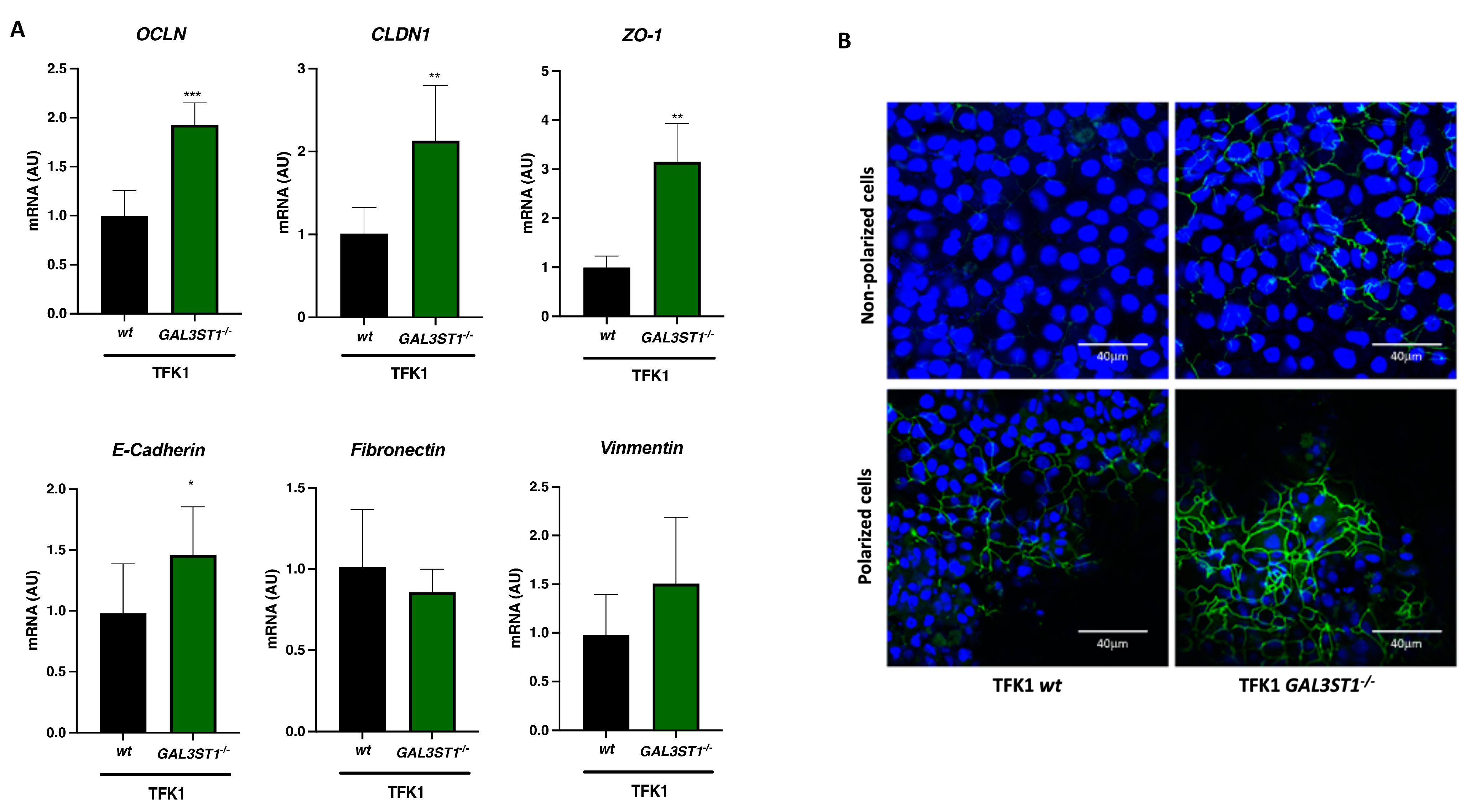
2.5. GAL3ST1 Deficiency Partially Restores Epithelial Identity in TFK1 Cells
2.6. Dependency on GAL3ST1 for the Growth of Human CCA Cell Lines
3. Discussion
4. Materials and Methods
4.1. Human Sample Collection
4.2. Hub Gene Validation by the TCGA and GEPIA Database
4.3. Cell Culture
4.4. Knockout of GAL3ST1 Genes by CRISPR-Cas9 in TFK1 Cells
4.5. MTS Proliferation Assay
4.6. Live-Cell Proliferation Assay
4.7. Colony Formation Assay
4.8. Cell Homogenate Preparation, Sulfatide Extraction, and Mass Spectrometry Analysis
4.9. Live-Cell Metabolic Analysis
4.10. Cell Differentiation, Transepithelial Electrical Resistance Assay, and Permeability Assay
4.11. Immunofluorescence Staining
4.12. RNA Isolation, cDNA Synthesis, and Quantitative Real-Time PCR Analysis
4.13. Gene Dependency Data in CCA Cell Lines
4.14. Western Immunoblotting
4.15. Wound Healing Assay
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Squadroni, M.; Tondulli, L.; Gatta, G.; Mosconi, S.; Beretta, G.; Labianca, R. Cholangiocarcinoma. Crit. Rev. Oncol. Hematol. 2017, 116, 11–31. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma-evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Oliveira, I.S.; Kilcoyne, A.; Everett, J.M.; Mino-Kenudson, M.; Harisinghani, M.G.; Ganesan, K. Cholangiocarcinoma: Classification, diagnosis, staging, imaging features, and management. Abdom. Radiol. 2017, 42, 1637–1649. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Bertuccio, P.; Bosetti, C.; Levi, F.; Decarli, A.; Negri, E.; La Vecchia, C. A comparison of trends in mortality from primary liver cancer and intrahepatic cholangiocarcinoma in Europe. Ann. Oncol. 2013, 24, 1667–1674. [Google Scholar] [CrossRef]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39 (Suppl. S1), 19–31. [Google Scholar] [CrossRef]
- Tam, P.K.H.; Yiu, R.S.; Lendahl, U.; Andersson, E.R. Cholangiopathies-Towards a molecular understanding. EBioMedicine 2018, 35, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Guedj, N.; Clapéron, A.; Nguyen Ho-Bouldoires, T.H.; Paradis, V.; Fouassier, L. Epithelial-mesenchymal transition in cholangiocarcinoma: From clinical evidence to regulatory networks. J. Hepatol. 2017, 66, 424–441. [Google Scholar] [CrossRef]
- Rao, R.K.; Samak, G. Bile duct epithelial tight junctions and barrier function. Tissue Barriers 2013, 1, e25718. [Google Scholar] [CrossRef]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef]
- Cao, X.; Surma, M.A.; Simons, K. Polarized sorting and trafficking in epithelial cells. Cell Res. 2012, 22, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Doussot, A.; Groot-Koerkamp, B.; Wiggers, J.K.; Chou, J.; Gonen, M.; DeMatteo, R.P.; Allen, P.J.; Kingham, T.P.; D’Angelica, M.I.; Jarnagin, W.R. Outcomes after Resection of Intrahepatic Cholangiocarcinoma: External Validation and Comparison of Prognostic Models. J. Am. Coll. Surg. 2015, 221, 452–461. [Google Scholar] [CrossRef]
- Sapisochin, G.; Facciuto, M.; Rubbia-Brandt, L.; Marti, J.; Mehta, N.; Yao, F.Y.; Vibert, E.; Cherqui, D.; Grant, D.R.; Hernandez-Alejandro, R.; et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016, 64, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Lunsford, K.E.; Javle, M.; Heyne, K.; Shroff, R.T.; Abdel-Wahab, R.; Gupta, N.; Mobley, C.M.; Saharia, A.; Victor, D.W.; Nguyen, D.T.; et al. Liver transplantation for locally advanced intrahepatic cholangiocarcinoma treated with neoadjuvant therapy: A prospective case-series. Lancet Gastroenterol. Hepatol. 2018, 3, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Flinders, B.; Huizing, L.R.S.; van Heerden, M.; Cuyckens, F.; Neumann, U.P.; van der Laan, L.J.W.; Olde Damink, S.W.M.; Heeren, R.M.A.; Schaap, F.G.; Vreeken, R.J. Cross-Species Molecular Imaging of Bile Salts and Lipids in Liver: Identification of Molecular Structural Markers in Health and Disease. Anal. Chem. 2018, 90, 11835–11846. [Google Scholar] [CrossRef] [PubMed]
- Huizing, L.; Chen, L.; Roeth, A.A.; Heij, L.R.; Flinders, B.; Bouwense, S.A.W.; Balluff, B.; Neumann, U.P.; Heeren, R.M.A.; Olde Damink, S.W.M.; et al. Tumor ratio of unsaturated to saturated sulfatide species is associated with disease-free survival in intrahepatic cholangiocarcinoma. Cell. Oncol. 2023, 46, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Nelson, L.J.; Ávila, M.A.; Cubero, F.J. Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link. Cells 2019, 8, 1172. [Google Scholar] [CrossRef]
- Seubwai, W.; Kraiklang, R.; Wongkham, C.; Wongkham, S. Hypoxia enhances aggressiveness of cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 2012, 13, 53–58. [Google Scholar]
- Pant, K.; Richard, S.; Peixoto, E.; Gradilone, S.A. Role of Glucose Metabolism Reprogramming in the Pathogenesis of Cholangiocarcinoma. Front. Med. 2020, 7, 113. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Masyuk, A.I.; Masyuk, T.V.; O’Hara, S.P.; LaRusso, N.F. Physiology of cholangiocytes. Compr. Physiol. 2013, 3, 541–565. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.; Zhao, Y. Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells. Biomed. Rep. 2018, 8, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Raggi, C.; Taddei, M.L.; Rae, C.; Braconi, C.; Marra, F. Metabolic reprogramming in cholangiocarcinoma. J. Hepatol. 2022, 77, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Colyn, L.; Alvarez-Sola, G.; Latasa, M.U.; Uriarte, I.; Herranz, J.M.; Arechederra, M.; Vlachogiannis, G.; Rae, C.; Pineda-Lucena, A.; Casadei-Gardini, A.; et al. New molecular mechanisms in cholangiocarcinoma: Signals triggering interleukin-6 production in tumor cells and KRAS co-opted epigenetic mediators driving metabolic reprogramming. J. Exp. Clin. Cancer Res. 2022, 41, 183. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; Lori, G.; Gentilini, A.; Taddei, M.L.; Di Maira, G.; Campani, C.; Recalcati, S.; Invernizzi, P.; Marra, F.; Raggi, C. Multifaceted Aspects of Metabolic Plasticity in Human Cholangiocarcinoma: An Overview of Current Perspectives. Cells 2020, 9, 596. [Google Scholar] [CrossRef]
- Takahashi, T.; Suzuki, T. Role of sulfatide in normal and pathological cells and tissues. J. Lipid Res. 2012, 53, 1437–1450. [Google Scholar] [CrossRef]
- Cao, Q.; Chen, X.; Wu, X.; Liao, R.; Huang, P.; Tan, Y.; Wang, L.; Ren, G.; Huang, J.; Dong, C. Inhibition of UGT8 suppresses basal-like breast cancer progression by attenuating sulfatide-αVβ5 axis. J. Exp. Med. 2018, 215, 1679–1692. [Google Scholar] [CrossRef]
- Singh, N.; Singh, A.K. A comprehensive review on structural and therapeutical insight of Cerebroside sulfotransferase (CST)—An important target for development of substrate reduction therapy against metachromatic leukodystrophy. Int. J. Biol. Macromol. 2024, 258, 128780. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed]
- Bartrons, R.; Caro, J. Hypoxia, glucose metabolism and the Warburg’s effect. J. Bioenerg. Biomembr. 2007, 39, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep. Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Poon, B.P.K.; Kano, Y.; Pluthero, F.G.; Kahr, W.H.A.; Ohh, M. A Hypoxia-Inducible HIF1-GAL3ST1-Sulfatide Axis Enhances ccRCC Immune Evasion via Increased Tumor Cell-Platelet Binding. Mol. Cancer Res. 2019, 17, 2306–2317. [Google Scholar] [CrossRef]
- Yoo, K.S.; Lim, W.T.; Choi, H.S. Biology of Cholangiocytes: From Bench to Bedside. Gut Liver 2016, 10, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, R. In Vitro Methods for Measuring the Permeability of Cell Monolayers. Methods Protoc. 2022, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Jiang, W.G. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta 2009, 1788, 872–891. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Mirzaian, M.; Kramer, G.; Poorthuis, B.J. Quantification of sulfatides and lysosulfatides in tissues and body fluids by liquid chromatography-tandem mass spectrometry. J. Lipid Res. 2015, 56, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Vaes, R.D.W.; van den Berk, L.; Boonen, B.; van Dijk, D.P.J.; Olde Damink, S.W.M.; Rensen, S.S. A novel human cell culture model to study visceral smooth muscle phenotypic modulation in health and disease. Am. J. Physiol.-Cell Physiol. 2018, 315, C598–C607. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Elizalde, M.; Dubois, L.J.; Roeth, A.A.; Neumann, U.P.; Olde Damink, S.W.M.; Schaap, F.G.; Alvarez-Sola, G. GAL3ST1 Deficiency Reduces Epithelial–Mesenchymal Transition and Tumorigenic Capacity in a Cholangiocarcinoma Cell Line. Int. J. Mol. Sci. 2024, 25, 7279. https://doi.org/10.3390/ijms25137279
Chen L, Elizalde M, Dubois LJ, Roeth AA, Neumann UP, Olde Damink SWM, Schaap FG, Alvarez-Sola G. GAL3ST1 Deficiency Reduces Epithelial–Mesenchymal Transition and Tumorigenic Capacity in a Cholangiocarcinoma Cell Line. International Journal of Molecular Sciences. 2024; 25(13):7279. https://doi.org/10.3390/ijms25137279
Chicago/Turabian StyleChen, Lin, Montserrat Elizalde, Ludwig J. Dubois, Anjali A. Roeth, Ulf P. Neumann, Steven W. M. Olde Damink, Frank G. Schaap, and Gloria Alvarez-Sola. 2024. "GAL3ST1 Deficiency Reduces Epithelial–Mesenchymal Transition and Tumorigenic Capacity in a Cholangiocarcinoma Cell Line" International Journal of Molecular Sciences 25, no. 13: 7279. https://doi.org/10.3390/ijms25137279
APA StyleChen, L., Elizalde, M., Dubois, L. J., Roeth, A. A., Neumann, U. P., Olde Damink, S. W. M., Schaap, F. G., & Alvarez-Sola, G. (2024). GAL3ST1 Deficiency Reduces Epithelial–Mesenchymal Transition and Tumorigenic Capacity in a Cholangiocarcinoma Cell Line. International Journal of Molecular Sciences, 25(13), 7279. https://doi.org/10.3390/ijms25137279