
3′-UTR Sequence of Exosomal NANOGP8 DNA as an Extracellular Vesicle-Localization Signal



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
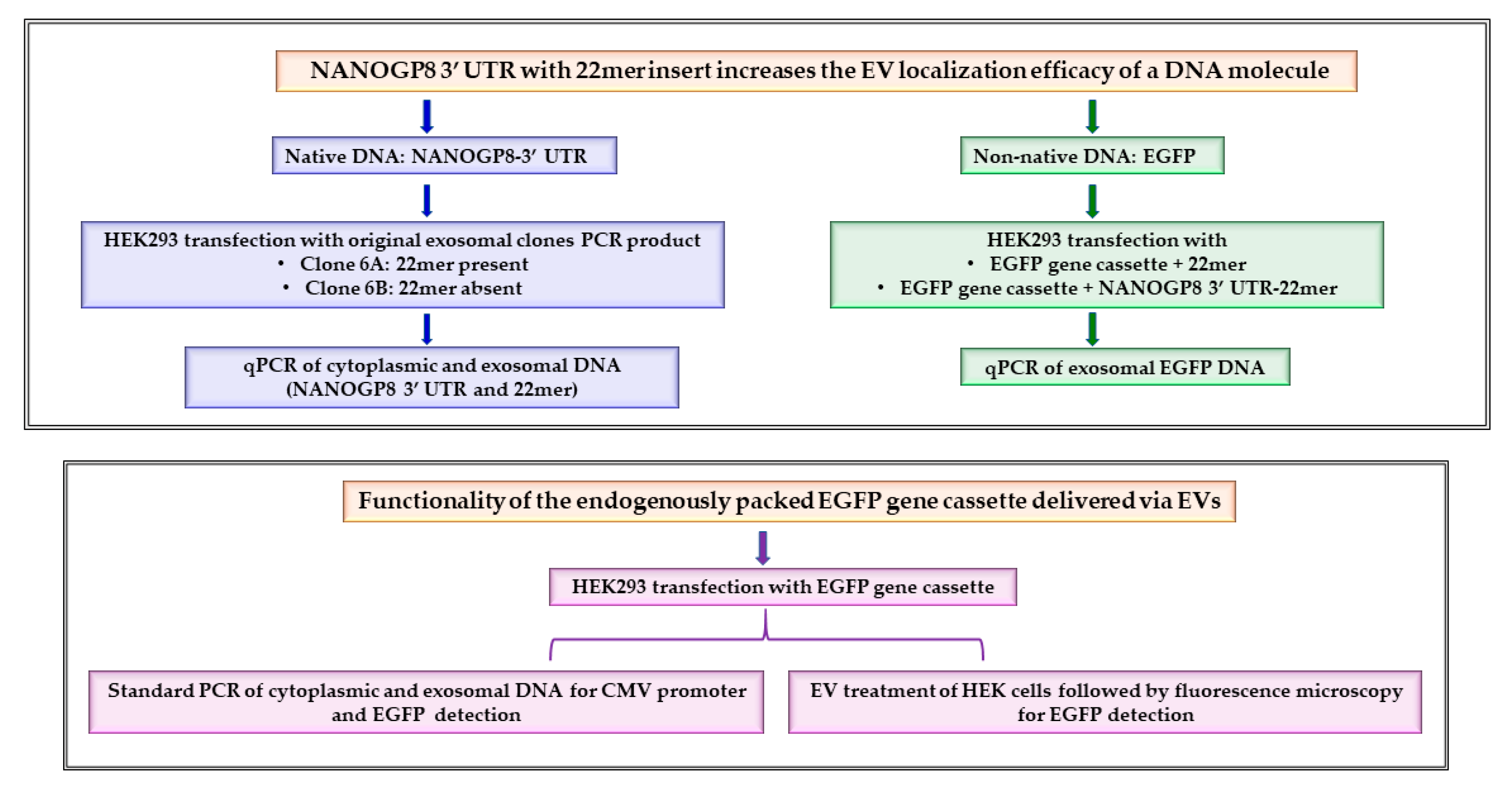
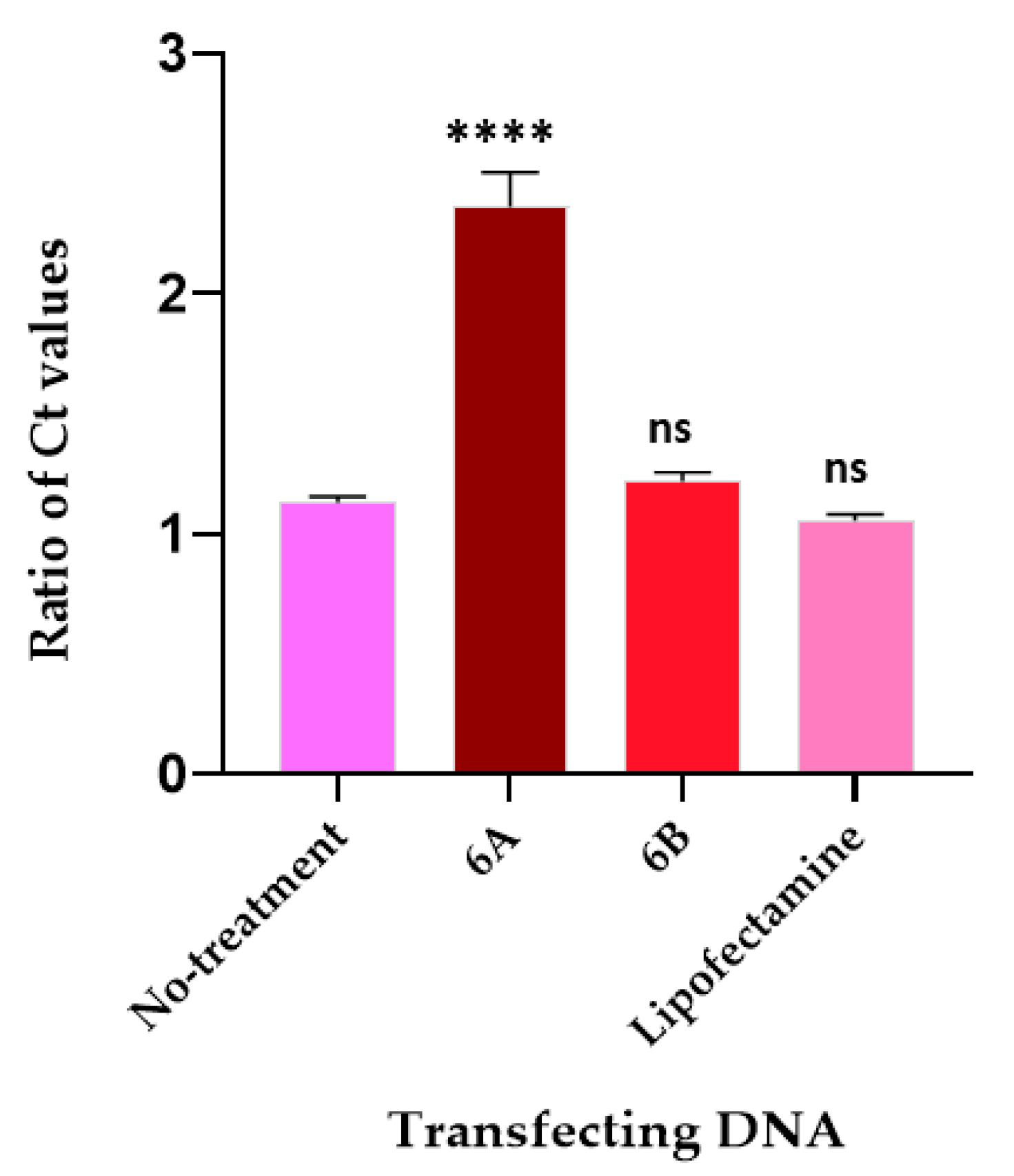
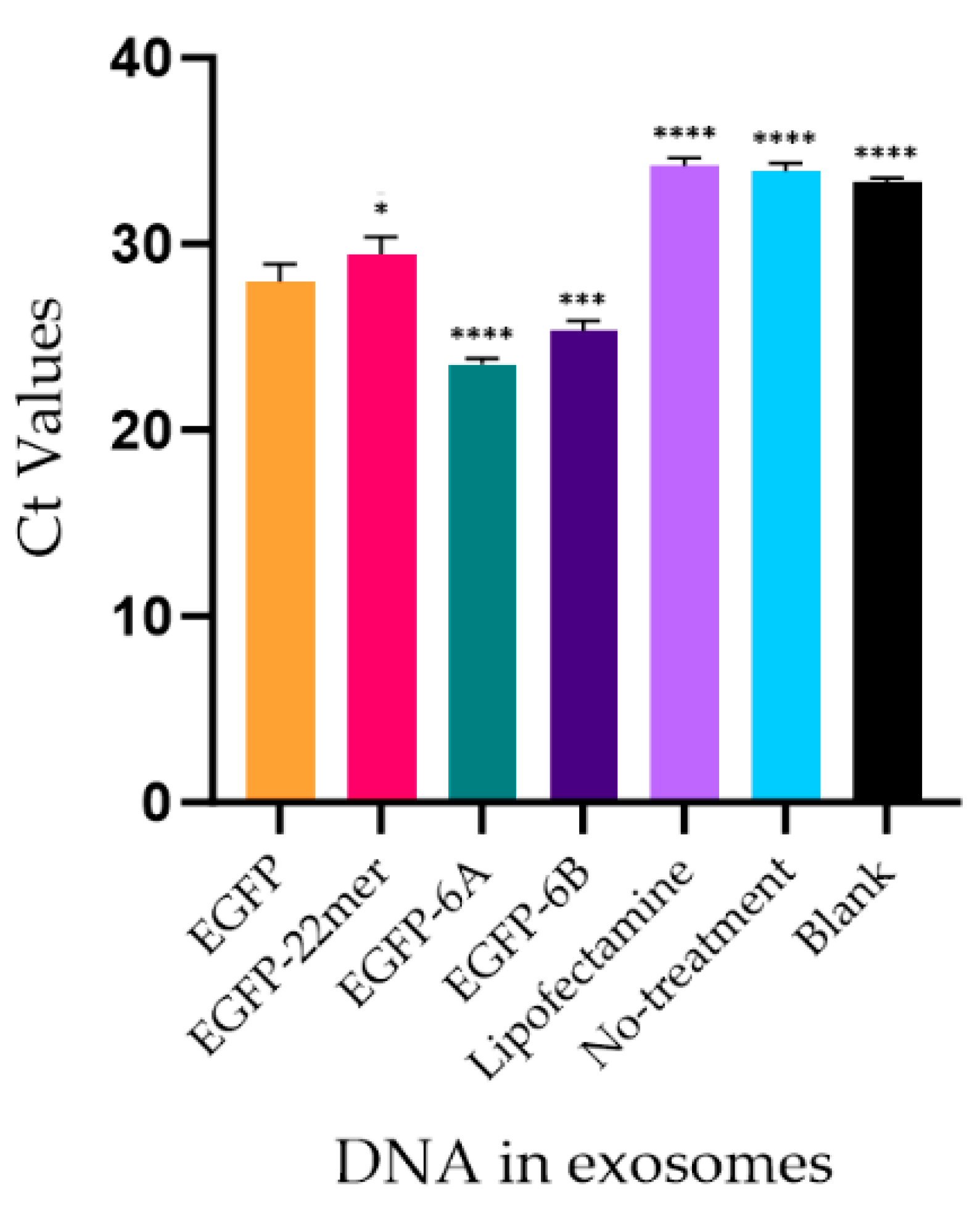
2.1. The 22mer Insert Does Not Affect the Localization Efficacy of the NANOGP8 3′-UTR DNA to Exosomes
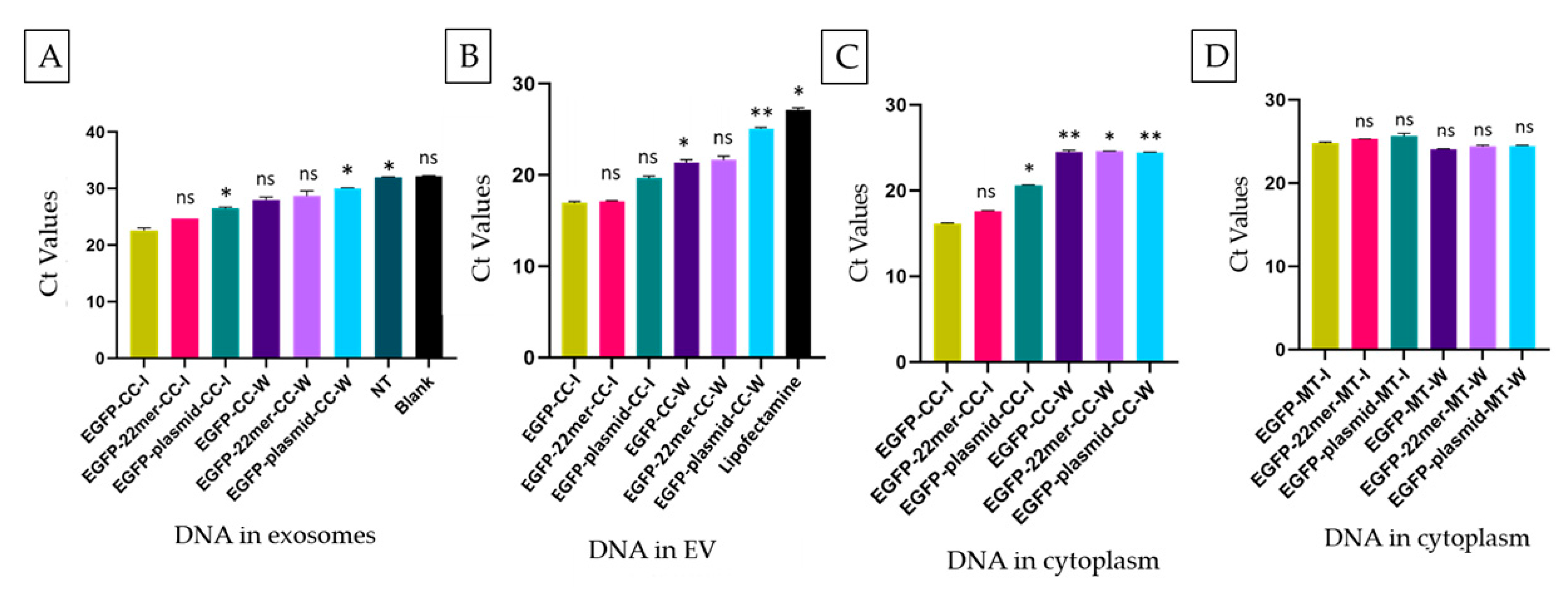
2.2. The EV-Localization of NANOGP8 3′-UTR-End Containing 22mer Is Independent of Its Cytosolic Concentration
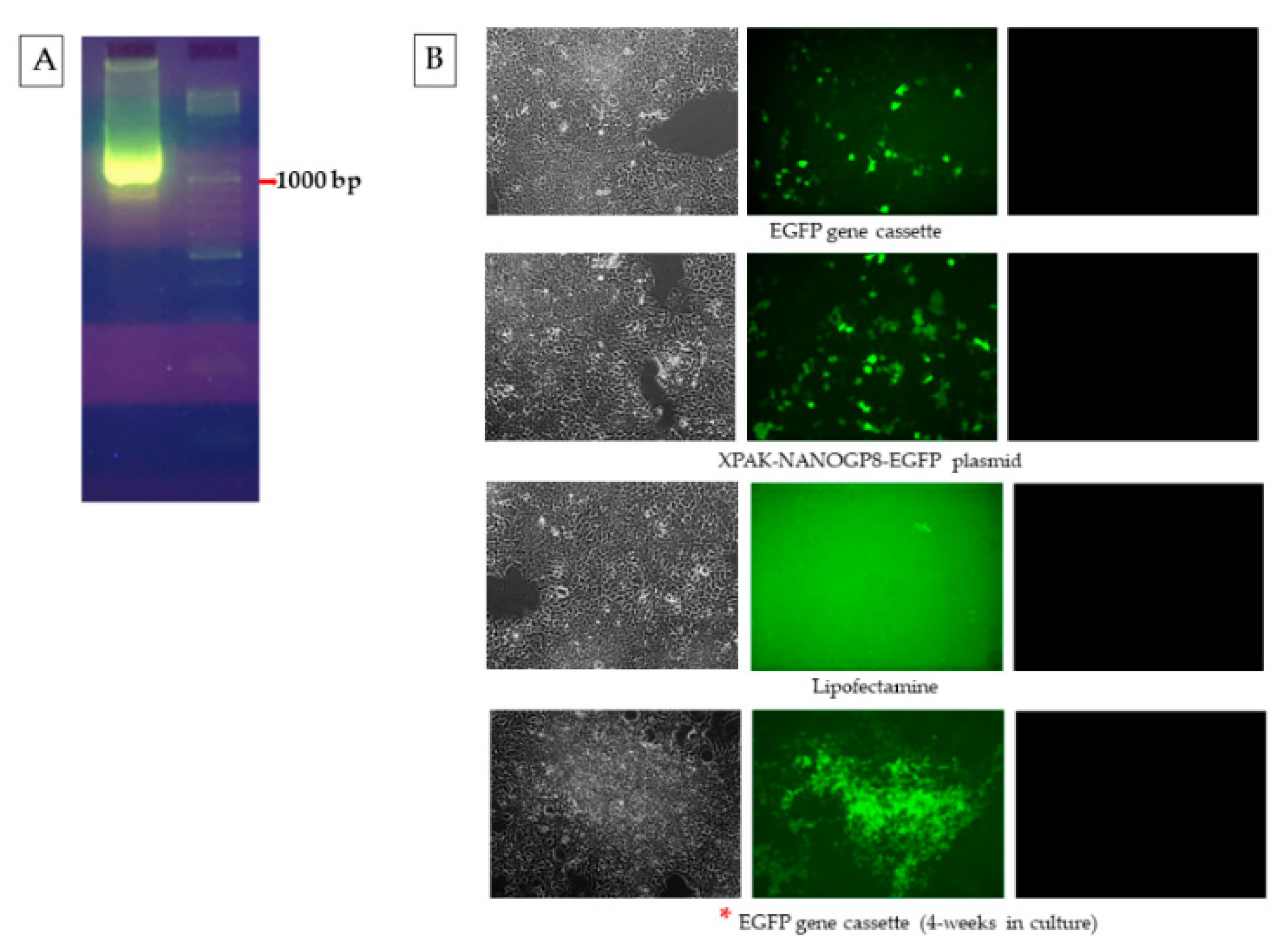
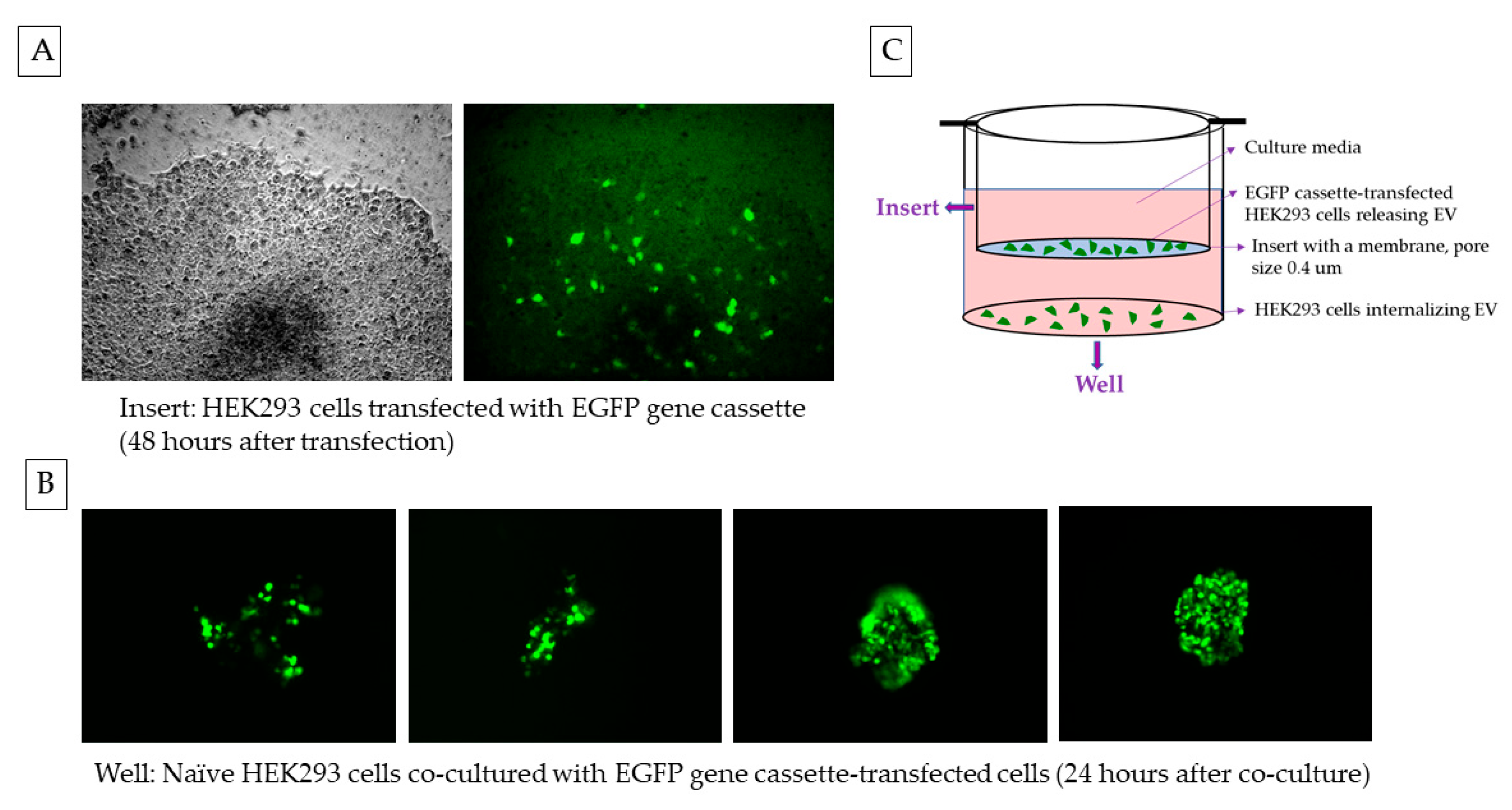
2.3. EGFP Gene Cassette Endogenously Packed in EVs Is Functional in the Recipient Cell
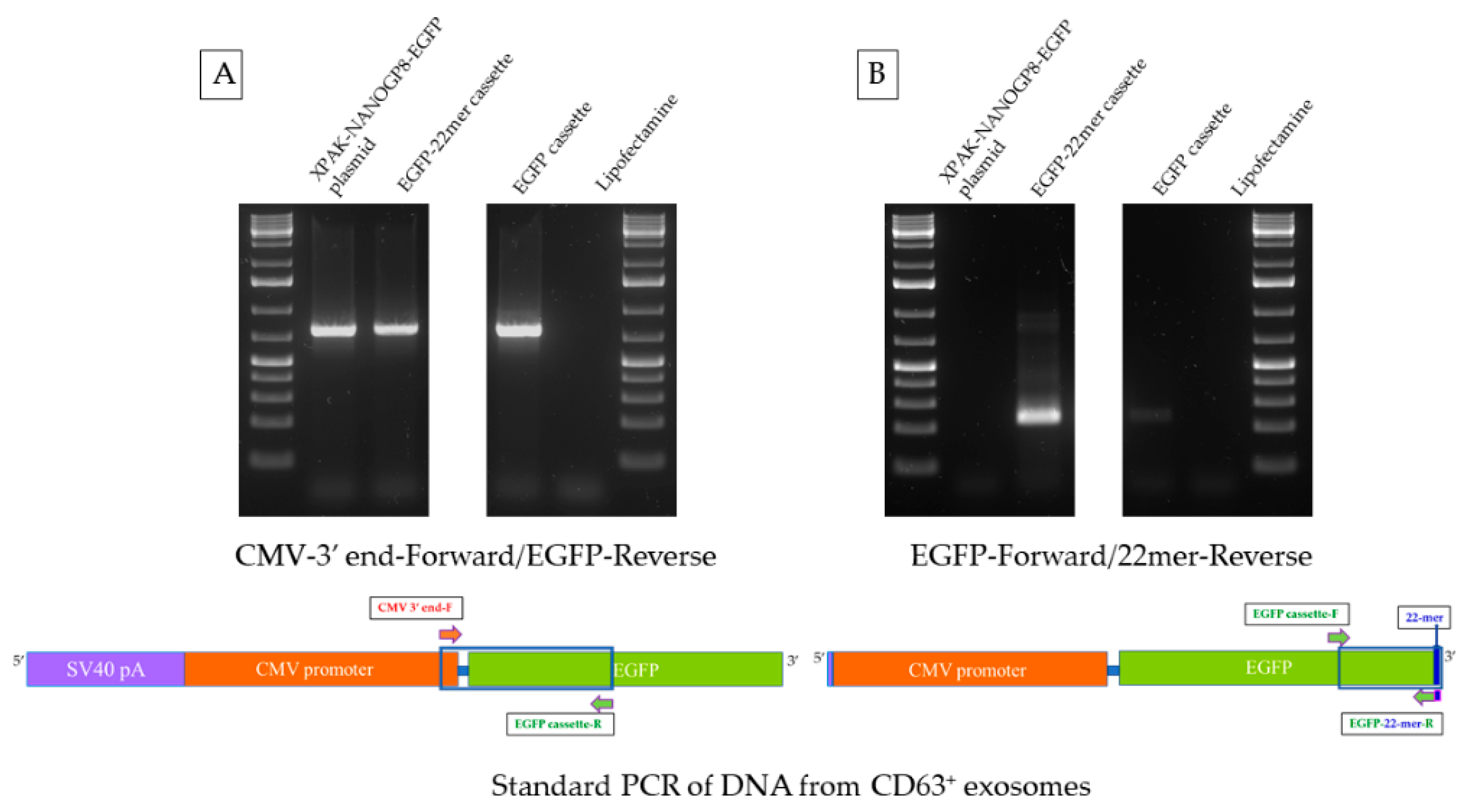
2.4. CMV-EGFP DNA Is Present in the Exosomes of HEK293 Cells after Transfection
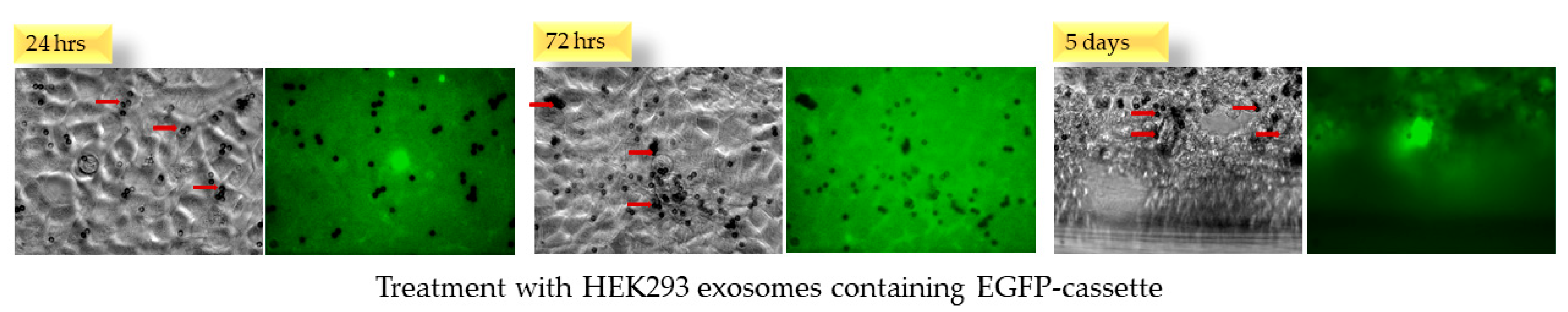
2.5. Naïve HEK293 Cells Express EGFP upon Treatment with Exosomes Containing CMV-EGFP DNA
2.6. Mer Insert Sequence Alone Is Not Enough for an Effective EV-Localization of A DNA
2.7. Attachment of 22mer Insert Containing NANOGP8 3′-UTR Improves the Localization of a Non-Native EGFP DNA to EVs
3. Discussion
4. Materials and Methods
4.1. Standard PCR Amplification of Exosomal NANOGP8 3′-UTR from pCR™4-TOPO™ TA Clones
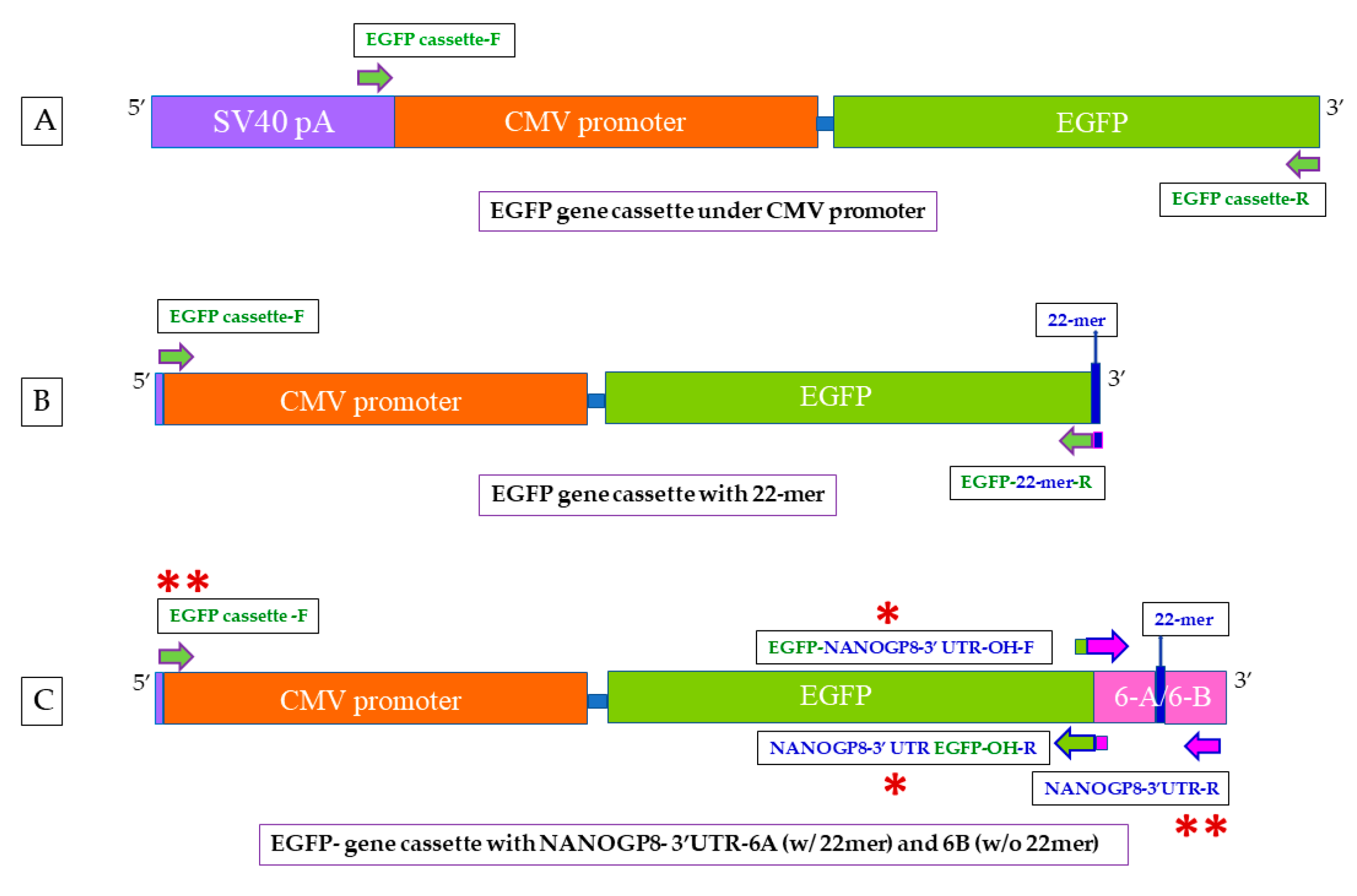
4.2. Creation of EGFP Gene Cassette
4.3. Addition of 22mer and NANOGP8-3′-UTR (PCR Products of Clones 6A and 6B) to the EGFP Gene Cassette
4.4. Cloning the EGFP-6A/6B into pCR™4-TOPO™ TA Vector for Sequencing
4.5. Restriction Enzyme (RE) Digestions of the pCR™4-TOPO™ TA-EGFP-6A/6B Clones
4.6. HEK293 Cell Culture
4.7. Testing the Functionality of the EGFP Gene Cassette
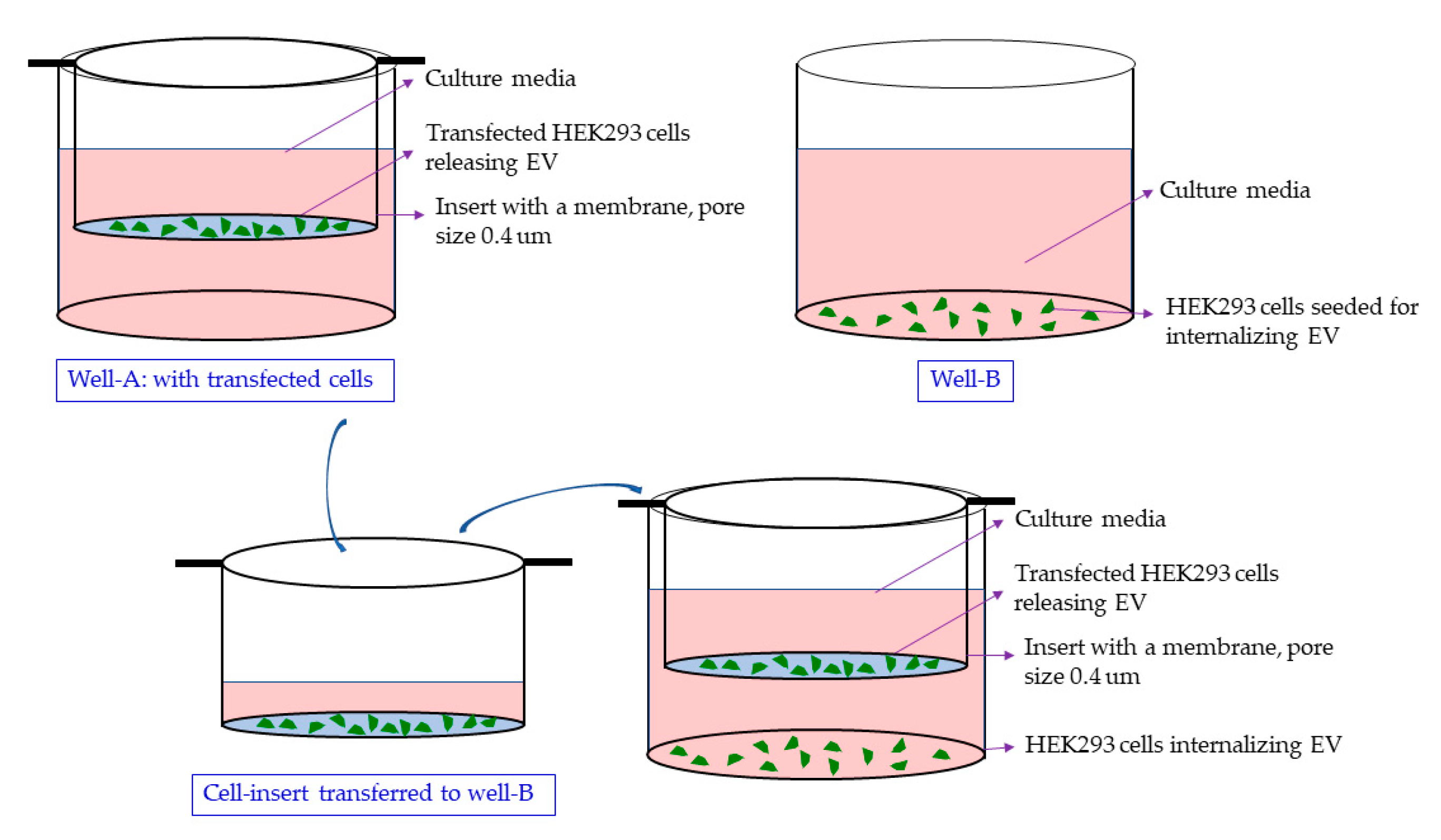
4.8. Delivery via EVs in a Co-Culture or the Conditioned Media Treatment of the HEK293 Naïve Cells
4.9. Treatment of Naïve HEK293 Cells with EGFP Cassette Containing Exosomes Containing
4.10. EV/Exosome Isolation
4.11. Standard PCR of Exosomal DNA Using CMV and EGFP Primers
4.12. Standard PCR Amplification of Cytoplasmic DNA Using CMV and EGFP Primers
4.13. qPCR of EGFP DNA in EV/Exosomes Using EGFP Probe
4.14. qPCR for the Quantification of NANOGP8 UTR in EV/Exosomes upon Transfection with the PCR Products of Clones 6A and 6B
4.15. qPCRs Quantification of Cytoplasmic DNA
4.16. qPCRs of Cytoplasmic and EV DNA for 22mer Quantification and Comparison
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; d’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Malkin, E.Z.; Bratman, S.V. Bioactive DNA from extracellular vesicles and particles. Cell Death Dis. 2020, 11, 584. [Google Scholar] [CrossRef]
- Vaidya, M.; Bacchus, M.; Sugaya, K. Differential sequences of exosomal NANOG DNA as a potential diagnostic cancer marker. PLoS ONE 2018, 13, e0197782. [Google Scholar] [CrossRef]
- Vaidya, M.; Sreerama, S.; Gaviria, M.; Sugaya, K. Exposure to a Pathological Condition May Be Required for the Cells to Secrete Exosomes Containing mtDNA Aberration. J. Nucleic Acids 2022, 2022, 7960198. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, M.; Sugaya, K. Differential sequences and single nucleotide polymorphism of exosomal SOX2 DNA in cancer. PLoS ONE 2020, 15, e0229309. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, M.; Sugaya, K. DNA associated with circulating exosomes as a biomarker for glioma. Genes 2020, 11, 1276. [Google Scholar] [CrossRef]
- Vaidya, M.; Sreerama, S.; Gonzalez-Vega, M.; Smith, J.; Field, M.; Sugaya, K. Coculture with Neural Stem Cells May Shift the Transcription Profile of Glioblastoma Multiforme towards Cancer-Specific Stemness. Int. J. Mol. Sci. 2023, 24, 3242. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, Y.; Zhao, M. Exosome-based cancer therapy: Implication for targeting cancer stem cells. Front. Pharmacol. 2017, 7, 533. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Soleymani, M.; Shahriyary, F.; Amirzargar, M.R.; Ofoghi, M.; Fattahi, M.D.; Safa, M. Viral vectors and extracellular vesicles: Innate delivery systems utilized in CRISPR/Cas-mediated cancer therapy. Cancer Gene Ther. 2023, 30, 936–954. [Google Scholar] [CrossRef]
- Sivanantham, A.; Jin, Y. Impact of storage conditions on EV Integrity/Surface markers and cargos. Life 2022, 12, 697. [Google Scholar] [CrossRef]
- Yuan, F.; Li, Y.-M.; Wang, Z. Preserving extracellular vesicles for biomedical applications: Consideration of storage stability before and after isolation. Drug Deliv. 2021, 28, 1501–1509. [Google Scholar] [CrossRef]
- Cheng, Y.; Zeng, Q.; Han, Q.; Xia, W. Effect of pH, temperature and freezing-thawing on quantity changes and cellular uptake of exosomes. Protein Cell 2019, 10, 295–299. [Google Scholar] [CrossRef]
- Chen, H.; Wang, L.; Zeng, X.; Schwarz, H.; Nanda, H.S.; Peng, X.; Zhou, Y. Exosomes, a new star for targeted delivery. Front. Cell Dev. Biol. 2021, 9, 751079. [Google Scholar] [CrossRef]
- Yoo, M.H.; Lee, A.-R.; Moon, K.-S. Characteristics of Extracellular Vesicles and Preclinical Testing Considerations Prior to Clinical Applications. Biomedicines 2022, 10, 869. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of extracellular vesicles across the blood-brain barrier: Brain pharmacokinetics and effects of inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef] [PubMed]
- García-Romero, N.; Carrión-Navarro, J.; Esteban-Rubio, S.; Lázaro-Ibáñez, E.; Peris-Celda, M.; Alonso, M.M.; Guzmán-De-Villoria, J.; Fernández-Carballal, C.; de Mendivil, A.O.; García-Duque, S.; et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget 2017, 8, 1416. [Google Scholar] [CrossRef]
- Liu, S.; Wu, X.; Chandra, S.; Lyon, C.; Ning, B.; Fan, J.; Hu, T.Y. Extracellular vesicles: Emerging tools as therapeutic agent carriers. Acta Pharm. Sin. B 2022, 12, 3822–3842. [Google Scholar] [CrossRef] [PubMed]
- Tutanov, O.; Shtam, T.; Grigor’eva, A.; Tupikin, A.; Tsentalovich, Y.; Tamkovich, S. Blood plasma exosomes contain circulating DNA in their crown. Diagnostics 2022, 12, 854. [Google Scholar] [CrossRef]
- Nordmeier, S.; Ke, W.; Afonin, K.A.; Portnoy, V. Exosome Mediated Delivery of Functional Nucleic Acid Nanoparticles (NANPs). In Therapeutic RNA Nanotechnology; Jenny Stanford Publishing: Singapore, 2021; pp. 539–564. [Google Scholar]
- Zhang, Y.; Liu, Q.; Zhang, X.; Huang, H.; Tang, S.; Chai, Y.; Xu, Z.; Li, M.; Chen, X.; Liu, J.; et al. Recent advances in exosome-mediated nucleic acid delivery for cancer therapy. J. Nanobiotechnol. 2022, 20, 279. [Google Scholar] [CrossRef] [PubMed]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Kandimalla, R.; Wallen, M.; Tyagi, N.; Wilcher, S.; Yan, J.; Schultz, D.J.; Spencer, W.; et al. Exosome-mediated delivery of RNA and DNA for gene therapy. Cancer Lett. 2021, 505, 58–72. [Google Scholar] [CrossRef]
- Chitti, S.V.; Gummadi, S.; Kang, T.; Shahi, S.; Marzan, A.L.; Nedeva, C.; Sanwlani, R.; Bramich, K.; Stewart, S.; Petrovska, M.; et al. Vesiclepedia 2024: An extracellular vesicles and extracellular particles repository. Nucleic Acids Res. 2023, 52, gkad1007. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Tanaka, K.; Kawasaki, Y. A novel sorting signal for RNA packaging into small extracellular vesicles. Sci. Rep. 2023, 13, 17436. [Google Scholar] [CrossRef]
- Mayr, C. Regulation by 3′-untranslated regions. Annu. Rev. Genet. 2017, 51, 171–194. [Google Scholar] [CrossRef]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef]
- Yokoi, A.; Villar-Prados, A.; Oliphint, P.A.; Zhang, J.; Song, X.; De Hoff, P.; Morey, R.; Liu, J.; Roszik, J.; Clise-Dwyer, K.; et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019, 5, eaax8849. [Google Scholar] [CrossRef]
- Sutaria, D.S.; Badawi, M.; Phelps, M.A.; Schmittgen, T.D. Achieving the promise of therapeutic extracellular vesicles: The devil is in details of therapeutic loading. Pharm. Res. 2017, 34, 1053–1066. [Google Scholar] [CrossRef]
- Wallen, M.; Aqil, F.; Spencer, W.; Gupta, R.C. Exosomes as an Emerging Plasmid Delivery Vehicle for Gene Therapy. Pharmaceutics 2023, 15, 1832. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C. What are 3′ UTRs doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef]
- Batagov, A.O.; Kurochkin, I.V. Exosomes secreted by human cells transport largely mRNA fragments that are enriched in the 3′-untranslated regions. Biol. Direct 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Szostak, N.; Royo, F.; Rybarczyk, A.; Szachniuk, M.; Blazewicz, J.; del Sol, A.; Falcon-Perez, J.M. Sorting signal targeting mRNA into hepatic extracellular vesicles. RNA Biol. 2014, 11, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y. Nucleic acid drugs—Current status, issues, and expectations for exosomes. Cancers 2021, 13, 5002. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Danhier, P.; Krishnamachary, B.; Bharti, S.; Kakkad, S.; Mironchik, Y.; Bhujwalla, Z.M. Combining optical reporter proteins with different half-lives to detect temporal evolution of hypoxia and reoxygenation in tumors. Neoplasia 2015, 17, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Sutaria, D.S.; Jiang, J.; Elgamal, O.A.; Pomeroy, S.M.; Badawi, M.; Zhu, X.; Pavlovicz, R.; Azevedo-Pouly, A.C.P.; Chalmers, J.; Li, C.; et al. Low active loading of cargo into engineered extracellular vesicles results in inefficient miRNA mimic delivery. J. Extracell. Vesicles 2017, 6, 1333882. [Google Scholar] [CrossRef]
- Cai, J.; Han, Y.; Ren, H.; Chen, C.; He, D.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227–238. [Google Scholar] [CrossRef]
- Fischer, S.; Cornils, K.; Speiseder, T.; Badbaran, A.; Reimer, R.; Indenbirken, D.; Grundhoff, A.; Brunswig-Spickenheier, B.; Alawi, M.; Lange, C. Indication of horizontal DNA gene transfer by extracellular vesicles. PLoS ONE 2016, 11, e0163665. [Google Scholar] [CrossRef]
- Ludwig, A.-K.; De Miroschedji, K.; Doeppner, T.R.; Börger, V.; Ruesing, J.; Rebmann, V.; Durst, S.; Jansen, S.; Bremer, M.; Behrmann, E.; et al. Precipitation with polyethylene glycol followed by washing and pelleting by ultracentrifugation enriches extracellular vesicles from tissue culture supernatants in small and large scales. J. Extracell. Vesicles 2018, 7, 1528109. [Google Scholar] [CrossRef]
- Vaidya, M.; Sayeed, N.; Hobson, C.; Sreerama, S.; Smith, J.; Shah, R.; Sugaya, K. Methods and Protocols for Using Extracellular Vesicles as Delivery Vehicles in Neuronal Research. In Cell-Secreted Vesicles; Methods and Protocols; Springer: New York, NY, USA, 2023; pp. 159–189. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaidya, M.; Kimura, A.; Bajaj, A.; Sugaya, K. 3′-UTR Sequence of Exosomal NANOGP8 DNA as an Extracellular Vesicle-Localization Signal. Int. J. Mol. Sci. 2024, 25, 7294. https://doi.org/10.3390/ijms25137294
Vaidya M, Kimura A, Bajaj A, Sugaya K. 3′-UTR Sequence of Exosomal NANOGP8 DNA as an Extracellular Vesicle-Localization Signal. International Journal of Molecular Sciences. 2024; 25(13):7294. https://doi.org/10.3390/ijms25137294
Chicago/Turabian StyleVaidya, Manjusha, Ayaka Kimura, Arjun Bajaj, and Kiminobu Sugaya. 2024. "3′-UTR Sequence of Exosomal NANOGP8 DNA as an Extracellular Vesicle-Localization Signal" International Journal of Molecular Sciences 25, no. 13: 7294. https://doi.org/10.3390/ijms25137294
APA StyleVaidya, M., Kimura, A., Bajaj, A., & Sugaya, K. (2024). 3′-UTR Sequence of Exosomal NANOGP8 DNA as an Extracellular Vesicle-Localization Signal. International Journal of Molecular Sciences, 25(13), 7294. https://doi.org/10.3390/ijms25137294