
Molecular Targeting of the Isocitrate Dehydrogenase Pathway and the Implications for Cancer Therapy



Abstract
1. Introduction
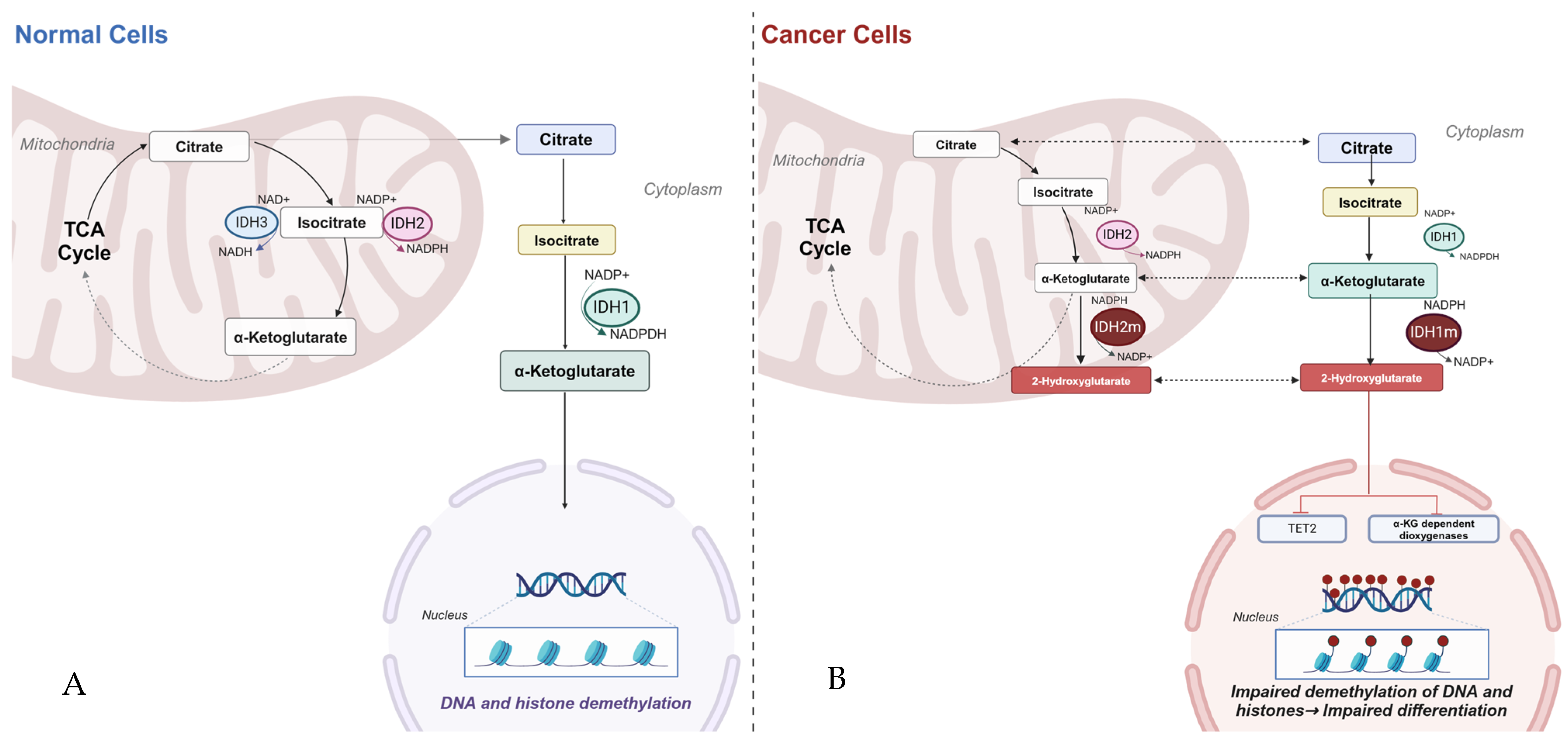
2. Biology of Isocitrate Dehydrogenase
3. Pathophysiology of IDH Mutants
4. Myeloid Neoplasms—Acute Myeloid Leukemia/Myelodysplastic Syndrome
4.1. Enasidenib
4.2. Ivosidenib
4.3. Olutasidenib
5. Glioblastoma Multiforme
5.1. Vorasidenib
5.2. Cholangiocarcinoma
6. Future Direction
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.W.; Jensen, M.V.; Ilkayeva, O.; Palmieri, F.; Alárcon, C.; Rhodes, C.J.; Newgard, C.B. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 35624–35632. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Dean, A.M.; Koshland, D.E., Jr.; Stroud, R.M. Catalytic mechanism of NADP(+)-dependent isocitrate dehydrogenase: Implications from the structures of magnesium-isocitrate and NADP+ complexes. Biochemistry 1991, 30, 8671–8678. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.J.; Lee, S.M.; Son, B.G.; Lee, S.H.; Ryoo, Z.Y.; Chang, K.T.; Park, J.W.; Park, D.C.; Song, B.J.; Veech, R.L.; et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J. Biol. Chem. 2004, 279, 39968–39974. [Google Scholar] [CrossRef]
- Badur, M.G.; Muthusamy, T.; Parker, S.J.; Ma, S.; McBrayer, S.K.; Cordes, T.; Magana, J.H.; Guan, K.L.; Metallo, C.M. Oncogenic R132 IDH1 mutations limit NADPH for de novo lipogenesis through (D)2-hydroxyglutarate production in fibrosarcoma sells. Cell Rep. 2018, 25, 1018.e4–1026.e4. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2- hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R (-)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Burgess, K.; Bhargava, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef]
- Jin, G.; Reitman, Z.J.; Duncan, C.G.; Spasojevic, I.; Gooden, D.M.; Rasheed, B.A.; Yang, R.; Lopez, G.Y.; He, Y.; McLendon, R.E.; et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013, 73, 496–501. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Macarulla, T.; Charlson, J.A.; Van Tine, B.A.; Goyal, L.; Italiano, A.; Massard, C.; Rosenthal, M.; De La Fuente, M.; Roxburgh, P.; et al. A phase Ib/II study of olutasidenib in patients with relapsed/refractory IDH1 mutant solid tumors: Safety and efficacy as single agent. J. Clin. Oncol. 2020, 38 (Suppl. S15), e16643. [Google Scholar] [CrossRef]
- Bunse, L.; Rupp, A.K.; Poschke, I.; Bunse, T.; Lindner, K.; Wick, A.; Blobner, J.; Misch, M.; Tabatabai, G.; Glas, M.; et al. AMPLIFY-NEOVAC: A randomized, 3-arm multicenter phase I trial to assess safety, tolerability and immunogenicity of IDH1-vac combined with an immune checkpoint inhibitor targeting programmed death-ligand 1 in isocitrate dehydrogenase 1 mutant gliomas. Neurol. Res. Pract. 2022, 4, 20. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First in-class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Venugopal, S.; Lachowiez, C.; Takahashi, K.; Loghavi, S.; Montalban-Bravo, G.; Wang, X.; Carraway, H.; Sekeres, M.; Sukkur, A.; et al. Targeted therapy with the mutant IDH2 inhibitor enasidenib for high-risk IDH2-mutant myelodysplastic syndrome. Blood Adv. 2023, 7, 2378–2387. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schuh, A.C.; Stein, E.M.; Montesinos, P.; Wei, A.H.; de Botton, S.; Zeidan, A.M.; Fathi, A.T.; Kantarjian, H.M.; Bennett, J.M.; et al. Effect of enasidenib (ENA) plus azacitidine (AZA) on complete remission and overall response versus AZA monotherapy in mutant-IDH2 (mIDH2) newly diagnosed acute myeloid leukemia (ND-AML). J. Clin. Oncol. 2020, 38, 7501. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Luo, L.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.L.; Sheth, C.M.; Charlab, R.; et al. FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia with an Isocitrate Dehydrogenase-1 Mutation. Clin. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef]
- Pasquier, F.; Lecuit, M.; Broutin, S.; Saada, S.; Jeanson, A.; Penard-Lacronique, V.; de Botton, S. Ivosidenib to treat adult patients with relapsed or refractory acute myeloid leukemia. Drugs Today 2020, 56, 21–32. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef]
- de Botton, S.; Fenaux, P.; Yee, K.; Récher, C.; Wei, A.H.; Montesinos, P.; Taussig, D.C.; Pigneux, A.; Braun, T.; Curti, A.; et al. Olutasidenib (FT-2102) induces durable complete remissions in patients with relapsed or refractory IDH1-mutated AML. Blood Adv. 2023, 7, 3117–3127. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; van den Bent, M.J.; Blumenthal, D.T.; Touat, M.; Peters, K.B.; Clarke, J.; Mendez, J.; Yust-Katz, S.; Welsh, L.; Mason, W.P.; et al. INDIGO Trial Investigators. Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma. N. Engl. J. Med. 2023, 389, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Ruth He, A.; Qin, S.; Chen, L.T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Ah Lee, M.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. ABC-06|A randomised phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin/5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem)chemotherapy. J. Clin. Oncol. 2019, 37 (Suppl. S15), 4003. [Google Scholar]
- Wintheiser, G.; Zemla, T.; Shi, Q.; Tran, N.; Prasai, K.; Tella, S.H.; Mody, K.; Ahn, D.; Borad, M.; Bekaii-Saab, T.; et al. Isocitrate Dehydrogenase-Mutated Cholangiocarcinoma: Natural History and Clinical Outcomes. JCO Precis. Oncol. 2022, 6, e2100156. [Google Scholar] [CrossRef]
- Xiang, X.; Liu, Z.; Zhang, C.; Li, Z.; Gao, J.; Zhang, C.; Cao, Q.; Cheng, J.; Liu, H.; Chen, D.; et al. IDH Mutation Subgroup Status Associates with Intratumor Heterogeneity and the Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Adv. Sci. 2021, 8, e2101230. [Google Scholar] [CrossRef]
- Salati, M.; Caputo, F.; Baldessari, C.; Galassi, B.; Grossi, F.; Dominici, M.; Ghidini, M. IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting. Cancers 2020, 12, 3310. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr. Treat. Options Oncol. 2016, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Burris, H.A., 3rd; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; Hollebecque, A.; et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: A phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients With Advanced Cholangiocarcinoma With IDH1 Mutation: The Phase 3 Randomized Clinical ClarIDHy Trial. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef]
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar]

{kind=link}
| Clinical Trial | Compound | Mechanism | Disease | Trial Phase |
|---|---|---|---|---|
| NCT04762602 | HMPL-306 | Dual IDH 1/2 inhibitor | Solid tumors | I |
| NCT02381886 | IDH 305 | Inhibition of IDH1 with R132 Mutations | Advanced malignancies that harbor IDH1R132 mutations | I |
| NCT02273739 | Enasidenib (AG-221) | IDH2 inhibitor | Advanced solid tumors with IDH2 mutation | I/II |
| NCT02193347 | PEPIDH1M vaccine | Vaccine | Gliomas, WHO grade 2, mIDH | I |
| NCT 03893903 | IDH-Vac + Avelumab | Vaccine | Glioma, mIDH | I |
| NCT02771301 | IDH1R132H-DC vaccine | Vaccine | Glioma, mIDH | I |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanov, S.; Nano, O.; Hana, C.; Bonano-Rios, A.; Hussein, A. Molecular Targeting of the Isocitrate Dehydrogenase Pathway and the Implications for Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 7337. https://doi.org/10.3390/ijms25137337
Ivanov S, Nano O, Hana C, Bonano-Rios A, Hussein A. Molecular Targeting of the Isocitrate Dehydrogenase Pathway and the Implications for Cancer Therapy. International Journal of Molecular Sciences. 2024; 25(13):7337. https://doi.org/10.3390/ijms25137337
Chicago/Turabian StyleIvanov, Stanislav, Olger Nano, Caroline Hana, Amalia Bonano-Rios, and Atif Hussein. 2024. "Molecular Targeting of the Isocitrate Dehydrogenase Pathway and the Implications for Cancer Therapy" International Journal of Molecular Sciences 25, no. 13: 7337. https://doi.org/10.3390/ijms25137337
APA StyleIvanov, S., Nano, O., Hana, C., Bonano-Rios, A., & Hussein, A. (2024). Molecular Targeting of the Isocitrate Dehydrogenase Pathway and the Implications for Cancer Therapy. International Journal of Molecular Sciences, 25(13), 7337. https://doi.org/10.3390/ijms25137337