
Pirfenidone Prevents Heart Fibrosis during Chronic Chagas Disease Cardiomyopathy
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
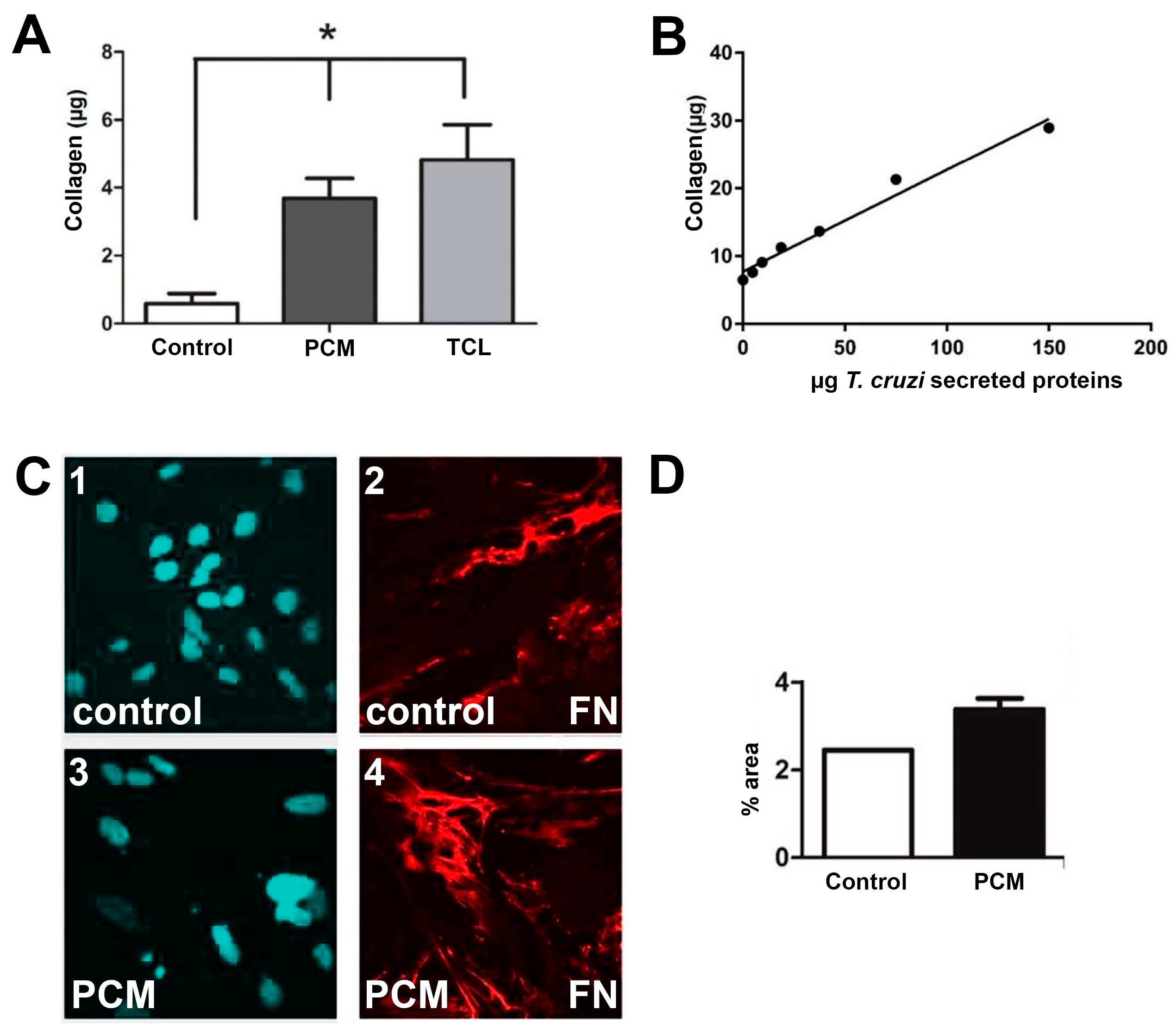
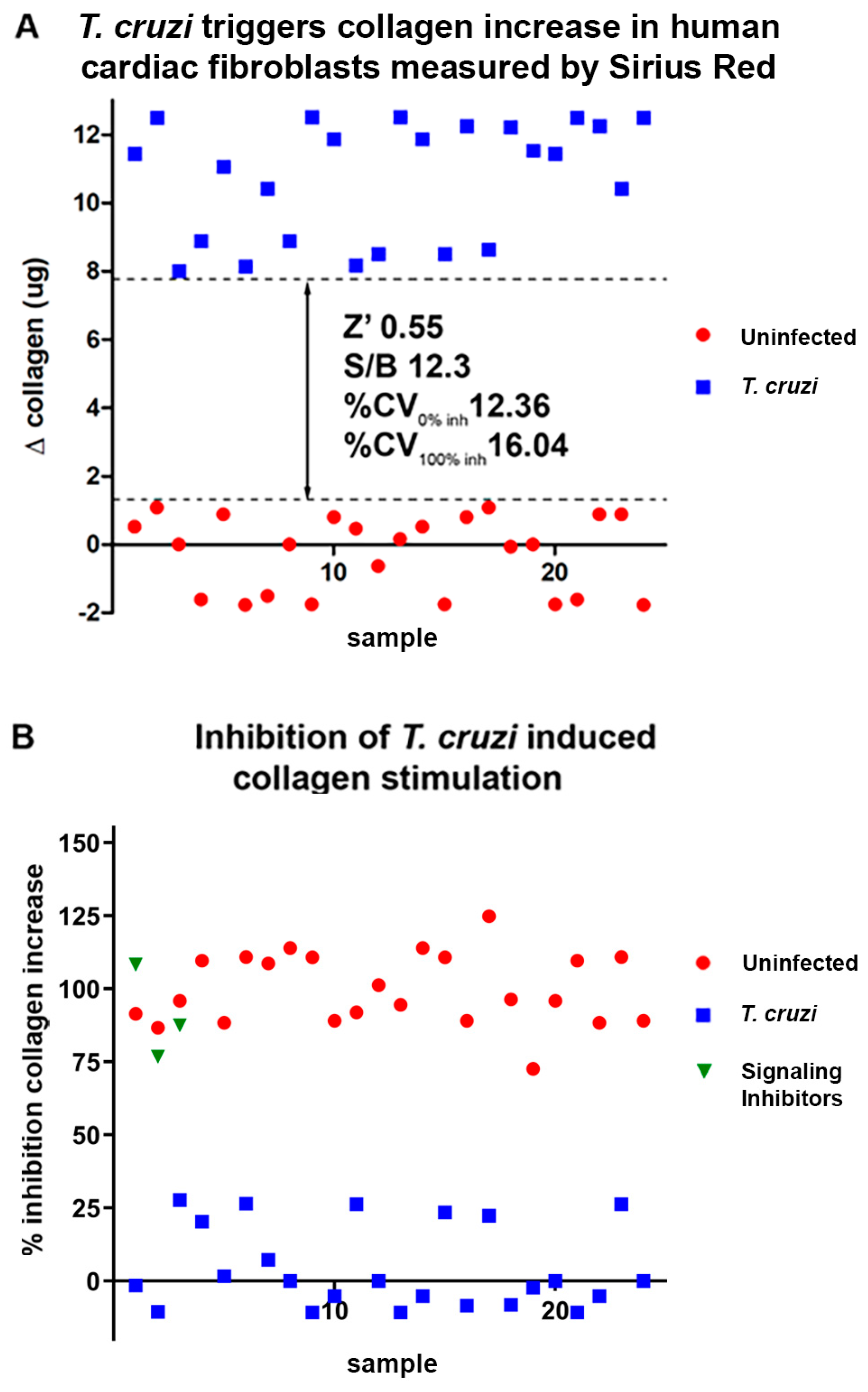
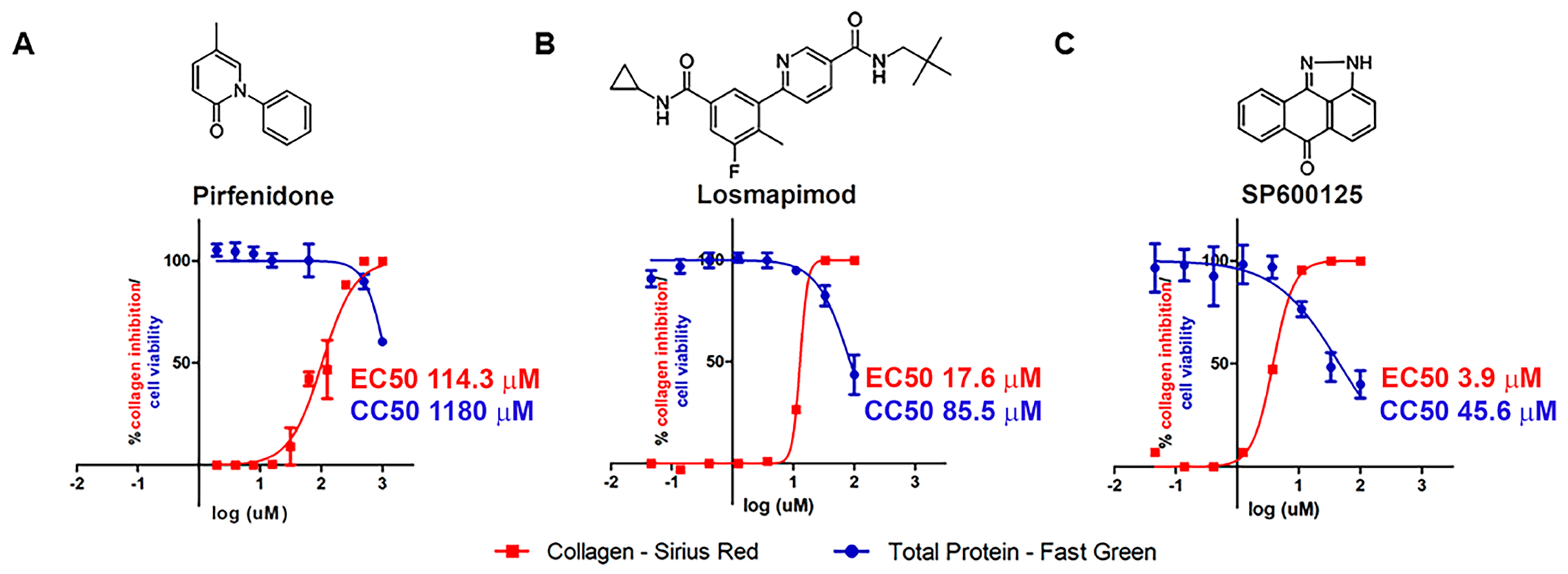
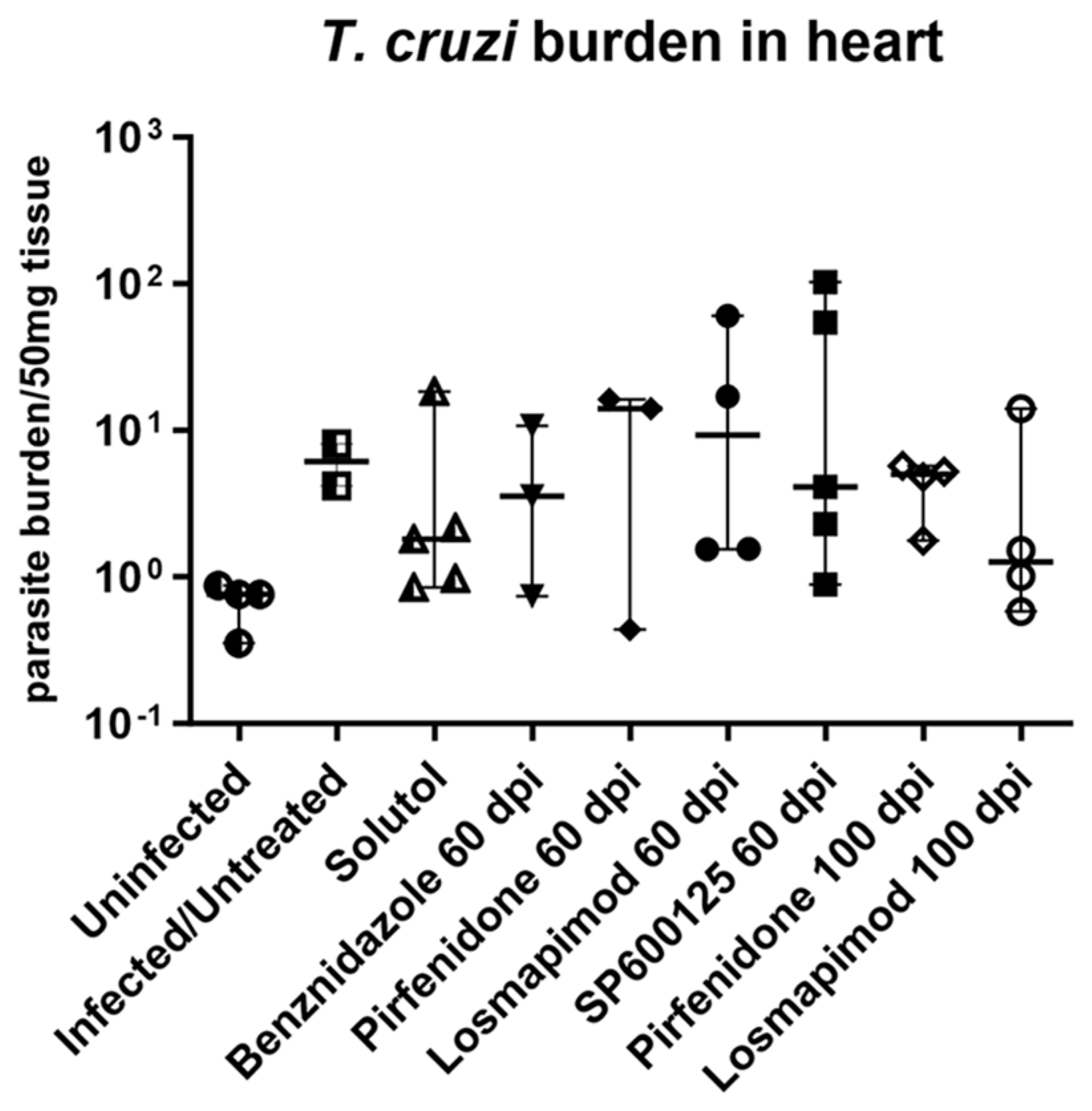
2. Results
2.1. In Vitro Assays
2.2. In Vivo Assays
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization Chagas Disease (Also Known as American Trypanosomiasis). Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 13 November 2023).
- Chagas, C. Nova Tripanozomiaze Humana. Mem. Inst. Oswaldo Cruz 1909, 1, 74–276. [Google Scholar] [CrossRef]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front Public Health 2019, 49, 166. [Google Scholar] [CrossRef]
- Andrade, D.V.; Gollob, K.J.; Dutra, W.O. Acute Chagas Disease: New Global Challenges for an Old Neglected Disease. PLoS Negl. Trop. Dis. 2014, 8, e3010. [Google Scholar] [CrossRef]
- Bern, C.; Kjos, S.; Yabsley, M.J.; Montgomery, S.P. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin. Microbiol. Rev. 2011, 24, 655–681. [Google Scholar] [CrossRef]
- Lee, B.Y.; Bacon, K.M.; Bottazzi, M.E.; Hotez, P.J. Global Economic Burden of Chagas Disease: A Computational Simulation Model. Lancet Infect. Dis. 2013, 13, 342–348. [Google Scholar] [CrossRef]
- Gómez-Ochoa, S.A.; Rojas, L.Z.; Echeverría, L.E.; Muka, T.; Franco, O.H. Global, Regional, and National Trends of Chagas Disease from 1990 to 2019: Comprehensive Analysis of the Global Burden of Disease Study. Glob. Heart 2022, 17, 59. [Google Scholar] [CrossRef]
- Bonney, K.M.; Luthringer, D.J.; Kim, S.A.; Garg, N.J.; Engman, D.M. Pathology and Pathogenesis of Chagas Heart Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 421–447. [Google Scholar] [CrossRef]
- Tassi, E.M.; Continentino, M.A.; Nascimento, E.M.D.; Pereira, B.D.B.; Pedrosa, R.C. Relationship between Fibrosis and Ventricular Arrhythmias in Chagas Heart Disease without Ventricular Dysfunction. Arq. Bras. Cardiol. 2014, 102, 456–464. [Google Scholar] [CrossRef]
- Barizon, G.C.; Simões, M.V.; Schmidt, A.; Gadioli, L.P.; Murta Junior, L.O. Relationship between Microvascular Changes, Autonomic Denervation, and Myocardial Fibrosis in Chagas Cardiomyopathy: Evaluation by MRI and SPECT Imaging. J. Nucl. Cardiol. 2018, 27, 434–444. [Google Scholar] [CrossRef]
- Melo, R.J.L.; Assunção, A.N.; Morais, T.C.; Nomura, C.H.; Scanavacca, M.I.; Martinelli-Filho, M.; Ramires, F.J.A.; Fernandes, F.; Ianni, B.M.; Mady, C.; et al. Detection of Early Diffuse Myocardial Fibrosis and Inflammation in Chagas Cardiomyopathy with T1 Mapping and Extracellular Volume. Radiol. Cardiothorac. Imaging 2023, 5, e220112. [Google Scholar] [CrossRef]
- Pecoul, B.; Batista, C.; Stobbaerts, E.; Ribeiro, I.; Vilasanjuan, R.; Gascon, J.; Pinazo, M.J.; Moriana, S.; Gold, S.; Pereiro, A.; et al. The BENEFIT Trial: Where Do We Go from Here? PLoS Negl. Trop. Dis. 2016, 10, e0004343. [Google Scholar] [CrossRef]
- Morillo, C.; Marin-Neto, J.; Avezum, A.; Sosa-Estani, S.; Rassi, A., Jr.; Rosas, F.; Vilhena, E.; Quiroz, R.; Bonilla, R.; Britto, C.; et al. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 2015, 373, 1295–1306. [Google Scholar] [CrossRef]
- Sánchez-Valdéz, F.J.; Padilla, A.; Wang, W.; Orr, D.; Tarleton, R. Spontaneous Dormancy Protects Trypanosoma cruzi during Extended Drug Exposure. Elife 2018, 7, e34039. [Google Scholar] [CrossRef]
- World Health Organization. Research Priorities for Chagas Disease, Human African Trypanosomiasis and Leishmaniasis; World Health Organ Technical Report Series; WHO: Geneva, Switzerland, 2012; Volume v–xii, pp. 1–100.
- Waghabi, M.C.; Ferreira, R.R.; Da Silva, A.R.; Degrave, W.; De Souza, E.M.; Bailly, S.; Feige, J.J.; De Araújo-Jorge, T.C. Transforming Growth Factor-ß as a Therapeutic Target for the Cardiac Damage of Chagas Disease. Mem. Inst. Oswaldo Cruz 2022, 117, e210395. [Google Scholar] [CrossRef]
- Torres, D.J.L.; De Arruda, T.R.; da Silva, B.M.; Gonçales, J.P.; Soares, A.K.A.; dos Santos, O.K.K.; Moreira, L.R.; Medeiros, C.; Cavalcanti, M.D.G.A.M.; Martins, S.M.; et al. Is a Negative Correlation between STNFR1 and TNF in Patients with Chronic Chagas Disease the Key to Clinical Progression? Immunobiology 2022, 227, 152166. [Google Scholar] [CrossRef]
- Cunha-Neto, E.; Dzau, V.J.; Allen, P.D.; Stamatiou, D.; Benvenutti, L.; Higuchi, M.L.; Koyama, N.S.; Silva, J.S.; Kalil, J.; Liew, C.-C. Cardiac Gene Expression Profiling Provides Evidence for Cytokinopathy as a Molecular Mechanism in Chagas’ Disease Cardiomyopathy. Am. J. Pathol. 2005, 167, 305–313. [Google Scholar] [CrossRef]
- Calvet, C.M.; Oliveira, F.O.R.; Araújo-Jorge, T.C.; Pereira, M.C.S. Regulation of Extracellular Matrix Expression and Distribution in Trypanosoma cruzi-Infected Cardiomyocytes. Int. J. Med. Microbiol. 2009, 299, 301–312. [Google Scholar] [CrossRef]
- Calvet, C.M.; Meuser, M.; Almeida, D.; Meirelles, M.N.L.; Pereira, M.C.S. Trypanosoma cruzi-Cardiomyocyte Interaction: Role of Fibronectin in the Recognition Process and Extracellular Matrix Expression In Vitro and In Vivo. Exp. Parasitol. 2004, 107, 20–30. [Google Scholar] [CrossRef]
- Silva, T.A.; Ferreira, L.F.; Pereira, M.C.d.S.; Calvet, C.M. Differential Role of TGF-Β in Extracellular Matrix Regulation during Trypanosoma cruzi-Host Cell Interaction. Int. J. Mol. Sci. 2019, 20, 4836. [Google Scholar] [CrossRef]
- Araújo-Jorge, T.C.; Waghabi, M.C.; Hasslocher-moreno, A.M.; Xavier, S.; Higuchi, M.D.L.; Keramidas, M.; Bailly, S.; Feige, J. Implication of Transforming Growth Factor–Β1 in Chagas Disease Myocardiopathy. J. Infect. Dis. 2002, 186, 1823–1828. [Google Scholar] [CrossRef]
- Nogueira, L.G.; Santos, R.H.B.; Fiorelli, A.I.; Mairena, E.C.; Benvenuti, L.A.; Bocchi, E.A.; Stolf, N.A.; Kalil, J.; Cunha-Neto, E.; Gabriel Nogueira, L.; et al. Myocardial Gene Expression of T-Bet, GATA-3, Ror-γt, FoxP3, and Hallmark Cytokines in Chronic Chagas Disease Cardiomyopathy: An Essentially Unopposed TH1-Type Response. Mediat. Inflamm. 2014, 2014, 914326. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad Signaling Pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C. Non-Smad TGF-β Signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic Activities of Pirfenidone in Animal Models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef]
- Aimo, A.; Spitaleri, G.; Panichella, G.; Lupón, J.; Emdin, M.; Bayes-Genis, A. Pirfenidone as a Novel Cardiac Protective Treatment. Heart Fail. Rev. 2022, 27, 525–532. [Google Scholar] [CrossRef]
- Bracco, G.T.C.L.; Crnko, S.; Leiteris, L.; van Adrichem, I.; van Laake, L.W.; Bouten, C.V.C.; Goumans, M.J.; Suyker, W.J.L.; Sluijter, J.P.G.; Hjortnaes, J. Pirfenidone Has Anti-Fibrotic Effects in a Tissue-Engineered Model of Human Cardiac Fibrosis. Front. Cardiovasc. Med. 2022, 9, 854314. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, H.; Huang, X.; Wang, S.; Ouyang, X.; Wang, Y.; Cao, Q.; Yang, L.; Tao, Y.; Lai, H. Pirfenidone Attenuates Cardiac Hypertrophy against Isoproterenol by Inhibiting Activation of the Janus Tyrosine Kinase-2/Signal Transducer and Activator of Transcription 3 (JAK-2/STAT3) Signaling Pathway. Bioengineered 2022, 13, 12772–12782. [Google Scholar] [CrossRef]
- Li, N.; Hang, W.; Shu, H.; Zhou, N. Pirfenidone Alleviates Cardiac Fibrosis Induced by Pressure Overload via Inhibiting TGF-Β1/Smad3 Signalling Pathway. J. Cell. Mol. Med. 2022, 26, 4548–4555. [Google Scholar] [CrossRef]
- Lee, K.W.; Everett, T.H., IV; Rahmutula, D.; Guerra, J.M.; Wilson, E.; Ding, C.; Olgin, J.E. Pirfenidone Prevents the Development of a Vulnerable Substrate for Atrial Fibrillation in a Canine Model of Heart Failure. Circulation 2006, 114, 1703–1712. [Google Scholar] [CrossRef]
- Lewis, G.A.; Rosala-Hallas, A.; Dodd, S.; Schelbert, E.B.; Williams, S.G.; Cunnington, C.; McDonagh, T.; Miller, C.A. Predictors of Myocardial Fibrosis and Response to Anti-Fibrotic Therapy in Heart Failure with Preserved Ejection Fraction. Int. J. Cardiovasc. Imaging 2022, 38, 1569–1578. [Google Scholar] [CrossRef]
- Newby, L.K.; Marber, M.S.; Melloni, C.; Sarov-Blat, L.; Aberle, L.H.; Aylward, P.E.; Cai, G.; de Winter, R.J.; Hamm, C.W.; Heitner, J.F.; et al. Losmapimod, a Novel P38 Mitogen-Activated Protein Kinase Inhibitor, in Non-ST-Segment Elevation Myocardial Infarction: A Randomised Phase 2 Trial. Lancet 2014, 384, 1187–1195. [Google Scholar] [CrossRef]
- Mellion, M.L.; Ronco, L.; Berends, C.L.; Pagan, L.; Brooks, S.; van Esdonk, M.J.; van Brummelen, E.M.J.; Odueyungbo, A.; Thompson, L.A.; Hage, M.; et al. Phase 1 Clinical Trial of Losmapimod in Facioscapulohumeral Dystrophy: Safety, Tolerability, Pharmacokinetics, and Target Engagement. Br. J. Clin. Pharmacol. 2021, 87, 4658–4669. [Google Scholar] [CrossRef]
- Willette, R.N.; Eybye, M.E.; Olzinski, A.R.; Behm, D.J.; Aiyar, N.; Maniscalco, K.; Bentley, R.G.; Coatney, R.W.; Zhao, S.; Westfall, T.D.; et al. Differential Effects of P38 Mitogen-Activated Protein Kinase and Cyclooxygenase 2 Inhibitors in a Model of Cardiovascular Disease. J. Pharmacol. Exp. Ther. 2009, 330, 964–970. [Google Scholar] [CrossRef]
- Barbour, A.M.; Sarov-Blat, L.; Cai, G.; Fossler, M.J.; Sprecher, D.L.; Graggaber, J.; Mcgeoch, A.T.; Maison, J.; Cheriyan, J. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Losmapimod Following a Single Intravenous or Oral Dose in Healthy Volunteers. Br. J. Clin. Pharmacol. 2013, 76, 99–106. [Google Scholar] [CrossRef]
- Elkhawad, M.; Rudd, J.H.F.; Sarov-Blat, L.; Cai, G.; Wells, R.; Ceri, D.L.; Collier, D.J.; Marber, M.S.; Choudhury, R.P.; Fayad, Z.A.; et al. Effects of P38 Mitogen-Activated Protein Kinase Inhibition on Vascular and Systemic Inflammation in Patients with Atherosclerosis. JACC Cardiovasc. Imaging 2012, 5, 911–922. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.A.; Gutierrez, J.A.T.; et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized with Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2016, 315, 1591–1599. [Google Scholar] [CrossRef]
- Li, T.; Jiang, S.; Ni, B.; Cui, Q.; Liu, Q.; Zhao, H. Discontinued Drugs for the Treatment of Cardiovascular Disease from 2016 to 2018. Int. J. Mol. Sci. 2019, 20, 4513. [Google Scholar] [CrossRef]
- Guo, W.; Cao, S.; Yan, B.; Zhang, G.; Li, J.; Zhao, Y.; Zhang, S. Myocardial Protective Effects of a C-Jun N-Terminal Kinase Inhibitor in Rats with Brain Death. J. Cell. Mol. Med. 2016, 20, 1214–1218. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an Anthrapyrazolone Inhibitor of Jun N-Terminal Kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Tsai, C.F.; Yang, S.F.; Lo, C.H.; Chu, H.J.; Ueng, K.C. Role of the Ros-Jnk Signaling Pathway in Hypoxia-Induced Atrial Fibrotic Responses in Hl-1 Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 3249. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, Y.; Zhao, Y.; Peng, K.; Li, W.; Wang, Y.; Zhang, J.; Zhou, S.; Liu, Q.; Li, X.; et al. Inhibition of JNK Phosphorylation by a Novel Curcumin Analog Prevents High Glucose-Induced Inflammation and Apoptosis in Cardiomyocytes and the Development of Diabetic Cardiomyopathy. Diabetes 2014, 63, 3497–3511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Wells, A.U. Pirfenidone for the Treatment of Idiopathic Pulmonary Fibrosis. Expert. Rev. Clin. Pharmacol. 2017, 10, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Tucci, A.R.; de Oliveira, F.O.R.; Lechuga, G.C.; Oliveira, G.M.; Eleuterio, A.C.; de Mesquita, L.B.; Farani, P.S.G.; Britto, C.; Moreira, O.C.; Pereira, M.C.S. Role of FAK Signaling in Chagasic Cardiac Hypertrophy. Braz. J. Infect. Dis. 2020, 24, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Jelicks, L.; Chandra, M.; Shirani, J.; Shtutin, V.; Tang, B.; Christ, G.J.; Factor, S.M.; Wittner, M.; Huang, H.; Weiss, L.M.; et al. Cardioprotective Effects of Phosphoramidon on Myocardial Structure and Function in Murine Chagas’ Disease. Int. J. Parasitol. 2002, 32, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Tanowitz, H.B.; Huang, H.; Jelicks, L.A.; Chandra, M.; Loredo, M.L.; Weiss, L.M.; Factor, S.M.; Shtutin, V.; Mukherjee, S.; Kitsis, R.N.; et al. Role of Endothelin 1 in the Pathogenesis of Chronic Chagasic Heart Disease. Infect. Immun. 2005, 73, 2496–2503. [Google Scholar] [CrossRef]
- Duschak, V.G. Targets and Patented Drugs for Chemotherapy of Chagas Disease in the Last 15 Years-Period. Recent. Pat. Antiinfect. Drug Discov. 2016, 11, 74–173. [Google Scholar] [CrossRef]
- Hochberg, N.S.; Montgomery, S.P. Chagas Disease. Ann. Intern. Med. 2023, 176, ITC17–ITC32. [Google Scholar] [CrossRef]
- Hall, C.L.; Wells, A.R.; Leung, K.P. Pirfenidone Reduces Profibrotic Responses in Human Dermal Myofibroblasts, In Vitro. Lab. Investig. 2018, 98, 640–655. [Google Scholar] [CrossRef]
- Kim, E.S.; Keating, G.M. Pirfenidone: A Review of Its Use in Idiopathic Pulmonary Fibrosis. Drugs 2015, 75, 219–230. [Google Scholar] [CrossRef]
- Lewis, M.D.; Francisco, A.F.; Taylor, M.C.; Jayawardhana, S.; Kelly, J.M. Host and Parasite Genetics Shape a Link between Trypanosoma cruzi Infection Dynamics and Chronic Cardiomyopathy. Cell Microbiol. 2016, 18, 1429–1443. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chan, J.; Wittner, M.; Jelicks, L.A.; Morris, S.A.; Factor, S.M.; Weiss, L.M.; Braunstein, V.L.; Bacchi, C.J.; Yarlett, N.; et al. Expression of Cardiac Cytokines and Inducible Form of Nitric Oxide Synthase (NOS2) in Trypanosoma cruzi-Infected Mice. J. Mol. Cell. Cardiol. 1999, 31, 75–88. [Google Scholar] [CrossRef]
- Roman-Campos, D.; Duarte, H.L.L.; Sales, P.A.; Natali, A.J.; Ropert, C.; Gazzinelli, R.T.; Cruz, J.S. Changes in Cellular Contractility and Cytokines Profile during Trypanosoma cruzi Infection in Mice. Basic. Res. Cardiol. 2009, 104, 238–246. [Google Scholar] [CrossRef]
- Machado, F.; Martins, G.; Aliberti, J.; Mestriner, F.; Cunha, F.; Silva, J. Trypanosoma cruzi-Infected Cardiomyocytes Produce Chemokines and Cytokines That Trigger Potent Nitric Oxide-Dependent Trypanocidal Activity. Circulation 2000, 102, 3003–3008. [Google Scholar] [CrossRef] [PubMed]
- Waghabi, M.C.; Coutinho-Silva, R.; Feige, J.-J.; Higuchi, M.D.L.; Becker, D.; Burnstock, G.; Araújo-Jorge, T.C. Gap Junction Reduction in Cardiomyocytes Following Transforming Growth Factor-Beta Treatment and Trypanosoma cruzi Infection. Mem. Inst. Oswaldo Cruz 2009, 104, 1083–1090. [Google Scholar] [CrossRef]
- Pinho, R.T.; Vannier-Santos, M.A.; Alves, C.R.; Marino, A.P.M.P.; Castello, B.L.R.R.; Lannes-Vieira, J. Effect of Trypanosoma cruzi Released Antigens Binding to Non-Infected Cells on Anti-Parasite Antibody Recognition and Expression of Extracellular Matrix Components. Acta Trop. 2002, 83, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Serra, N.; Gualdron-Lopez, M.; Pinazo, M.J.; Torrecilhas, A.C.; Fernandez-Becerra, C. Extracellular Vesicles in Trypanosoma cruzi Infection: Immunomodulatory Effects and Future Perspectives as Potential Control Tools against Chagas Disease. J. Immunol. Res. 2022, 2022, 5230603. [Google Scholar] [CrossRef] [PubMed]
- Brossas, J.Y.; Gulin, J.E.N.; Bisio, M.M.C.; Chapelle, M.; Marinach-Patrice, C.; Bordessoules, M.; Ruiz, G.P.; Vion, J.; Paris, L.; Altcheh, J.; et al. Secretome Analysis of Trypanosoma cruzi by Proteomics Studies. PLoS ONE 2017, 12, e0185504. [Google Scholar] [CrossRef]
- Nogueira, P.M.; Ribeiro, K.; Silveira, A.C.O.; Campos, J.H.; Martins-Filho, O.A.; Bela, S.R.; Campos, M.A.; Pessoa, N.L.; Colli, W.; Alves, M.J.M.; et al. Vesicles from Different Trypanosoma cruzi Strains Trigger Differential Innate and Chronic Immune Responses. J. Extracell. Vesicles 2015, 4, 28734. [Google Scholar] [CrossRef]
- Moraes, A.V.d.; Esteves-Filho, A.; Barbero-Marcial, M.; Belloti, G.; Kalil, R.; Bocchi, E.A.; Higuchi, M.d.L.; Weiss, R.; Sosa, E.; Jatene, A.; et al. In Vivo Detection of Trypanosoma cruzi Antigens in Hearts of Patients with Chronic Chagas’ Heart Disease. Am. Heart J. 2004, 131, 301–307. [Google Scholar] [CrossRef]
- Ferrão, P.M.; d’Avila-Levy, C.M.; Araujo-Jorge, T.C.; Degrave, W.M.; Gonçalves, A.D.S.; Garzoni, L.R.; Lima, A.P.; Feige, J.J.; Bailly, S.; Mendonça-Lima, L.; et al. Cruzipain Activates Latent TGF-β from Host Cells during T. cruzi Invasion. PLoS ONE 2015, 10, e0124832. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, X.; Bai, Y.; Cui, C.; Li, J.; Li, Y.; Hu, S.; Wei, Y. In Vitro Effects of Pirfenidone on Cardiac Fibroblasts: Proliferation, Myofibroblast Differentiation, Migration and Cytokine Secretion. PLoS ONE 2011, 6, e28134. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Muchir, A.; Shan, J.; Bonne, G.; Worman, H.J. Mitogen-Activated Protein Kinase Inhibitors Improve Heart Function and Prevent Fibrosis in Cardiomyopathy Caused by Mutation in Lamin A/C Gene. Circulation 2011, 123, 53–61. [Google Scholar] [CrossRef]
- Xu, X.; Jiang, R.; Chen, M.; Dong, M.; Liu, Q.; Cheng, H.; Zhou, K.; Chen, L.; Li, M.; Jiang, C. Puerarin Decreases Collagen Secretion in AngII-Induced Atrial Fibroblasts through Inhibiting Autophagy Via the JNK-Akt-MTOR Signaling Pathway. J. Cardiovasc. Pharmacol. 2019, 73, 373–382. [Google Scholar] [CrossRef]
- Lu, C.; Yang, Y.; Zhu, Y.; Lv, S.; Zhang, J. An Intervention Target for Myocardial Fibrosis: Autophagy. BioMed. Res. Int. 2018, 2018, 6215916. [Google Scholar] [CrossRef] [PubMed]
- Schieven, G.L. The Biology of P38 Kinase: A Central Role in Inflammation. Curr. Top. Med. Chem. 2005, 5, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Kojonazarov, B.; Novoyatleva, T.; Boehm, M.; Happe, C.; Sibinska, Z.; Tian, X.; Sajjad, A.; Luitel, H.; Kriechling, P.; Posern, G.; et al. P38 Mapk Inhibition Improves Heart Function in Pressure-Loaded Right Ventricular Hypertrophy. Am. J. Respir. Cell Mol. Biol. 2017, 57, 603–614. [Google Scholar] [CrossRef]
- Arabacilar, P.; Marber, M. The Case for Inhibiting p38 Mitogen-Activated Protein Kinase in Heart Failure. Front. Pharmacol. 2015, 6, 102. [Google Scholar] [CrossRef]
- De Souza, A.P.; Tanowitz, H.B.; Chandra, M.; Shtutin, V.; Weiss, L.M.; Morris, S.A.; Factor, S.M.; Huang, H.; Wittner, M.; Shirani, J.; et al. Effects of Early and Late Verapamil Administration on the Development of Cardiomyopathy in Experimental Chronic Trypanosoma cruzi (Brazil Strain) Infection. Parasitol. Res. 2004, 92, 496–501. [Google Scholar] [CrossRef]
- Araujo-Jorge, T.C.; Rivera, M.T.; Vanderpas, J.; Garzoni, L.R.; Carvalho, A.C.C.; Waghabi, M.C.; Holanda, M.T.; Mediano, M.F.F.; Hasslocher-Moreno, A.M.; Bonecini-Almeida, M.d.G.; et al. Selenium, TGF-Beta and Infectious Endemic Cardiopathy: Lessons from Benchwork to Clinical Application in Chagas Disease. Biomolecules 2022, 12, 349. [Google Scholar] [CrossRef]
- de Oliveira, F.L.; Araújo-Jorge, T.C.; de Souza, E.M.; de Oliveira, G.M.; Degrave, W.M.; Feige, J.-J.; Bailly, S.; Waghabi, M.C. Oral Administration of GW788388, an Inhibitor of Transforming Growth Factor Beta Signaling, Prevents Heart Fibrosis in Chagas Disease. PLoS Negl. Trop. Dis. 2012, 6, e1696. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.R.; Abreu, S.; Vilar-Pereira, G.; Ferreira, C.; Degrave, W.; Meuser-Batista, M.; Vale, N.; Souza, E.M.D.; Ramos, I.P.; Moreira, C.; et al. TGF-β Inhibitor Therapy Decreases Fibrosis and Stimulates Cardiac Improvement in a Pre-Clinical Study of Chronic Chagas’ Heart Disease. PLoS Negl. Trop. Dis. 2019, 13, e0007602. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.R.; de Souza, E.M.; Vilar-Pereira, G.; Degrave, W.M.S.; Abreu, R.d.S.; Meuser-Batista, M.; Ferreira, N.V.C.; Ledbeter, S.; Barker, R.H.; Bailly, S.; et al. In Chagas Disease, Transforming Growth Factor Beta Neutralization Reduces Trypanosoma cruzi Infection and Improves Cardiac Performance. Front. Cell. Infect. Microbiol. 2022, 12, 1017040. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Y.; Chen, J.; Zhao, S.; Li, H. Pirfenidone Attenuates Cardiac Fibrosis in a Mouse Model of TAC-Induced Left Ventricular Remodeling by Suppressing NLRP3 Inflammasome Formation. Cardiology 2013, 126, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, X.; Wang, B.; Nie, Y.; Wen, J.; Wang, Q.; Gu, C. Pirfenidone Suppresses MAPK Signalling Pathway to Reverse Epithelial-Mesenchymal Transition and Renal Fibrosis. Nephrology 2017, 22, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, Y.; Chi, P. Pirfenidone Suppresses TGF-Β1-Induced Human Intestinal Fibroblasts Activities by Regulating Proliferation and Apoptosis via the Inhibition of the Smad and PI3K/AKT Signaling Pathway. Mol. Med. Rep. 2018, 18, 3907–3913. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.A.; Rosala-Hallas, A.; Dodd, S.; Schelbert, E.B.; Williams, S.G.; Cunnington, C.; McDonagh, T.; Miller, C.A. Characteristics Associated with Growth Differentiation Factor 15 in Heart Failure with Preserved Ejection Fraction and the Impact of Pirfenidone. J. Am. Heart Assoc. 2022, 11, e024668. [Google Scholar] [CrossRef] [PubMed]
- Gunatilleke, S.S.; Calvet, C.M.; Johnston, J.B.; Chen, C.-K.; Erenburg, G.; Gut, J.; Engel, J.C.; Ang, K.K.H.; Mulvaney, J.; Chen, S.; et al. Diverse Inhibitor Chemotypes Targeting Trypanosoma cruzi CYP51. PLoS Negl. Trop. Dis. 2012, 6, e1736. [Google Scholar] [CrossRef]
- Houghton, P.E.; Keefer, K.A.; Diegelmann, R.F.; Krummel, T.M. A Simple Method to Assess the Relative Amount of Collagen Deposition in Wounded Fetal Mouse Limbs. Wound Repair Regen. 1996, 4, 489–495. [Google Scholar] [CrossRef]
- Calvet, C.M.; Silva, T.A.; Thomas, D.; Suzuki, B.; Hirata, K.; Siqueira-Neto, J.L.; McKerrow, J.H. Long Term Follow-up of Trypanosoma cruzi Infection and Chagas Disease Manifestations in Mice Treated with Benznidazole or Posaconazole. PLoS Negl. Trop. Dis. 2020, 14, e0008726. [Google Scholar] [CrossRef]
- McCall, L.I.; Morton, J.T.; Bernatchez, J.A.; De Siqueira-Neto, J.L.; Knight, R.; Dorrestein, P.C.; McKerrow, J.H. Mass Spectrometry-Based Chemical Cartography of a Cardiac Parasitic Infection. Anal. Chem. 2017, 89, 10414–10421. [Google Scholar] [CrossRef] [PubMed]
- Hossain, E.; Khanam, S.; Dean, D.A.; Wu, C.; Lostracco-Johnson, S.; Thomas, D.; Kane, S.S.; Parab, A.R.; Flores, K.; Katemauswa, M.; et al. Mapping of Host-Parasite-Microbiome Interactions Reveals Metabolic Determinants of Tropism and Tolerance in Chagas Disease. Sci. Adv. 2020, 6, eaaz2015. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, Y.; Muraski, J.; Shiraishi, I.; Kato, T.; Kajstura, J.; Anversa, P.; Sussman, M.A. Nuclear Targeting of Akt Antagonizes Aspects of Cardiomyocyte Hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11946–11951. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, T.A.; Thomas, D.; Siqueira-Neto, J.L.; Calvet, C.M. Pirfenidone Prevents Heart Fibrosis during Chronic Chagas Disease Cardiomyopathy. Int. J. Mol. Sci. 2024, 25, 7302. https://doi.org/10.3390/ijms25137302
Silva TA, Thomas D, Siqueira-Neto JL, Calvet CM. Pirfenidone Prevents Heart Fibrosis during Chronic Chagas Disease Cardiomyopathy. International Journal of Molecular Sciences. 2024; 25(13):7302. https://doi.org/10.3390/ijms25137302
Chicago/Turabian StyleSilva, Tatiana Araújo, Diane Thomas, Jair L. Siqueira-Neto, and Claudia Magalhaes Calvet. 2024. "Pirfenidone Prevents Heart Fibrosis during Chronic Chagas Disease Cardiomyopathy" International Journal of Molecular Sciences 25, no. 13: 7302. https://doi.org/10.3390/ijms25137302