
Structural Studies of the Taurine Transporter: A Potential Biological Target from the GABA Transporter Subfamily in Cancer Therapy


Abstract
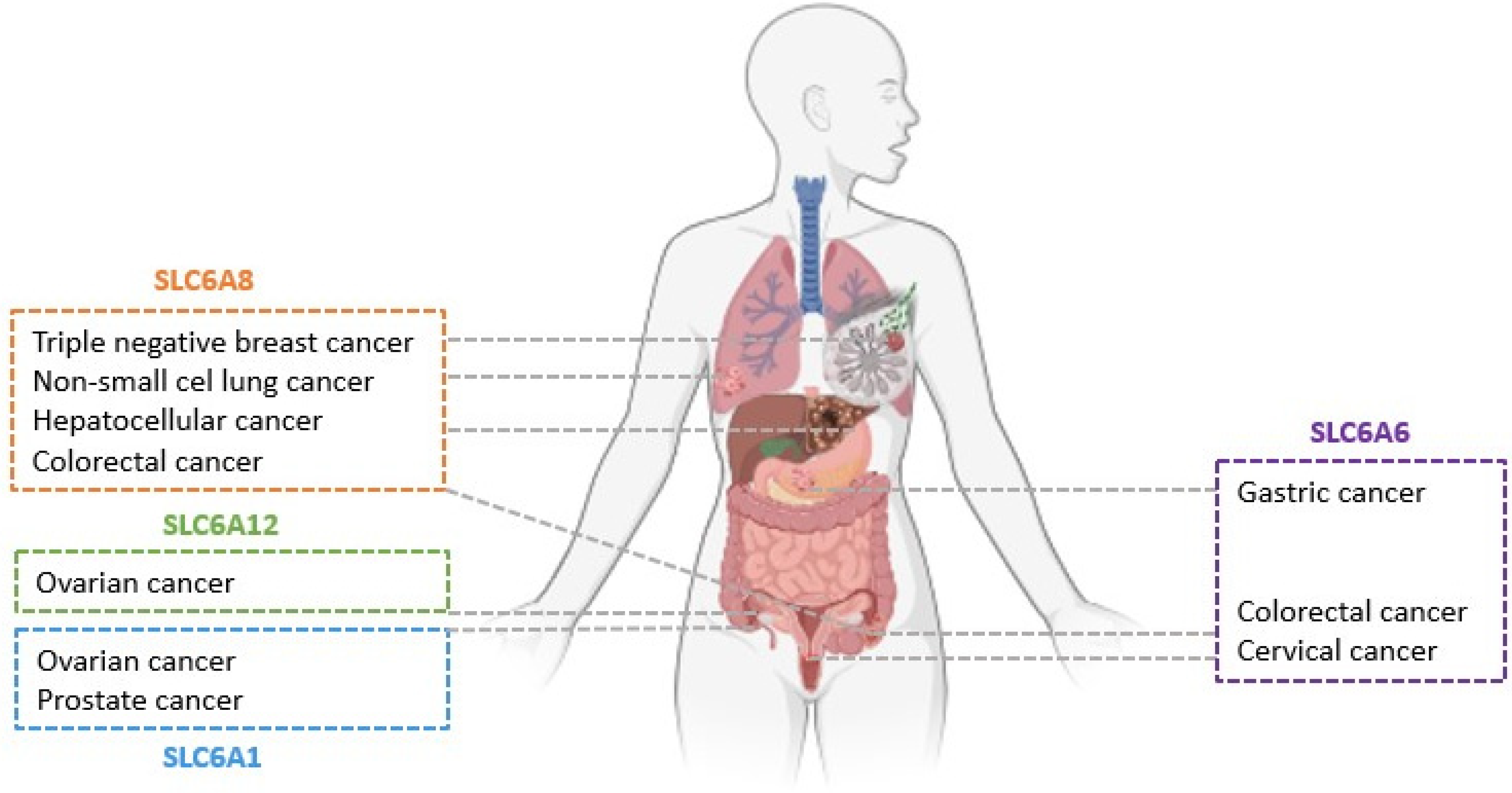
:1. Introduction
2. Results and Discussion
2.1. Model Building and Evaluation
2.2. Structure of Taurine Transporter
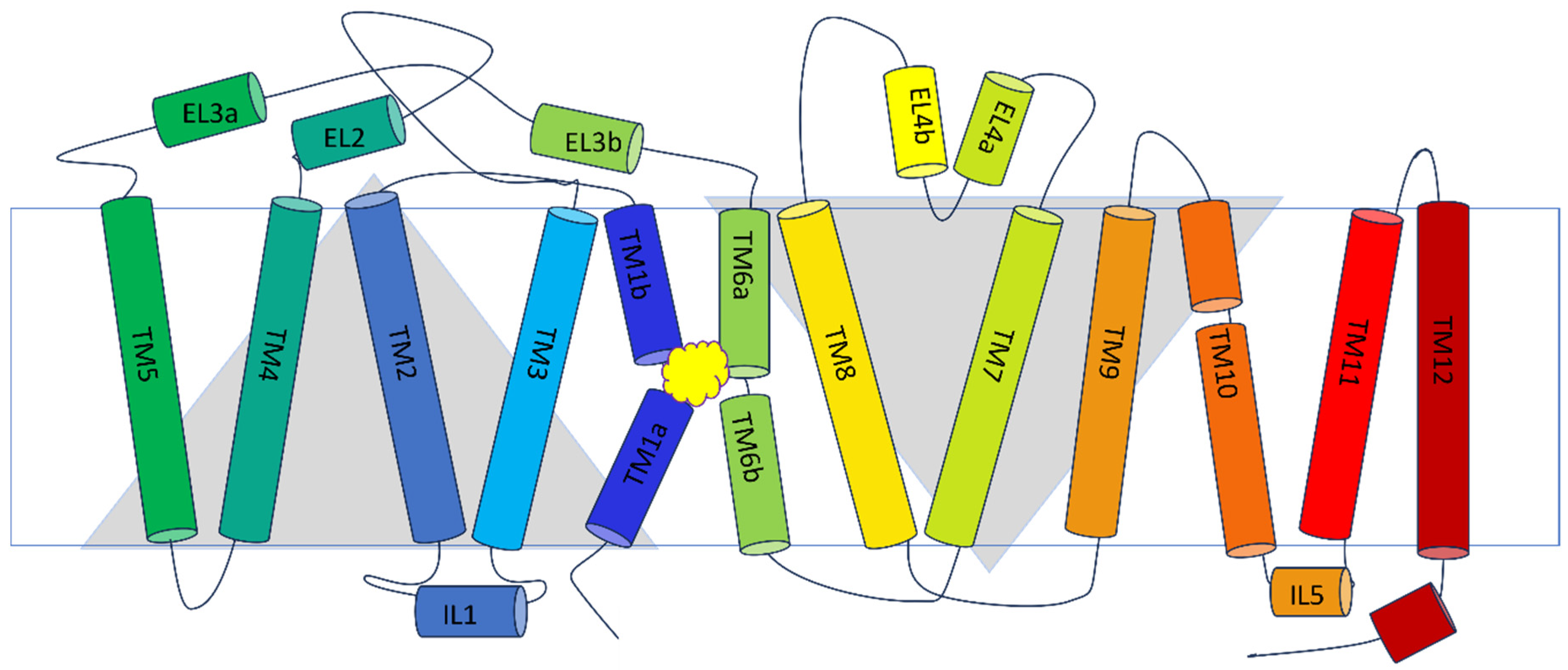
2.2.1. Overall Structure
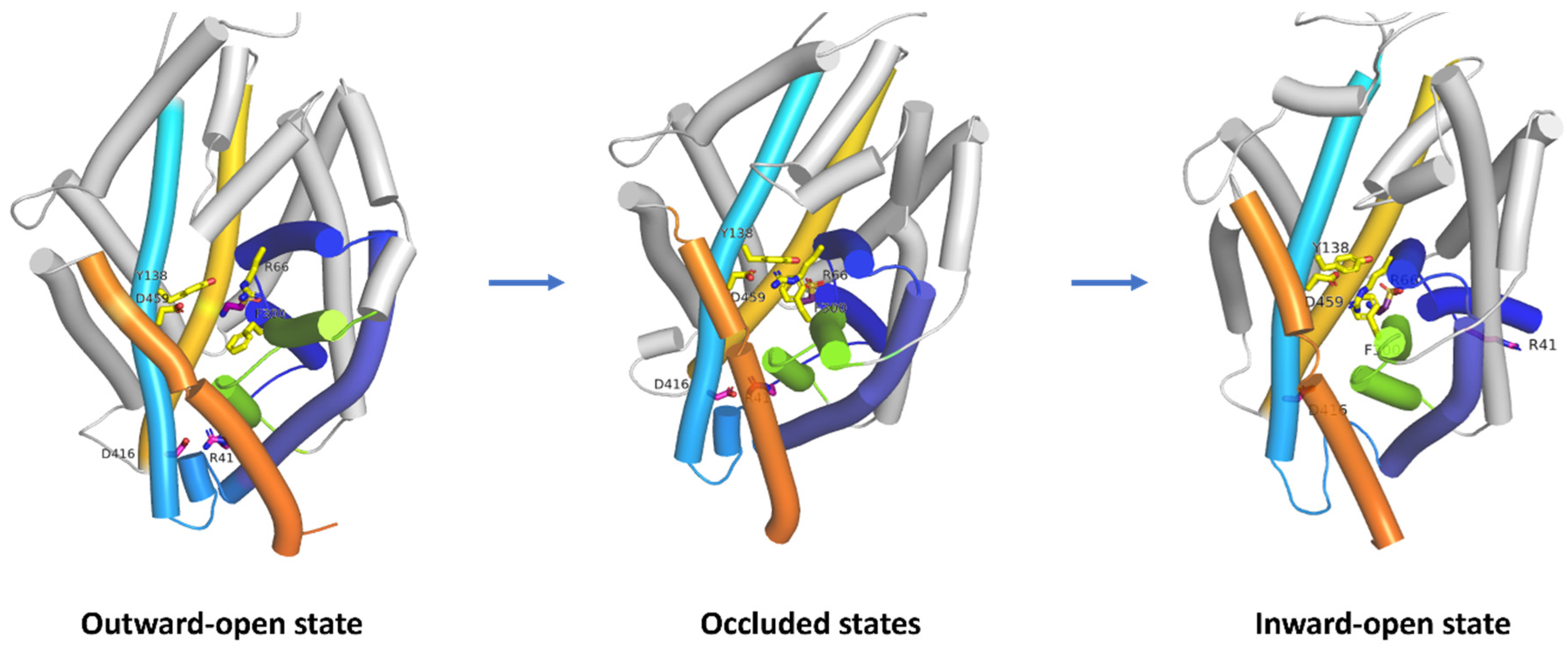
2.2.2. Mechanism of Action
2.2.3. Binding Sites
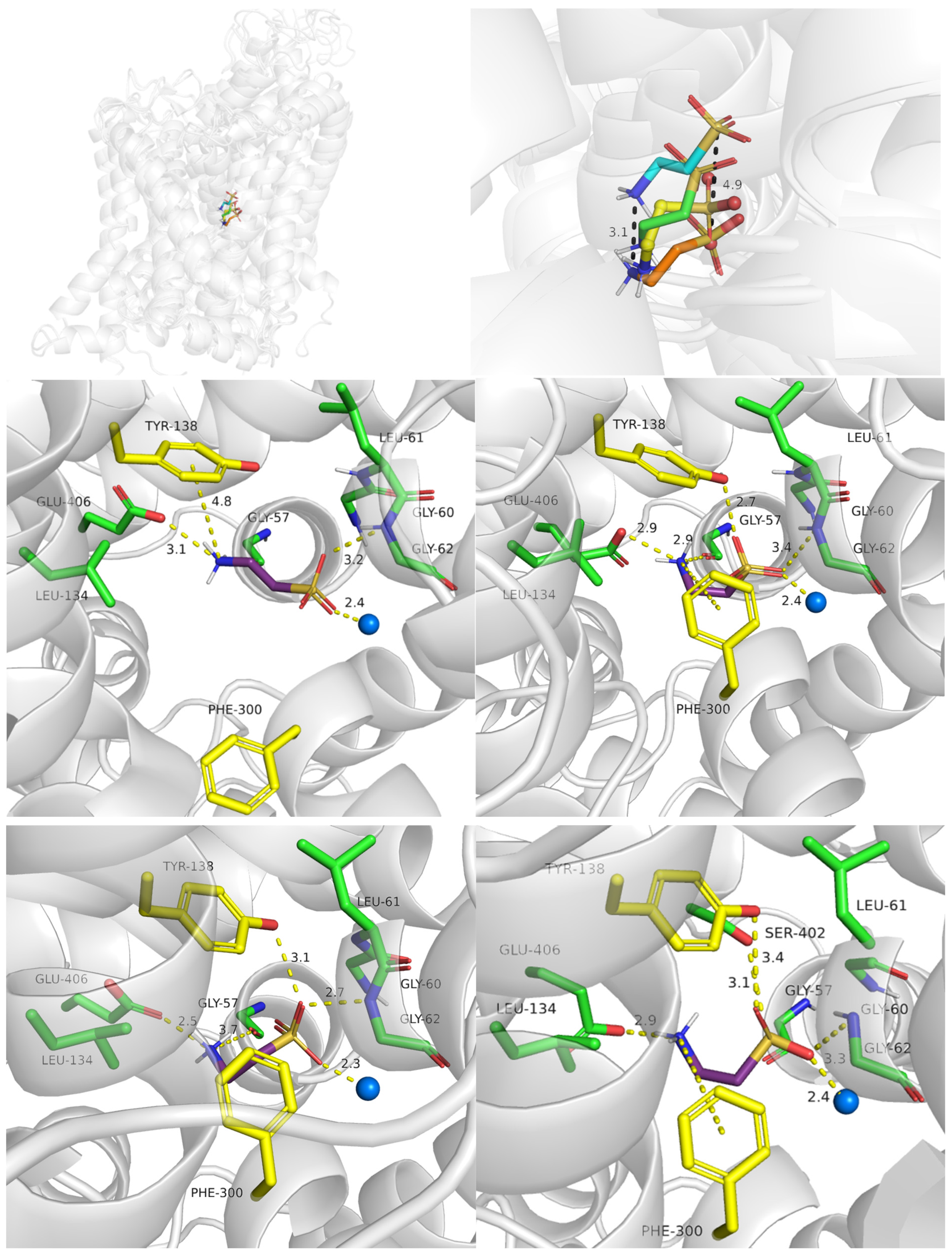
2.2.4. S1 Binding Site Analysis
2.3. TauT Docking Studies
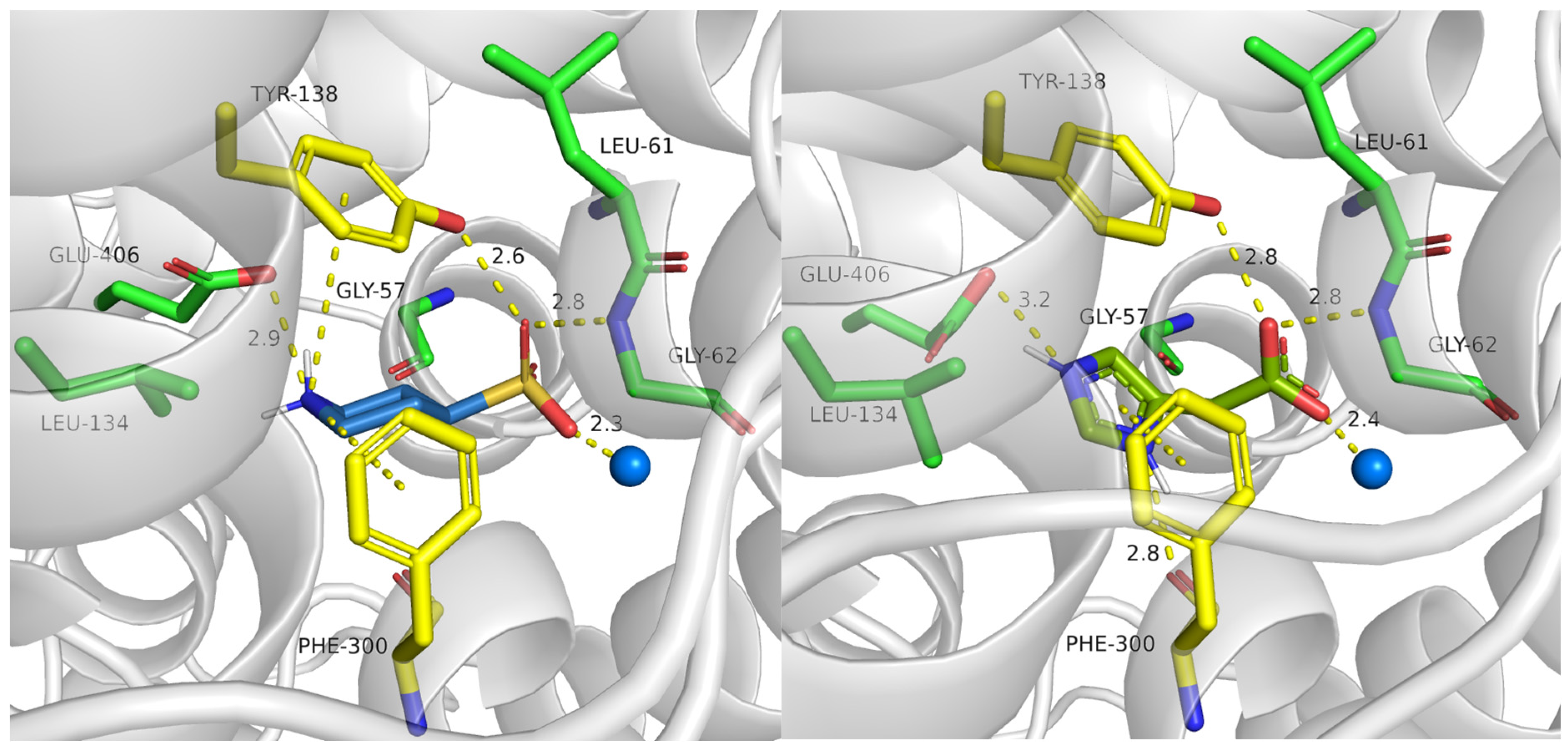
2.3.1. TauT Ligands Binding Mode
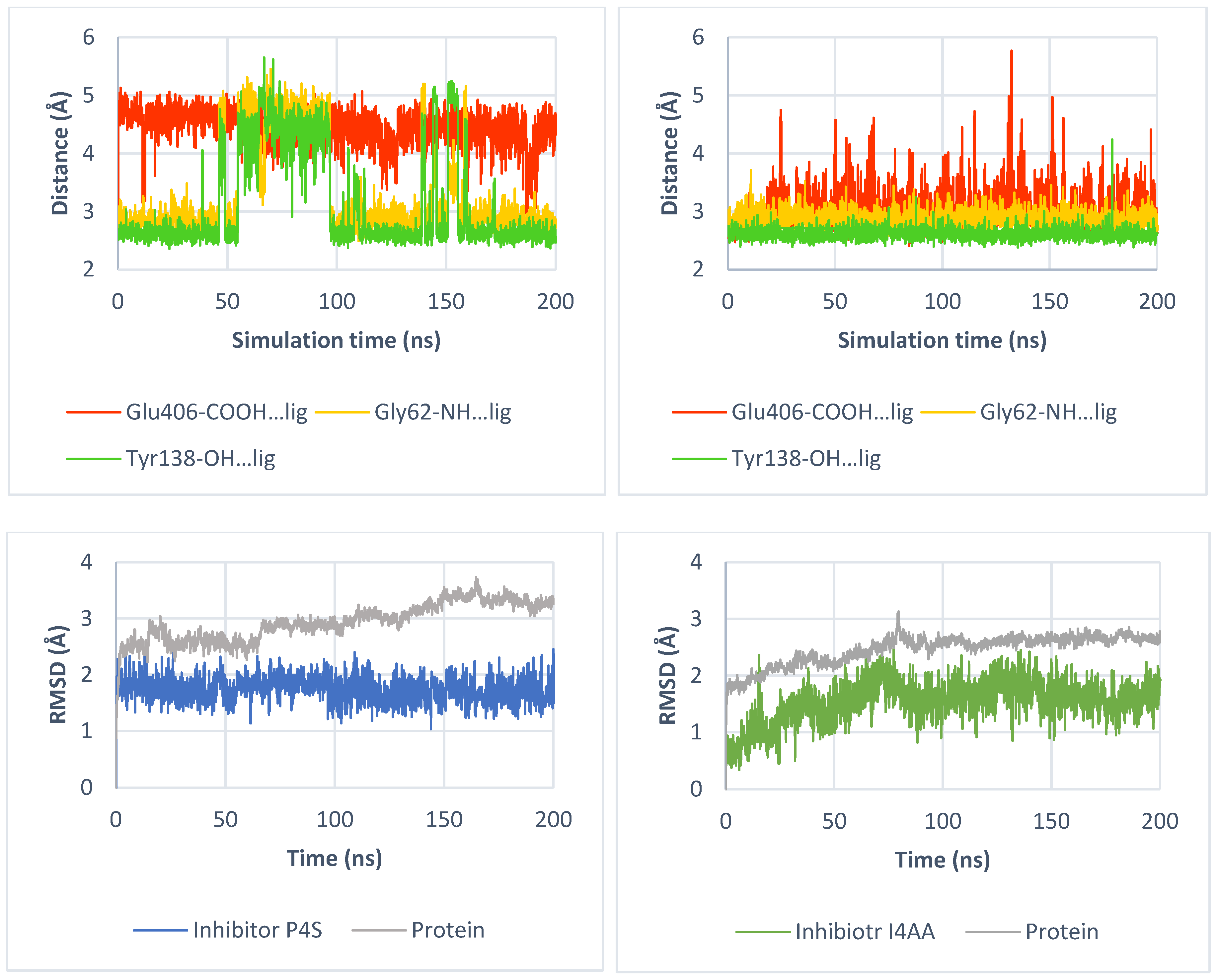
2.3.2. Binding of Taurine Transporter Inhibitors
2.4. Role of Mutations in Taurine Transporter
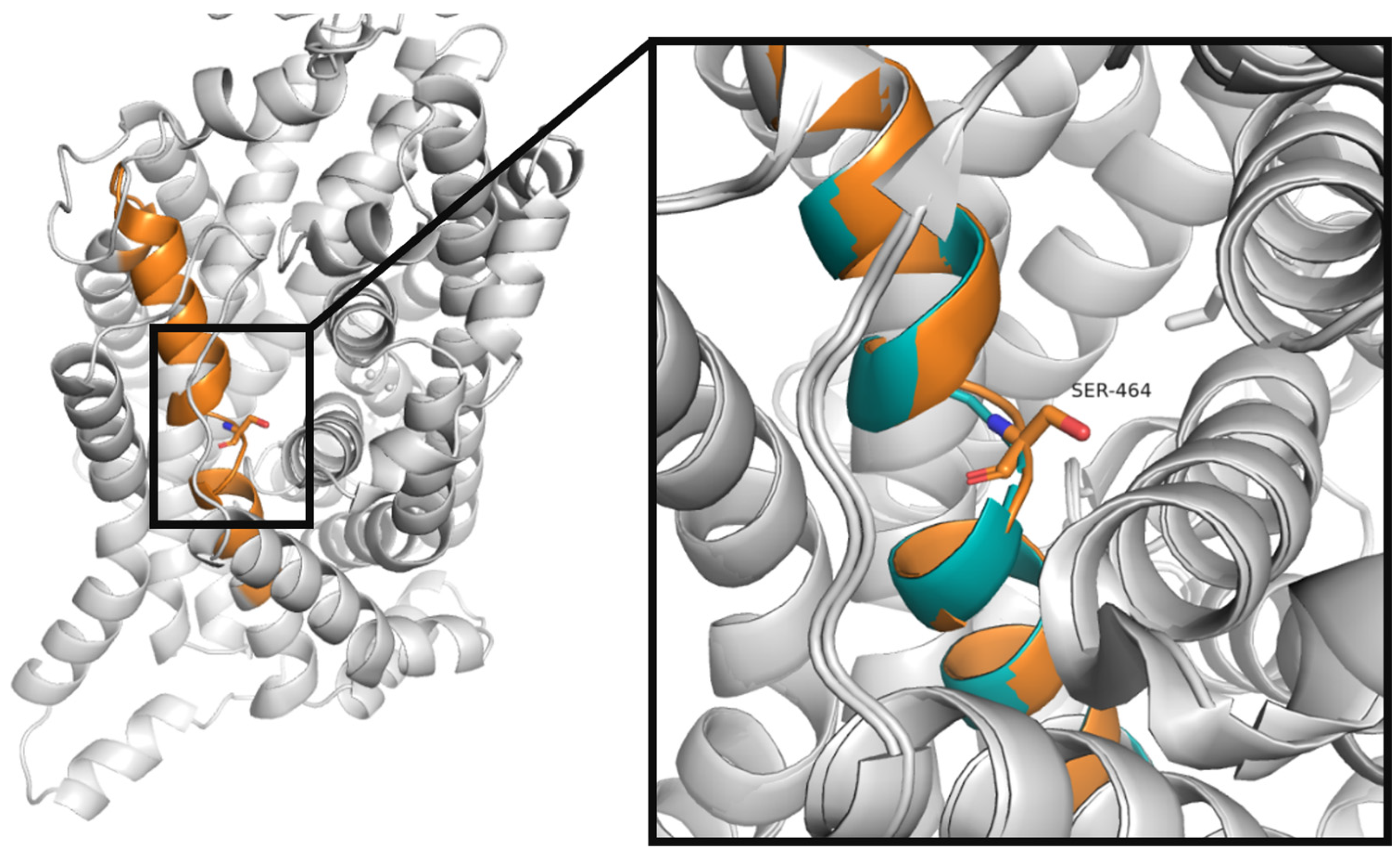
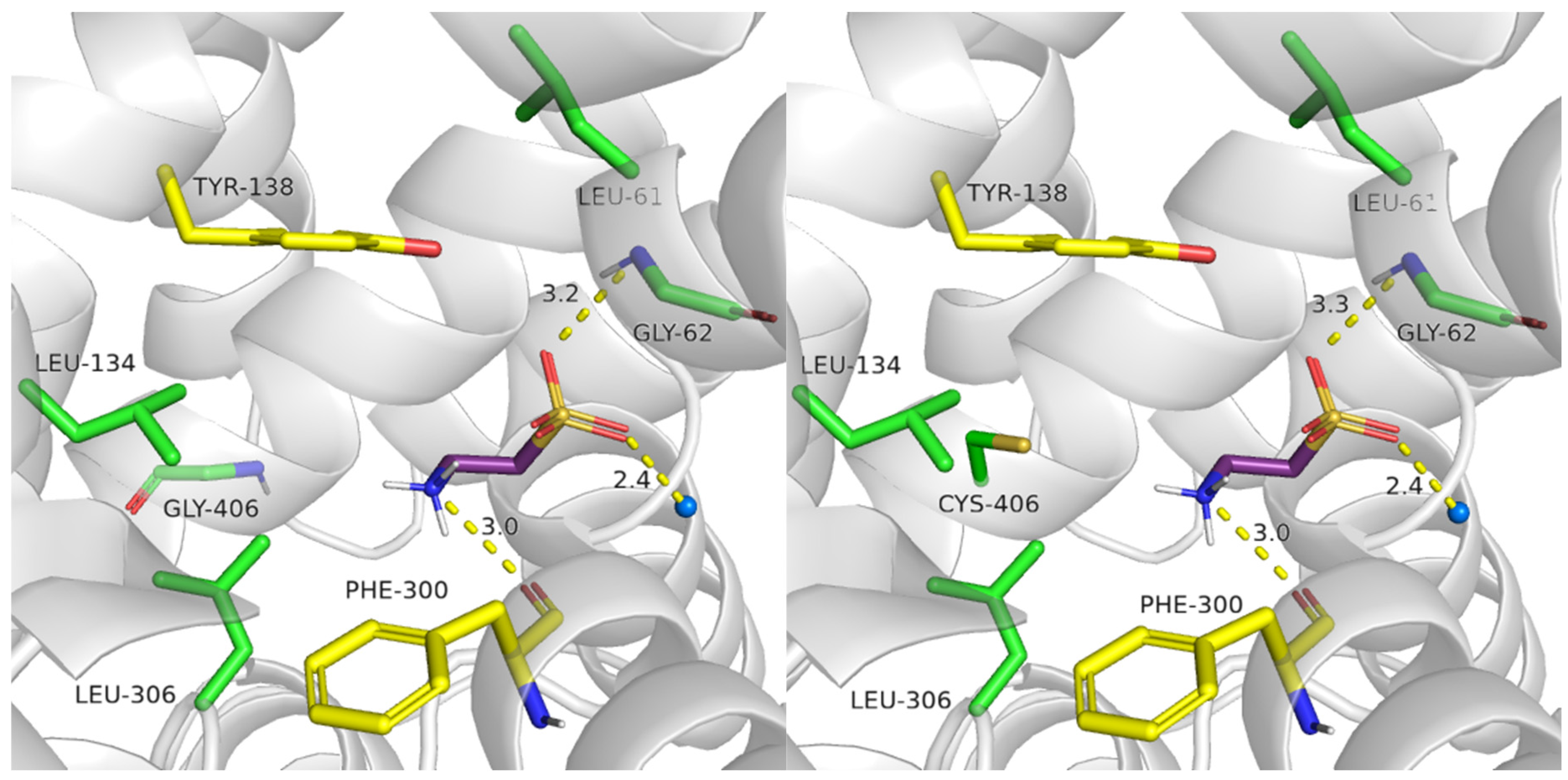
2.4.1. Glu406 Is Crucial for SLC6A6 Transporter Activity
2.4.2. Importance of Gly57 and Leu306 for SLC6A6 Activity
3. Materials and Methods
3.1. Homology Modelling
3.2. Docking Studies
3.3. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pramod, A.B.; Foster, J.; Carvelli, L.; Henry, L.K. SLC6 transporters: Structure, function, regulation, disease association and therapeutics. Mol. Aspects Med. 2013, 34, 197–219. [Google Scholar] [CrossRef]
- Saks, V.; Kaambre, T.; Guzun, R.; Anmann, T.; Sikk, P.; Schlattner, U.; Wallimann, T.; Aliev, M.; Vendelin, M. Creatine and Creatine Kinase in Health and Disease. Subcell. Biochem. 2007, 46, 27–65. Available online: http://www.researchgate.net/publication/240997256_The_Creatine_Kinase_Phosphotransfer_Network_Thermodynamic_andKinetic_Considerations_the_Impact_of_the_Mitochondrial_OuterMembrane_and_Modelling_Approaches (accessed on 18 May 2021). [PubMed]
- SHarding, D.; Armstrong, J.F.; Faccenda, E.; Southan, C.; Alexander, S.P.H.; Davenport, A.P.; Spedding, M.; Davies, J.A. The IUPHAR/BPS Guide to PHARMACOLOGY in 2024. Nucleic Acids Res 2024, 52, D1438–D1449. [Google Scholar] [CrossRef] [PubMed]
- Jangra, A.; Gola, P.; Singh, J.; Gond, P.; Ghosh, S.; Rachamalla, M.; Dey, A.; Iqbal, D.; Kamal, M.; Sachdeva, P.; et al. Emergence of taurine as a therapeutic agent for neurological disorders. Neural Regen. Res. 2024, 19, 62–68. [Google Scholar] [CrossRef]
- García-Ayuso, D.; Di Pierdomenico, J.; Martínez-Vacas, A.; Vidal-Sanz, M.; Picaud, S.; Villegas-Pérez, M.P. Taurine: A promising nutraceutic in the prevention of retinal degeneration. Neural Regen. Res. 2024, 19, 606–610. [Google Scholar] [CrossRef]
- Ito, T.; Murakami, S. Taurine deficiency associated with dilated cardiomyopathy and aging. J. Pharmacol. Sci. 2024, 154, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Ansar, M.; Ranza, E.; Shetty, M.; Paracha, S.A.; Azam, M.; Kern, I.; Iwaszkiewicz, J.; Farooq, O.; Pournaras, C.J.; Malcles, A.; et al. Taurine treatment of retinal degeneration and cardiomyopathy in a consanguineous family with SLC6A6 taurine transporter deficiency. Hum. Mol. Genet. 2020, 29, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Preising, M.N.; Görg, B.; Friedburg, C.; Qvartskhava, N.; Budde, B.S.; Bonus, M.; Toliat, M.R.; Pfleger, C.; Altmüller, J.; Herebian, D.; et al. Biallelic mutation of human SLC6A6 encoding the taurine transporter TAUT is linked to early retinal degeneration. FASEB J. 2019, 33, 11507–11527. [Google Scholar] [CrossRef]
- Garnier, S.; Harakalova, M.; Weiss, S.; Mokry, M.; Regitz-Zagrosek, V.; Hengstenberg, C.; Cappola, T.P.; Isnard, R.; Arbustini, E.; Cook, S.A.; et al. Genome-wide association analysis in dilated cardiomyopathy reveals two new players in systolic heart failure on chromosomes 3p25.1 and 22q11.23. Eur. Heart J. 2021, 42, 2000–2011. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, X.; He, Y.; Liao, C. SLC6A1-miR133a-CDX2 loop regulates SK-OV-3 ovarian cancer cell proliferation, migration and invasion. Oncol. Lett. 2018, 16, 4977–4983. [Google Scholar] [CrossRef]
- Chen, C.; Cai, Z.; Zhuo, Y.; Xi, M.; Lin, Z.; Jiang, F.; Liu, Z.; Wan, Y.; Zheng, Y.; Li, J.; et al. Overexpression of SLC6A1 associates with drug resistance and poor prognosis in prostate cancer. BMC Cancer 2020, 20, 289. [Google Scholar] [CrossRef]
- Sung, H.Y.; Yang, S.-D.; Park, A.K.; Ju, W.; Ahn, J.-H. Aberrant Hypomethylation of Solute Carrier Family 6 Member 12 Promoter Induces Metastasis of Ovarian Cancer. Yonsei Med. J. 2017, 58, 27. [Google Scholar] [CrossRef]
- Wang, D.; Du, J.; Ren, C.; Zhou, M.; Xia, Z. Elevated SLC6A6 expression drives tumorigenesis and affects clinical outcomes in gastric cancer. Biomark. Med. 2019, 13, 95–104. [Google Scholar] [CrossRef]
- Yasunaga, M.; Matsumura, Y. Role of SLC6A6 in promoting the survival and multidrug resistance of colorectal cancer. Sci. Rep. 2014, 4, 4852. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.F.; Pei, G.H.; Wang, N.; Che, Y.C.; Yu, F.S.; Yin, F.F.; Liu, H.X.; Luo, B.; Wang, Y.K. MiR-3156-3p is downregulated in HPV-positive cervical cancer and performs as a tumor-suppressive miRNA. Virol. J. 2017, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Guo, X.; Tang, H. SLC6A8 is involved in the progression of non-small cell lung cancer through the Notch signaling pathway. Ann. Transl. Med. 2021, 9, 264. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhou, Y.; Lou, M.; Gao, Z.; Li, X.; Yuan, K. SLC6A8 is a Potential Biomarker for Poor Prognosis in Lung Adenocarcinoma. Front. Genet. 2022, 13, 845373. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, M.; Sun, Y.; Jin, T.; Zhu, P.; Wan, X.; Hou, Y.; Tu, G. SLC6A8-mediated intracellular creatine accumulation enhances hypoxic breast cancer cell survival via ameliorating oxidative stress. J. Exp. Clin. Cancer Res. 2021, 40, 168. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Wu, X.J.; Li, W.C.; Zhuo, C.; Xu, Z.M.; Tan, C.; Ma, R.H.; Wang, J.C.; Pu, J. SLC6A8 Knockdown Suppresses the Invasion and Migration of Human Hepatocellular Carcinoma Huh-7 and Hep3B Cells. Technol. Cancer Res. Treat. 2020, 19, 1533033820983029. [Google Scholar] [CrossRef]
- Kurth, I.; Yamaguchi, N.; Andreu-Agullo, C.; Tian, H.S.; Sridhar, S.; Takeda, S.; Gonsalves, F.C.; Loo, J.M.; Barlas, A.; Manova-Todorova, K.; et al. Therapeutic targeting of SLC6A8 creatine transporter suppresses colon cancer progression and modulates human creatine levels. Sci. Adv. 2021, 7, eabi7511. [Google Scholar] [CrossRef]
- Stary, D.; Bajda, M. Taurine and Creatine Transporters as Potential Drug Targets in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 3788. [Google Scholar] [CrossRef] [PubMed]
- Study Details|A Study of RGX-202-01 (Ompenaclid) as Combination Therapy in 2nd Line RAS Mutant Advanced Colorectal Cancer|ClinicalTrials.gov, (n.d.). Available online: https://clinicaltrials.gov/study/NCT03597581?cond=ompenaclid&rank=2#more-information (accessed on 14 May 2024).
- Łątka, K.; Jończyk, J.; Bajda, M. Structure modeling of γ-aminobutyric acid transporters—Molecular basics of ligand selectivity. Int. J. Biol. Macromol. 2020, 158, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Beuming, T.; Shi, L.; Javitch, J.A.; Weinstein, H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 2006, 70, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Banci, G.; Martini, R.; Ecker, G.F. Studies of structural determinants of substrate binding in the Creatine Transporter (CreaT, SLC6A8) using molecular models. Sci. Rep. 2020, 10, 6241. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Joseph, D.; Pidathala, S.; Mallela, A.K.; Penmatsa, A. Structure and Gating Dynamics of Na+/Cl− Coupled Neurotransmitter Transporters. Front. Mol. Biosci. 2019, 6, 80. [Google Scholar] [CrossRef]
- Yahara, T.; Tachikawa, M.; Akanuma, S.I.; Kubo, Y.; Hosoya, K.I. Amino acid residues involved in the substrate specificity of TauT/SLC6A6 for taurine and γ-aminobutyric acid. Biol. Pharm. Bull. 2014, 37, 817–825. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Fabbro, D.; Kelly, E.; Mathie, A.A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Davies, J.A.; et al. The Concise Guide to PHARMACOLOGY 2023/24: Transporters. Br. J. Pharmacol 2023, 180, S374–S469. [Google Scholar] [CrossRef]
- Sakai, S.; Tosaka, T.; Tasaka, J.; Hashiguchi, T.; Yoshihama, I. Taurine uptake by glial cells in the bullfrog sympathetic ganglia. Neurochem. Int. 1989, 14, 193–198. [Google Scholar] [CrossRef]
- Scimemi, A. Structure, function, and plasticity of GABA transporters. Front. Cell Neurosci. 2014, 8, 161. [Google Scholar] [CrossRef]
- UniProt, (n.d.). Available online: https://www.uniprot.org/ (accessed on 14 October 2021).
- Bourne, P.E.; Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Motiwala, Z.; Aduri, N.G.; Shaye, H.; Han, G.W.; Lam, J.H.; Katritch, V.; Cherezov, V.; Gati, C. Structural basis of GABA reuptake inhibition. Nature 2022, 606, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef]
- Zafra, F.; Giménez, C. Glycine transporters and synaptic function. IUBMB Life 2008, 60, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Harsing, L.; Zsilla, G.; Matyus, P.; Nagy, K.; Marko, B.; Gyarmati, Z.; Timar, J. Interactions between glycine transporter type 1 (GlyT-1) and some inhibitor molecules—Glycine transporter type 1 and its inhibitors (Review). Acta Physiol. Hung. 2012, 99, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Łątka, K.; Jończyk, J.; Bajda, M. γ-Aminobutyric acid transporters as relevant biological target: Their function, structure, inhibitors and role in the therapy of different diseases. Int. J. Biol. Macromol. 2020, 158, 750–772. [Google Scholar] [CrossRef] [PubMed]
- Łątka, K.; Bajda, M. Analysis of Different Binding Modes for Tiagabine within the GAT-1 Transporter. Biomolecules 2022, 12, 1663. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Moroniak, S.J.; Michel, H. Identification of competitive inhibitors of the human taurine transporter TauT in a human kidney cell line. Pharmacol. Rep. 2019, 71, 121–127. [Google Scholar] [CrossRef]
- Rasmussen, R.N.; Lagunas, C.; Plum, J.; Holm, R.; Nielsen, C.U. Interaction of GABA-mimetics with the taurine transporter (TauT, Slc6a6) in hyperosmotic treated Caco-2, LLC-PK1 and rat renal SKPT cells. Eur. J. Pharm. Sci. 2016, 82, 138–146. [Google Scholar] [CrossRef]
- Valembois, S.; Krall, J.; Frølund, B.; Steffansen, B. Imidazole-4-acetic acid, a new lead structure for interaction with the taurine transporter in outer blood-retinal barrier cells. Eur. J. Pharm. Sci. 2017, 103, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Ishizuka, S.; Ito, T.; Yoneyama, D.; Akanuma, S.-I.; Hosoya, K.-I. Involvement of TauT/SLC6A6 in Taurine Transport at the Blood–Testis Barrier. Metabolites 2022, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Bioinformatics Tools for Multiple Sequence Alignment < EMBL-EBI, (n.d.). Available online: https://www.ebi.ac.uk/Tools/msa/ (accessed on 22 November 2021).
- Clamp, M.; Cuff, J.; Searle, S.M.; Barton, G.J. The Jalview Java alignment editor. Bioinformatics 2004, 20, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Piscitelli, C.L.; Yamashita, A.; Gouaux, E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 2008, 322, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Goehring, A.; Wang, K.H.; Penmatsa, A.; Ressler, R.; Gouaux, E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 2013, 503, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef]
- Pidathala, S.; Mallela, A.K.; Joseph, D.; Penmatsa, A. Structural basis of norepinephrine recognition and transport inhibition in neurotransmitter transporters. Nat. Commun. 2021, 12, 2199. [Google Scholar] [CrossRef]
- Shahsavar, A.; Stohler, P.; Bourenkov, G.; Zimmermann, I.; Siegrist, M.; Guba, W.; Pinard, E.; Sinning, S.; Seeger, M.A.; Schneider, T.R.; et al. Structural insights into the inhibition of glycine reuptake. Nature 2021, 591, 677–681. [Google Scholar] [CrossRef]
- Zhu, A.; Huang, J.; Kong, F.; Tan, J.; Lei, J.; Yuan, Y.; Yan, C. Molecular basis for substrate recognition and transport of human GABA transporter GAT1. Nat. Struct. Mol. Biol. 2023, 30, 1012–1022. [Google Scholar] [CrossRef]

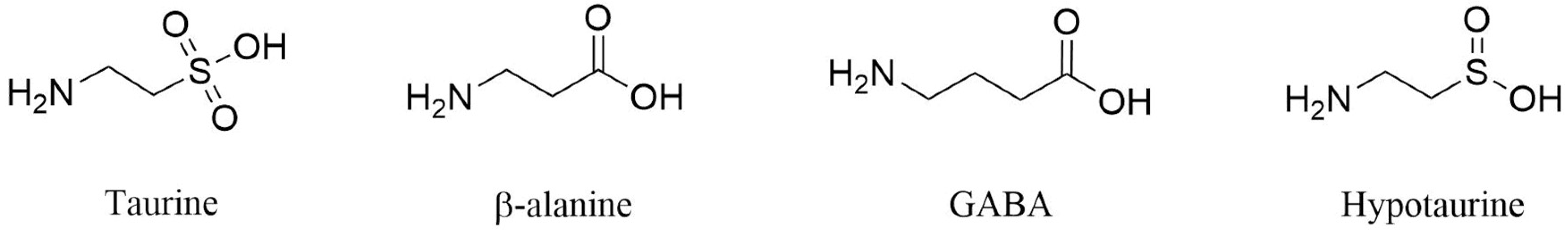




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamilies | Human Gene Name | Transporter Name |
|---|---|---|
| GABA (γ-aminobutyric acid) transporter subfamily | SLC6A1 SLC6A6 SLC6A8 SLC6A10 SLC6A11 SLC6A12 SLC6A13 | GAT1 TauT CT1 CT2 GAT3 BGT1 GAT2 |
| Monoamine transporter subfamily | SLC6A2 SLC6A3 SLC6A4 | NET DAT SERT |
| Glycine transporter subfamily | SLC6A5 SLC6A7 SLC6A9 SLC6A14 | GlyT2 PROT GlyT1 ATB0,+ |
| Neutral amino acid transporter subfamily | SLC6A15 SLC6A16 SLC6A17 SLC6A18 SLC6A19 SLC6A20 | B0AT2 NTT5 NTT4 B0AT3 B0AT1 SIT1 |
| Sequences | dDAT | aLeuT | hSERT | hGlyT1 | hGAT1 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Identity (I.), similarity (S.) | I. [%] | S. [%] | I. [%] | S. [%] | I. [%] | S. [%] | I. [%] | S. [%] | I. [%] | S. [%] |
| hGAT1 | 40.3 | 58.8 | 21.2 | 36.4 | 39.5 | 59.0 | 36.4 | 53.2 | 100.0 | 100.0 |
| hBGT1 | 41.6 | 58.6 | 21.0 | 36.5 | 38.7 | 57.9 | 35.0 | 50.5 | 47.6 | 66.4 |
| hGAT2 | 42.1 | 60.1 | 21.4 | 36.1 | 40.7 | 56.7 | 35.6 | 50.7 | 49.5 | 67.5 |
| hGAT3 | 39.6 | 55.2 | 19.4 | 36.3 | 38.0 | 56.1 | 36.9 | 54.4 | 49.0 | 64.5 |
| hTauT | 41.2 | 58.2 | 21.5 | 34.1 | 38.1 | 53.4 | 35.8 | 53.6 | 46.5 | 63.1 |
| hCT1 | 39.6 | 57.2 | 20.5 | 33.7 | 38.6 | 57.1 | 35.9 | 51.3 | 48.2 | 64.7 |
| Protein | Extracellular Gate | S1 Site | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GAT1 | Arg69 | Tyr140 | Phe294 | Asp451 | Tyr60 | Gly63 | Leu64 | Gly65 | Leu136 | Gly297 | Leu300 | Thr400 |
| BGT1 | Arg61 | Tyr133 | Phe293 | Asp452 | Glu52 | Gly55 | Leu56 | Gly57 | Leu129 | Ala296 | Gln299 | Cys399 |
| GAT2 | Arg57 | Tyr129 | Phe288 | Asp447 | Glu48 | Gly51 | Leu52 | Gly53 | Leu125 | Ala291 | Leu294 | Cys394 |
| GAT3 | Arg75 | Tyr147 | Phe308 | Asp467 | Glu66 | Gly69 | Leu70 | Gly71 | Leu143 | Ala311 | Leu314 | Cys414 |
| TauT | Arg66 | Tyr138 | Phe300 | Asp459 | Gly57 | Gly60 | Leu61 | Gly62 | Leu134 | Ala303 | Leu306 | Glu406 |
| CT1 | Arg77 | Tyr148 | Phe315 | Asp474 | Phe68 | Gly71 | Leu72 | Gly73 | Cys144 | Ala318 | Leu321 | Gly421 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stary, D.; Bajda, M. Structural Studies of the Taurine Transporter: A Potential Biological Target from the GABA Transporter Subfamily in Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 7339. https://doi.org/10.3390/ijms25137339
Stary D, Bajda M. Structural Studies of the Taurine Transporter: A Potential Biological Target from the GABA Transporter Subfamily in Cancer Therapy. International Journal of Molecular Sciences. 2024; 25(13):7339. https://doi.org/10.3390/ijms25137339
Chicago/Turabian StyleStary, Dorota, and Marek Bajda. 2024. "Structural Studies of the Taurine Transporter: A Potential Biological Target from the GABA Transporter Subfamily in Cancer Therapy" International Journal of Molecular Sciences 25, no. 13: 7339. https://doi.org/10.3390/ijms25137339