
Thyroid Hormone Signaling in Retinal Development and Function: Implications for Diabetic Retinopathy and Age-Related Macular Degeneration
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TH Signaling
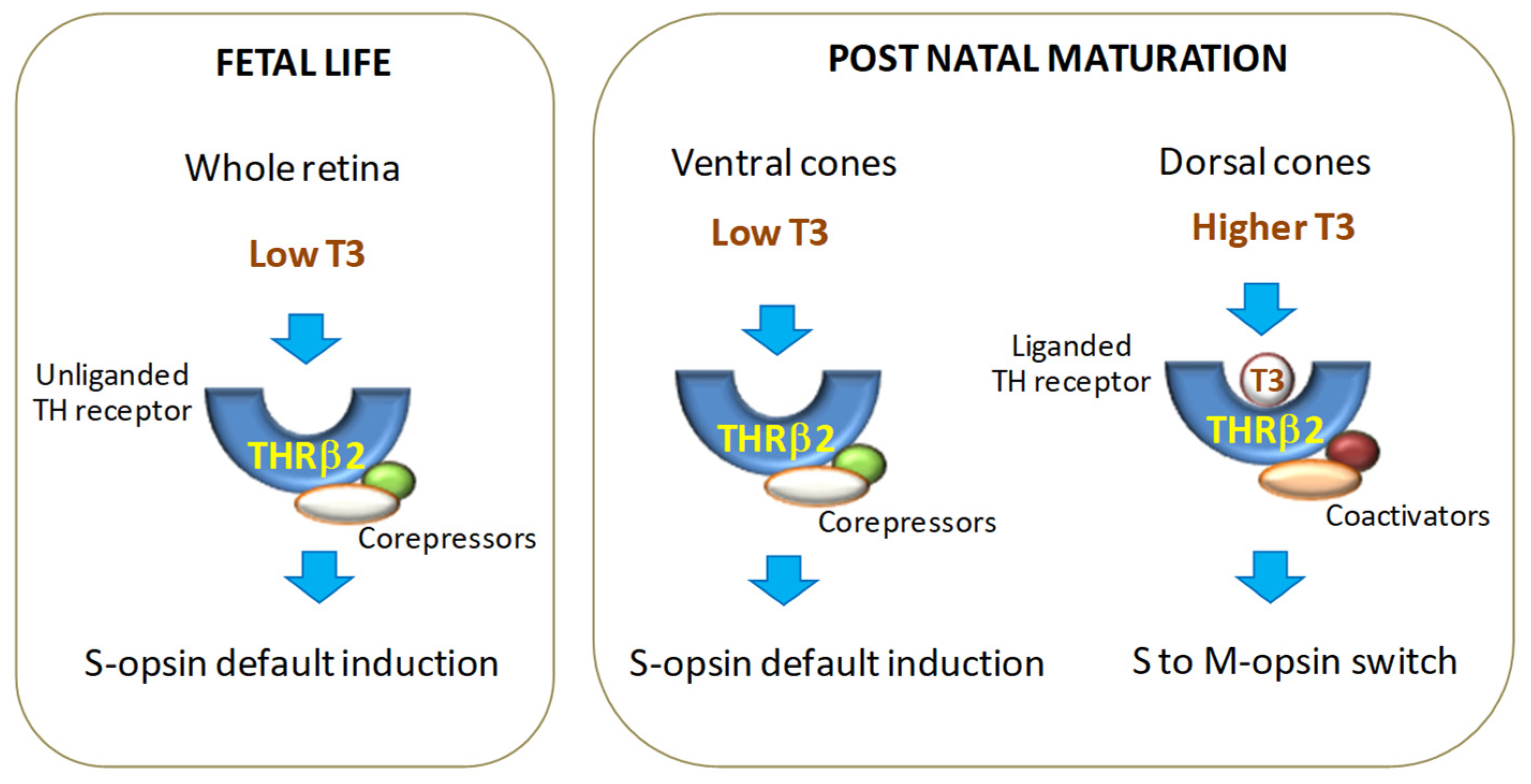
3. Role of Thyroid Hormone Signaling in Retinal Development and Function
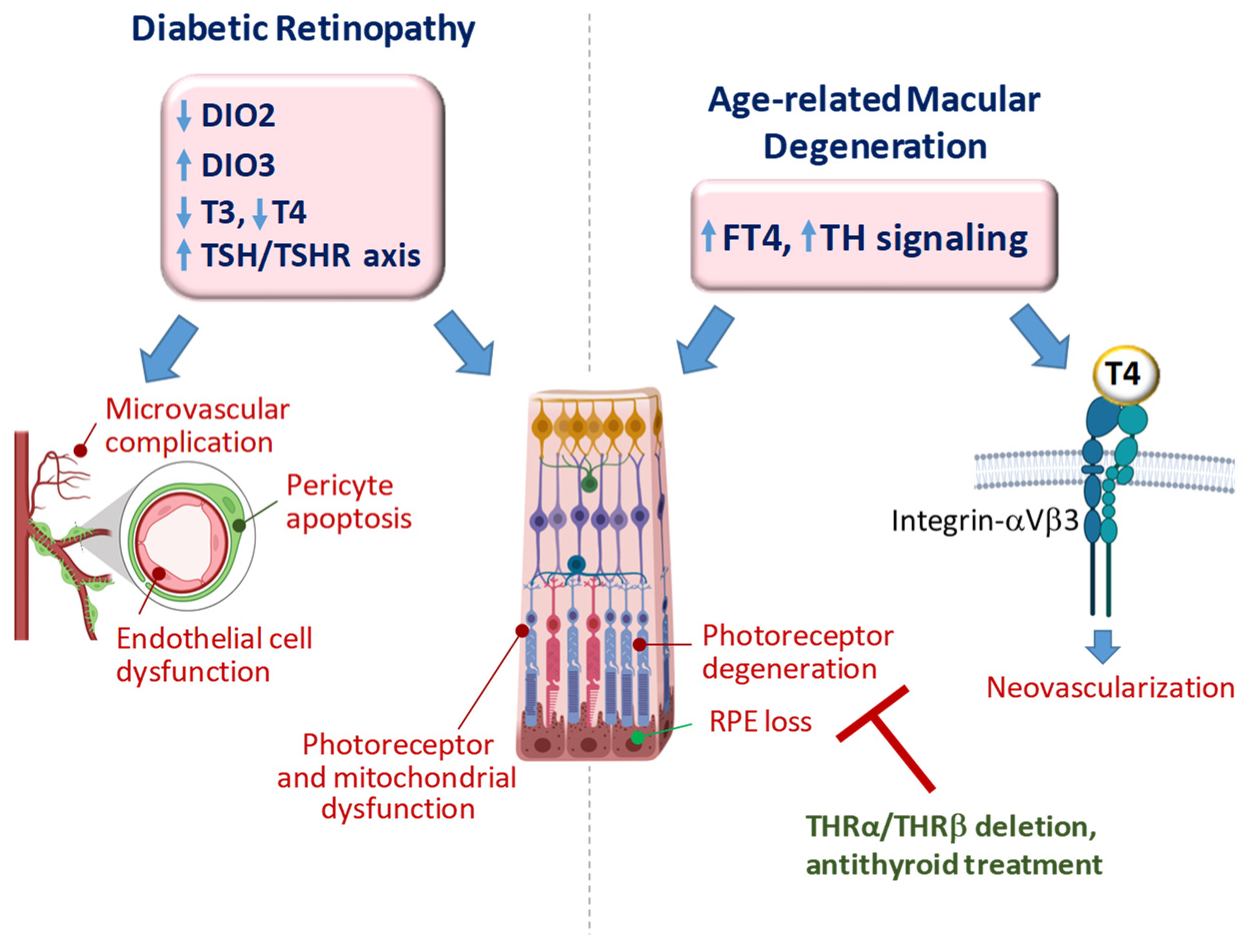
4. TH Signaling in Diabetic Retinopathy
4.1. DR: Disease Overview
4.2. Impact of TH Signaling on DR: Clinical Evidence
4.3. Impact of TH Signaling on DR: Experimental Evidence
5. TH Signaling in Age-Related Macular Degeneration
5.1. AMD: Disease Overview
5.2. Impact of TH Signaling in AMD: Clinical Evidence
5.3. Impact of TH Signaling in AMD: Experimental Evidence
6. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- García-Aldea, Á.; Guillén-Yunta, M.; Valcárcel-Hernández, V.; Montero-Pedrazuela, A.; Guadaño-Ferraz, A.; Bárez-López, S. Insights on the role of thyroid hormone transport in neurosensory organs and implication for the Allan-Herndon-Dudley syndrome. Eur. Thyroid. J. 2024, 13, e230241. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Ng, L.; Ma, M.; Kefas, B.; Davies, T.F.; Hernandez, A.; Forrest, D. Retarded developmental expression and patterning of retinal cone opsins in hypothyroid mice. Endocrinology 2009, 150, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Vancamp, P.; Bourgeois, N.M.A.; Houbrechts, A.M.; Darras, V.M. Knockdown of the thyroid hormone transporter MCT8 in chicken retinal precursor cells hampers early retinal development and results in a shift towards more UV/blue cones at the expense of green/red cones. Exp. Eye Res. 2019, 178, 135–147. [Google Scholar] [CrossRef] [PubMed]
- McNerney, C.; Johnston, R.J., Jr. Thyroid hormone signaling specifies cone photoreceptor subtypes during eye development: Insights from model organisms and human stem cell-derived retinal organoids. Vitam. Horm. 2021, 116, 51–90. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Sun, Y.; Li, S.; Chen, Y.; Li, L.; Fang, M.; Shi, R.; Tong, D.; Chen, J.; Ma, Y.; et al. Single-cell profiling reveals Müller glia coordinate retinal intercellular communication during light/dark adaptation via thyroid hormone signaling. Protein Cell. 2023, 14, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Pinazo-Durán, M.D.; Pons-Vázquez, S.; Gallego-Pinazo, R.; Galbis Estrada, C.; Zanón-Moreno, V.; Vila Bou, V.; Sanz Solana, P. Thyroid hormone deficiency disrupts rat eye neurodevelopment. Brain Res. 2011, 1392, 16–26, Erratum in Brain Res. 2012, 1446, 156. [Google Scholar] [CrossRef] [PubMed]
- Pinazo-Durán, M.D.; Iborra, F.J.; Pons, S.; Sevilla-Romero, E.; Gallego-Pinazo, R.; Muñoz, A. Postnatal thyroid hormone supplementation rescues developmental abnormalities induced by congenital-neonatal hypothyroidism in the rat retina. Ophthalmic Res. 2005, 37, 225–234. [Google Scholar] [CrossRef]
- Boyes, W.K.; Degn, L.; George, B.J.; Gilbert, M.E. Moderate perinatal thyroid hormone insufficiency alters visual system function in adult rats. Neurotoxicology 2018, 67, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Gamborino, M.J.; Sevilla-Romero, E.; Muñoz, A.; Hernández-Yago, J.; Renau-Piqueras, J.; Pinazo-Durán, M.D. Role of thyroid hormone in craniofacial and eye development using a rat model. Ophthalmic Res. 2001, 33, 283–291. [Google Scholar] [CrossRef]
- Bianco, A.C.; Dumitrescu, A.; Gereben, B.; Ribeiro, M.O.; Fonseca, T.L.; Fernandes, G.W.; Bocco, B.M.L.C. Paradigms of Dynamic Control of Thyroid Hormone Signaling. Endocr. Rev. 2019, 40, 1000–1047. [Google Scholar] [CrossRef]
- El-Eshmawy, M.M.; Shahin, M. Thyroid and Eye: Where They Meet in Clinical Practice. Endocr. Metab. Immun. Disord. Drug. Targets 2018, 20, 39–49. [Google Scholar] [CrossRef]
- Wu, J.; Yue, S.; Geng, J.; Liu, L.; Teng, W.; Liu, L.; Chen, L. Relationship between Diabetic Retinopathy and Subclinical Hypothyroidism: A meta-analysis. Sci. Rep. 2015, 5, 12212. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, Z.; Tang, W.; Liu, W.; Wu, X.; Pan, C. Association of Thyroid Hormone Levels with Microvascular Complications in Euthyroid Type 2 Diabetes Mellitus Patients. Diab. Metab. Syndr. Obes. 2022, 15, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, J.; Gao, G.; Tan, M.; Ding, B.; Li, H.; Ma, J. Association between Free Thyroxine Levels and Diabetic Retinopathy in Euthyroid Patients with Type 2 Diabetes Mellitus. Endocr. Res. 2020, 45, 111–118. [Google Scholar] [CrossRef]
- Chaker, L.; Buitendijk, G.H.; Dehghan, A.; Medici, M.; Hofman, A.; Vingerling, J.R.; Franco, O.H.; Klaver, C.C.; Peeters, R.P. Thyroid function and age-related macular degeneration: A prospective population-based cohort study--the Rotterdam Study. BMC Med. 2015, 13, 94. [Google Scholar] [CrossRef]
- Forini, F.; Nicolini, G.; Amato, R.; Balzan, S.; Saba, A.; Bertolini, A.; Andreucci, E.; Marracci, S.; Melecchi, A.; Terlizzi, D.; et al. Local modulation of thyroid hormone signaling in the retina affects the development of diabetic retinopathy. Biochim. Biophys. Acta. Mol. Basis. Dis. 2024, 1870, 166892. [Google Scholar] [CrossRef] [PubMed]
- Ortiga-Carvalho, T.M.; Chiamolera, M.I.; Pazos-Moura, C.C.; Wondisford, F.E. Hypothalamus-Pituitary-Thyroid Axis. Compr. Physiol. 2016, 6, 1387–1428. [Google Scholar] [CrossRef]
- Flier, E.; Unmehopa, U.A.; Alkemade, A. Functional neuroanatomy of thyroid hormone feedback in the human hypothalamus and pituitary gland. Mol. Cell. Endocrinol. 2006, 251, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gereben, B.; Zavacki, A.M.; Ribich, S.; Kim, B.W.; Huang, S.A.; Simonides, W.S.; Zeöld, A.; Bianco, A.C. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. 2008, 29, 898–938. [Google Scholar] [CrossRef]
- Müller, J.; Mayerl, S.; Visser, T.J.; Darras, V.M.; Boelen, A.; Frappart, L.; Mariotta, L.; Verrey, F.; Heuer, H. Tissue-specific alterations in thyroid hormone homeostasis in combined Mct10 and Mct8 deficiency. Endocrinology 2014, 155, 315–325. [Google Scholar] [CrossRef]
- Galton, V.A. The ups and downs of the thyroxine pro-hormone hypothesis. Mol. Cell. Endocr. 2017, 458, 105–111. [Google Scholar] [CrossRef]
- Larsen, P.R.; Zavacki, A.M. The role of the iodothyronine deiodinases in the physiology and pathophysiology of thyroid hormone action. Eur. Thyroid. J. 2012, 1, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Luongo, C.; Dentice, M.; Salvatore, D. Deiodinases and their intricate role in thyroid hormone homeostasis. Nat. Rev. Endocrinol. 2019, 15, 479–488. [Google Scholar] [CrossRef]
- Bianco, A.C.; Kim, B.W. Deiodinases: Implications of the local control of thyroid hormone action. J. Clin. Investig. 2006, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Bren, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef]
- Flamant, F.; Baxter, J.D.; Forrest, D.; Refetoff, S.; Samuels, H.; Scanlan, T.S.; Vennström, B.; Samarut, J. International Union of Pharmacology. LIX. The pharmacology and classification of the nuclear receptor superfamily: Thyroid hormone receptors. Pharmacol. Rev. 2006, 58, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Forrest, D.; Reh, T.A.; Rüsch, A. Neurodevelopmental control by thyroid hormone receptors. Curr. Opin. Neurobiol. 2002, 12, 49–56. [Google Scholar] [CrossRef]
- Minakhina, S.; Bansal, S.; Zhang, A.; Brotherton, M.; Janodia, R.; De Oliveira, V.; Tadepalli, S.; Wondisford, F.E. A Direct Comparison of Thyroid Hormone Receptor Protein Levels in Mice Provides Unexpected Insights into Thyroid Hormone Action. Thyroid 2020, 30, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Hönes, G.S.; Härting, N.; Mittag, J.; Kaiser, F.J. TRα2-An Untuned Second Fiddle or Fine-Tuning Thyroid Hormone Action? Int. J. Mol. Sci. 2022, 23, 6998. [Google Scholar] [CrossRef]
- Grøntved, L.; Waterfall, J.J.; Kim, D.W.; Baek, S.; Sung, M.H.; Zhao, L.; Park, J.W.; Nielsen, R.; Walker, R.L.; Zhu, Y.J.; et al. Transcriptional activation by the thyroid hormone receptor through ligand-dependent receptor recruitment and chromatin remodelling. Nat. Commun. 2015, 6, 7048. [Google Scholar] [CrossRef]
- Forini, F.; Nicolini, G.; Pitto, L.; Iervasi, G. Novel Insight into the Epigenetic and Post-transcriptional Control of Cardiac Gene Expression by Thyroid Hormone. Front. Endocrinol. 2019, 10, 601. [Google Scholar] [CrossRef] [PubMed]
- Astapova, I. Role of co-regulators in metabolic and transcriptional actions of thyroid hormone. J. Mol. Endocrinol. 2016, 56, 73–97. [Google Scholar] [CrossRef] [PubMed]
- Göthe, S.; Wang, Z.; Ng, L.; Kindblom, J.M.; Barros, A.C.; Ohlsson, C.; Forrest, D. Mice devoid of all known TH receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev. 1999, 13, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M.; Feng, X.; Flamant, F.; Chen, Y.; Walker, R.L.; Weiss, R.E.; Chassande, O.; Samarut, J.; Refetoff, S.; Meltzer, P.S. Status and TH Receptor (TR) isoforms on hepatic gene expression profiles in TR knockout mice. EMBO Rep. 2003, 4, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Samarut, J.; Plateroti, M. Thyroid Hormone Receptors: Several Players for One Hormone and Multiple Functions; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1801, pp. 1–8. [Google Scholar] [CrossRef]
- Tagami, T.; Madison, L.D.; Nagaya, T.; Jameson, J.L. Nuclear receptor corepressors activate rather than suppress basal transcription of genes that are negatively regulated by thyroid hormone. Mol. Cell. Biol. 1997, 17, 2642–2648. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, P.; Abraham, B.J.; Tsai, L.; Zhou, Y.; Costa-e-Sousa, R.H.; Ye, F.; Bilban, M.; Zhao, K.; Hollenberg, A.N. Novel mechanism of positive versus negative regulation by thyroid hormone receptor β1 (TRβ1) identified by genome-wide profiling of binding sites in mouse liver. J. Biol. Chem. 2014, 289, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Hercbergs, A. Clinical Implications and Impact of Discovery of the Thyroid Hormone Receptor on Integrin αvβ3-A Review. Front. Endocrinol. 2019, 10, 565. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Davis, F.B.; Mousa, S.A.; Luidens, M.K.; Lin, H.Y. Membrane receptor for thyroid hormone: Physiologic and pharmacologic implications. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Grossniklaus, H.E.; Geisert, E.E.; Nickerson, J.M. Introduction to the Retina. Prog. Mol. Biol. Transl. Sci. 2005, 134, 383–396. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, J.; Li, D. Functions and Diseases of the Retinal Pigment Epithelium. Front. Pharmacol. 2021, 12, 727870. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Tachibanaki, S. Rod and cone photoreceptors: Molecular basis of the difference in their physiology. Compar. Biochem. Physiol. Molec. Integr. Physiol. 2008, 150, 369–377. [Google Scholar] [CrossRef]
- Nathans, J.; Thomas, D.; Hogness, D.S. Molecular genetics of human color vision: The genes encoding blue, green, and red pigments. Science 1986, 232, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S. Molecular evolution of vertebrate visual pigments. Prog. Retin. Eye Res. 2000, 19, 385–419. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S. The molecular basis of variation in human color vision. Clin. Genet. 2005, 67, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Nathans, J.; Davenport, C.M.; Maumenee, I.H.; Lewis, R.A.; Hejtmancik, J.F.; Litt, M.; Lovrien, E.; Weleber, R.; Bachynski, B.; Zwas, F.; et al. Molecular genetics of human blue cone monochromacy. Science 1989, 245, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Nicolás, F.M.; Kunze, V.P.; Ball, J.M.; Peng, B.T.; Krishnan, A.; Zhou, G.; Dong, L.; Li, W. True S-cones are concentrated in the ventral mouse retina and wired for color detection in the upper visual field. Elife 2020, 9, e56840. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.R.; Srinivas, M.; Forrest, D.; Morreale de Escobar, G.; Reh, T.A. Making the gradient: Thyroid hormone regulates cone opsin expression in the developing mouse retina. Proc. Natl. Acad. Sci. USA 2006, 103, 6218–6223. [Google Scholar] [CrossRef]
- Eldred, K.C.; Hadyniak, S.E.; Hussey, K.A.; Brenerman, B.; Zhang, P.W.; Chamling, X.; Sluch, V.M.; Welsbie, D.S.; Hattar, S.; Taylor, J.; et al. Thyroid hormone signaling specifies cone subtypes in human retinal organoids. Science 2018, 362, eaau6348. [Google Scholar] [CrossRef]
- Glaschke, A.; Glosmann, M.; and Peichl, L. Developmental changes of cone opsin expression but not retinal morphology in the hypothyroid Pax8 knockout mouse. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1719–1727. [Google Scholar] [CrossRef]
- Glaschke, A.; Weiland, J.; Del Turco, D.; Steiner, M.; Peichl, L.; Glosmann, M. Thyroid hormone controls cone opsin expression in the retina of adult rodents. J. Neurosci. 2011, 31, 4844–4851. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.E.; Pinz, I.; Preda, M.; Norton, C.R.; Gridley, T.; Hernandez, A. DIO3 protects against thyrotoxicosis-derived cranio-encephalic and cardiac congenital abnormalities. JCI Insight 2022, 7, e161214. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Lyubarsky, A.; Nikonov, S.S.; Ma, M.; Srinivas, M.; Kefas, B.; Germain, D.L.S.; Hernandez, A.; Pugh, E.N., Jr.; Forrest, D. Type 3 deiodinase, a thyroid-hormone-inactivating enzyme, controls survival and maturation of cone photoreceptors. J. Neurosci. 2010, 30, 3347–3357. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Martinez, M.E.; Ng, L.; Forrest, D. Thyroid Hormone Deiodinases: Dynamic Switches in Developmental Transitions. Endocrinology 2021, 162, bqab091. [Google Scholar] [CrossRef] [PubMed]
- Sawant, O.B.; Horton, A.M.; Zucaro, O.F.; Chan, R.; Bonilha, V.L.; Samuels, I.S.; Rao, S. The Circadian Clock Gene Bmal1 Controls Thyroid Hormone-Mediated Spectral Identity and Cone Photoreceptor Function. Cell. Rep. 2017, 21, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lu, A.; Kelley, K.A.; Forrest, D. Noncoding Mutations in a Thyroid Hormone Receptor Gene That Impair Cone Photoreceptor Function. Endocrinology 2023, 164, bqad006. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, M.; Vennström, B.; Forrest, D. Thyroid hormone receptors in chick retinal development: Differential expression of mRNAs for alpha and N-terminal variant beta receptors. Development 1992, 114, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Hurley, J.B.; Dierks, B.; Srinivas, M.; Salto, C.; Vennstrom, B.; Reh, T.A.; Forrest, D. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat. Genet. 2001, 27, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Campi, I.; Cammarata, G.; Bianchi Marzoli, S.; Beck-Peccoz, P.; Santarsiero, D.; Dazzi, D.; Bottari de Castello, A.; Taroni, E.G.; Viola, F.; Mian, C.; et al. Retinal Photoreceptor Functions Are Compromised in Patients with Resistance to Thyroid Hormone Syndrome (RTHβ). J. Clin. Endocrinol. Metab. 2017, 102, 2620–2627. [Google Scholar] [CrossRef]
- Weiss, A.H.; Kelly, J.P.; Bisset, D.; Deeb, S.S. Reduced L- and M- and increased S-cone functions in an infant with thyroid hormone resistance due to mutations in the THRβ2 gene. Ophthalmic. Genet. 2012, 3, 187–195. [Google Scholar] [CrossRef]
- Applebury, M.L.; Farhangfar, F.; Glosmann, M.; Hashimoto, K.; Kage, K.; Robbins, J.T.; Shibusawa, N.; Wondisford, F.E.; Zhang, H. Transient expression of thyroid hormone nuclear receptor TRbeta2 sets S opsin patterning during cone photoreceptor genesis. Develop. Dynam. 2007, 236, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Hernandez, A.; He, W.; Ren, T.; Srinivas, M.; Ma, M.; St Germain, D.L.; Forrest, D. A protective role for type 3 deiodinase, a thyroid hormone-inactivating enzyme, in cochlear development and auditory function. Endocrinology 2009, 150, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Lu, A.; Swaroop, A.; Sharlin, D.S.; Swaroop, A.; Forrest, D. Two transcription factors can direct three photoreceptor outcomes from rod precursor cells in mouse retinal development. J. Neurosci. 2011, 31, 11118–11125. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, C.N.; Santiago, L.A.; Santiago, D.A.; Machado, D.S.; Rocha, F.A.; Ventura, D.F.; Hokoç, J.N.; Pazos-Moura, C.C.; Wondisford, F.E.; Gardino, P.F.; et al. Thyroid hormone action is required for normal cone opsin expression during mouse retinal development. Investig. Ophthalmol. Visual Sci. 2008, 49, 2039–2045. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, M.; Wu, X.; Liu, H.; Liu, Y.; Cho, Y.W.; Song, M.; Fu, Y.; Ng, L.; Forrest, D. Transcriptional control of cone photoreceptor diversity by a thyroid hormone receptor. Proc. Natl. Acad. Sci. USA 2022, 119, e2209884119. [Google Scholar] [CrossRef]
- Sawant, O.; Horton, A.M.; Shukla, M.; Rayborn, M.E.; Peachey, N.S.; Hollyfield, J.G.; Rao, S. Light-Regulated Thyroid Hormone Signaling Is Required for Rod Photoreceptor Development in the Mouse Retina. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8248–8257. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Liu, H.; St Germain, D.L.; Hernandez, A.; Forrest, D. Deletion of the Thyroid Hormone-Activating Type 2 Deiodinase Rescues Cone Photoreceptor Degeneration but Not Deafness in Mice Lacking Type 3 Deiodinase. Endocrinology 2017, 158, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Yang, F.; York, L.R.; Li, S.; Ding, X.Q. Excessive Thyroid Hormone Signaling Induces Photoreceptor Degeneration in Mice. Eneuro 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Liu, H.; Liu, Y.; Forrest, D. Biphasic expression of thyroid hormone receptor TRβ1 in mammalian retina and anterior ocular tissues. Front. Endocrinol. 2023, 14, 1174600. [Google Scholar] [CrossRef]
- Kocaturk, T.; Ergin, K.; Cesur, G.; Evlicoglu, G.E.; Cakmak, H. The effect of methimazole-induced postnatal hypothyroidism on the retinal maturation and on the Sirtuin 2 level. Cutan. Ocul. Toxicol. 2016, 35, 36–40. [Google Scholar] [CrossRef]
- Ittermann, T.; Dörr, M.; Völzke, H.; Tost, F.; Lehmphul, I.; Köhrle, J.; Jürgens, C. High serum thyrotropin levels are associated with retinal arteriolar narrowing in the general population. Thyroid 2014, 24, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Fani, L.; Roa Dueñas, O.; Bos, D.; Vernooij, M.W.; Klaver, C.C.W.; Ikram, M.K.; Peeters, R.P.; Ikram, M.A.; Chaker, L. Thyroid Status and Brain Circulation: The Rotterdam Study. J. Clin. Endocrinol. Metab. 2022, 107, e1293–e1302. [Google Scholar] [CrossRef] [PubMed]
- Mookadam, M.; Leske, D.A.; Fautsch, M.P.; Lanier, W.L.; Holmes, J.M. The anti-thyroid drug methimazole induces neovascularization in the neonatal rat analogous to ROP. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4145–4150. [Google Scholar] [CrossRef] [PubMed]
- Mutapcic, L.; Wren, S.M.; Leske, D.A.; Fautsch, M.P.; Holmes, J.M. The effect of L-thyroxine supplementation on retinal vascular development in neonatal rats. Curr. Eye. Res. 2005, 30, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Wren, S.M.; Leske, D.A.; Mutapcic, L.; Fautsch, M.P.; Holmes, J.M. The effect of L-thyroxine supplementation in a neonatal rat model of ROP. Curr. Eye Res. 2006, 31, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, B.A.; Luan, H.; Roberts, R.L. Effect of methylimidazole induced hypothyroidism in a model of low retinal neovascular incidence. Investig. Ophthalmol. Vis. Sci. 2004, 45, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz-Rutkowska, M.M.; Baranowska-Jurkun, A.; Matuszewski, W.; Bandurska-Stankiewicz, E.M. Thyroid dysfunction in patients with diabetic retinopathy. Endokrynol. Pol. 2020, 71, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Kropp, M.; Golubnitschaja, O.; Mazurakova, A.; Koklesova, L.; Sargheini, N.; Vo, T.K.S.; de Clerck, E.; Polivka, J., Jr.; Potuznik, P.; Polivka, J.; et al. Diabetic retinopathy as the leading cause of blindness and early predictor of cascading complications-risks and mitigation. EPMA J. 2023, 14, 21–42. [Google Scholar] [CrossRef]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes. Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Stitt, A.W.; Lois, N.; Medina, R.J.; Adamson, P.; Curtis, T.M. Advances in our understanding of diabetic retinopathy. Clin. Sci. 2013, 125, 1–17. [Google Scholar] [CrossRef]
- Luo, W.; Hu, L.; Li, W.; Xu, G.; Xu, L.; Zhang, C.; Wang, F. Epo inhibits the fibrosis and migration of Müller glial cells induced by TGF-beta and high glucose. Graefes. Arch. Clin. Exp. Ophthalmol. 2016, 254, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Kamboj, A.; Lause, M.; Kumar, P. Ophthalmic manifestations of endocrine disorders-endocrinology and the eye. Transl. Pediatr. 2017, 6, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.M.; Gardiner, T.A.; Stitt, A.W. Microvascular lesions of diabetic retinopathy: Clues towards understanding pathogenesis? Eye 2009, 23, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes. Res. 2007, 2007, 43603. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Che, D.; Lan, Y.; Fang, Z.; Xie, J.; Gong, H.; Li, C.; Feng, J.; Hong, H.; Qi, W.; et al. Mesenchymal marker expression is elevated in Müller cells exposed to high glucose and in animal models of diabetic retinopathy. Oncotarget 2017, 8, 4582–4594. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Nawaz, M.I.; Siddiquei, M.M.; Al-Amro, S.; Abu El-Asrar, A.M. Recent advances in understanding the biochemical and molecular mechanism of diabetic retinopathy. J. Diab. Complic. 2012, 26, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, J.; Chen, Y.; Wang, J.J.; Ratan, R.; Zhang, S.X. Activation of endoplasmic reticulum stress by hyperglycemia is essential for Müller cell-derived inflammatory cytokine production in diabetes. Diabetes 2012, 61, 492–504. [Google Scholar] [CrossRef]
- Guidry, C.; King, J.L.; Mason, J.O. Fibrocontractive Müller cell phenotypes in proliferative diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1929–1939. [Google Scholar] [CrossRef]
- Sramek, S.J.; Wallow, I.H.; Stevens, T.S.; Nork, T.M. Immunostaining of preretinal membranes for actin, fibronectin, and glial fibrillary acidic protein. Ophthalmology 1989, 96, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Müller cell changes in human diabetic retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Guidry, C. The role of Müller cells in fibrocontractive retinal disorders. Prog. Retin. Eye Res. 2005, 24, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.; Catalani, E.; Dal Monte, M.; Cammalleri, M.; Cervia, D.; Casini, G. Morpho-functional analysis of the early changes induced in retinal ganglion cells by the onset of diabetic retinopathy: The effects of a neuroprotective strategy. Pharmacol. Res. 2022, 185, 106516. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.H.; van Dijk, H.W.; Jiao, C.; Kok, P.H.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; van Velthoven, M.E.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.; O’Leary, O.E.; Stitt, A.W. The pathology associated with diabetic retinopathy. Vision. Res. 2017, 139, 7–14. [Google Scholar] [CrossRef]
- Lange, C.; Storkebaum, E.; de Almodóvar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.; Biagioni, M.; Cammalleri, M.; Dal Monte, M.; Casini, G. VEGF as a Survival Factor in Ex Vivo Models of Early Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3066–3076. [Google Scholar] [CrossRef] [PubMed]
- Rossino, M.G.; Lulli, M.; Amato, R.; Cammalleri, M.; Monte, M.D.; Casini, G. Cells. Oxidative Stress Induces a VEGF Autocrine Loop in the Retina: Relevance for Diabetic. Retinopathy 2020, 9, 1452. [Google Scholar] [CrossRef]
- Rossino, M.G.; Dal Monte, M.; Casini, G. Relationships Between Neurodegeneration and Vascular Damage in Diabetic Retinopathy. Front. Neurosci. 2019, 13, 1172. [Google Scholar] [CrossRef]
- Reddy, N.; Pradeep, T.V.S.; Tirupati, S.; Sarathi, V.; Kumar, D. Thyroid dysfunction and its association with microvascular complications in patients with type 2 diabetes mellitus in south India. Diabetes Metab. Syndr. 2020, 14, 615–617. [Google Scholar] [CrossRef]
- Biondi, B.; Kahaly, G.J.; Robertson, R.P. Thyroid dysfunction and diabetes mellitus: Two closely associated disorders. Endocr. Rev. 2019, 40, 789–824. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, M.; Dong, S.; Zhang, S.; Dong, A.; Zhang, M. Assessment of the association between genetic factors regulating thyroid function and microvascular complications in diabetes: A two-sample Mendelian randomization study in the European population. Front. Endocrinol. 2023, 14, 1126339. [Google Scholar] [CrossRef]
- Fang, T.; Deng, X.; Wang, J.; Han, F.; Liu, X.; Liu, Y.; Sun, B.; Chen, L. The effect of hypothyroidism on the risk of diabetes and its microvascular complications: A Mendelian randomization study. Front. Endocrinol. 2023, 14, 1288284. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.K.; Liu, W.; Shi, J.; Li, Y.-B. An association between subclinical hypothyroidism and sight-threatening diabetic retinopathy in type 2 diabetic patients. Diabetes Care 2010, 33, 1018–1020. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Jeong, H.S.; Kim, K.J.; Han, M.H.; Lee, E.H.; Lee, K.; Cho, C.H. β-Adrenergic receptor agonists attenuate pericyte loss in diabetic retinas through Akt activation. FASEB J. 2018, 32, 2324–2338. [Google Scholar] [CrossRef]
- Han, C.; He, X.; Xia, X.; Li, Y.; Shi, X.; Shan, Z.; Teng, W. Subclinical hypothyroidism and type 2 diabetes: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0135233. [Google Scholar] [CrossRef]
- El-Sehrawy, A.A.; Elkhamisy, E.M.; Badawi, A.E.; Elshahawy, H.A.; Elsayed, E.; Mohammed, N.T.; El-Eshmawy, M.M. Subclinical Hypothyroidism in Patients with Diabetic Retinopathy: Role of Vascular Endothelial Growth Factor. Endocr. Metab. Immune. Disord. Drug. Targets. 2022, 22, 502–509. [Google Scholar] [CrossRef]
- Sailesh, S. The THOR Effect: Thyroid Hormone Offsets Retinopathy. J. Endocrinol. Thyroid. Res. 2018, 3, 555605. [Google Scholar] [CrossRef]
- Lin, D.; Qin, R.; Guo, L. Thyroid stimulating hormone aggravates diabetic retinopathy through the mitochondrial apoptotic pathway. J. Cell. Physiol. 2022, 237, 868–880. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, J.; Xu, X.; Xin, S.; Zhang, X. The effect of Central and peripheral thyroid resistance indices on diabetic retinopathy: A study of hospitalized euthyroid patients with T2DM in China. Ann. Med. 2023, 55, 2249017. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Lionetti, V.; Ardehali, H.; Pucci, A.; Cecchetti, F.; Ghanefar, M.; Nicolini, G.; Ichikawa, Y.; Nannipieri, M.; Recchia, F.A.; et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J. Cell. Mol. Med. 2011, 15, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, G.; Forini, F.; Kusmic, C.; Pitto, L.; Mariani, L.; Iervasi, G. Early and Short-term Triiodothyronine Supplementation Prevents Adverse Postischemic Cardiac Remodeling: Role of Transforming Growth Factor-β1 and Antifibrotic miRNA Signaling. Mol. Med. 2016, 21, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Kusmic, C.; Nicolini, G.; Mariani, L.; Zucchi, R.; Matteucci, M.; Iervasi, G.; Pitto, L. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology 2014, 155, 4581–4590. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Nicolini, G.; Kusmic, C.; D’Aurizio, R.; Rizzo, M.; Baumgart, M.; Groth, M.; Doccini, S.; Iervasi, G.; Pitto, L. Integrative analysis of differentially expressed genes and miRNAs predicts complex T3-mediated protective circuits in a rat model of cardiac ischemia reperfusion. Sci. Rep. 2018, 8, 3870. [Google Scholar] [CrossRef] [PubMed]
- Rodacki, M.; Zajdenverg, L.; Dantas, J.R.; Palma, C.C.; Negrato, C.A.; Gomes, M.B. Should thyroid-stimulating hormone goals be reviewed in patients with type 1 diabetes mellitus? Results from the Brazilian Type 1 Diabetes Study Group. Diabet. Med. 2014, 31, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, B.; Rogowicz-Frontczak, A.; Grzelka, A.; Uruska, A.; Schlaffke, J.; Araszkiewicz, A.; Zozulinska-Ziolkiewicz, D. Higher free triiodothyronine concentration is associated with lower prevalence of microangiopathic complications and better metabolic control in adult euthyroid people with type 1 diabetes. Endocrine 2018, 60, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Barker, J.M.; Yu, J.; Yu, L.; Wang, J.; Miao, D.; Bao, F.; Hoffenberg, E.; Nelson, J.C.; Gottlieb, P.A.; Rewers, M.; et al. Autoantibody “subspecificity” in type 1 diabetes: Risk for organ-specific autoimmunity clusters in distinct groups. Diabetes Care 2005, 28, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Kordonouri, O.; Klinghammer, A.; Lang, E.B.; Gruters-Kieslich, A.; Grabert, M.; Holl, R.W. Thyroid autoimmunity in children and adolescents with type 1 diabetes: A multicenter survey. Diabetes Care 2002, 25, 1346–1350. [Google Scholar] [CrossRef]
- Wysocka-Mincewicz, M.; Baszyńska-Wilk, M.; Gołębiewska, J.; Olechowski, A.; Byczyńska, A.; Hautz, W.; Szalecki, M. The effect of coexisting autoimmune thyroiditis in children with Type 1 diabetes on optical coherence tomography results. Pediatr. Diab. 2021, 22, 329–334. [Google Scholar] [CrossRef]
- Rogowicz-Frontczak, A.; Pilacinski, S.; Chwialkowska, A.T.; Wierusz-Wysocka, B.; Zozulińska-Ziółkiewicz, D. Patients with diabetes type 1 and thyroid autoimmunity have low prevalence of microangiopathic complications. Endocrine 2016, 51, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Waber, S.; Meister, V.; Rossi, G.L.; Mordasini, R.C.; Riesen, W.F. Studies on retinal microangiopathy and coronary macroangiopathy in rats with streptozotocin-induced diabetes. Virchows. Arch. Cell. Pathol. Incl. Mol. Pathol. 1981, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Énzsöly, A.; Hajdú, R.I.; Turóczi, Z.; Szalai, I.; Tátrai, E.; Pálya, F.; Nagy, Z.Z.; Mátyás, C.; Oláh, A.; Radovits, T.; et al. The Predictive Role of Thyroid Hormone Levels for Early Diabetic Retinal Changes in Experimental Rat and Human Diabetes. Investig. Ophthalmol. Vis. Sci. 2021, 62, 20. [Google Scholar] [CrossRef] [PubMed]
- Bapputty, R.; Sapa, H.; Masaru, M.; Gubitosi-Klug, R.A. Diabetes Modulates Iodothyronine Deiodinase 2 Expression in the Mouse Retina: A Role for Thyroid Hormone in the Pathogenesis of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2023, 64, 3. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Bodó, E.; Kromminga, A.; Bíró, T.; Borbíró, I.; Gáspár, E.; Zmijewski, M.A. Human female hair follicles are a direct, nonclassical target for thyroid-stimulating hormone. J. Investig. Dermatol. 2009, 129, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tian, L.M.; Han, Y.; Ma, H.Y.; Wang, L.C.; Guo, J.; Gao, L.; Zhao, J.J. Presence of thyrotropin receptor in hepatocytes: Not a case of illegitimate transcription. J. Cell. Mol. Med. 2009, 13, 4636–4642. [Google Scholar] [CrossRef]
- Chen, J.; Ren, J.; Jing, Q.; Lu, S.; Zhang, Y.; Liu, Y.; Yu, C.; Gao, P.; Zong, C.; Li, X.; et al. TSH/TSHR Signaling Suppresses Fatty Acid Synthase (FASN) Expression in Adipocytes. J. Cell Physiol. 2015, 230, 2233–2239. [Google Scholar] [CrossRef]
- Balzan, S.; Nicolini, G.; Forini, F.; Boni, G.; Del Carratore, R.; Nicolini, A.; Carpi, A.; Iervasi, G. Presence of a functional TSH receptor on human erythrocytes. Biomed. Pharmacother. 2007, 61, 463–467. [Google Scholar] [CrossRef]
- Balzan, S.; Del Carratore, R.; Nicolini, G.; Beffy, P.; Lubrano, V.; Forini, F.; Iervasi, G. Proangiogenic effect of TSH in human microvascular endothelial cells through its membrane receptor. J. Clin. Endocrinol. Metab. 2012, 97, 1763–1770. [Google Scholar] [CrossRef]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, M.; Schmitz-Valckenberg, S.; Chakravarthy, U. Age-Related Macular Degeneration: A Review. JAMA 2024, 331, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2021, 9, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.S.; Ziemssen, F.; Bartz-Schmidt, K.U.; Gelisken, F.; Szurman, P. Treatment of age-related macular degeneration: Focus on ranibizumab. Clin. Ophthalmol. 2008, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Little, K.; Ma, J.H.; Yang, N.; Chen, M.; Xu, H. Myofibroblasts in macular fibrosis secondary to neovascular age-related macular degeneration—The potential sources and molecular cues for their recruitment and activation. EBio Med. 2018, 38, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Yamanaka, O.; Okada, Y.; Tanaka, S.; Miyamoto, T.; Sumioka, T.; Kitano, A.; Shirai, K.; Ikeda, K. TGF beta in fibroproliferative diseases in the eye. Front. Biosci. (Schol. Ed.) 2009, 1, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Zachary, I.; Jia, H. Mechanisms of Acquired Resistance to Anti-VEGF Therapy for Neovascular Eye Diseases. Investig. Ophthalmol. Vis. Sci. 2023, 64, 28. [Google Scholar] [CrossRef] [PubMed]
- Tenbrock, L.; Wolf, J.; Boneva, S.; Schlecht, A.; Agostini, H.; Wieghofer, P.; Schlunck, G.; Lange, C. Subretinal fibrosis in neovascular age-related macular degeneration: Current concepts, therapeutic avenues, and future perspectives. Cell Tissue Res. 2022, 387, 361–375. [Google Scholar] [CrossRef]
- Bromfield, S.; Keenan, J.; Jolly, P.; McGwin, G. A suggested association between hypothyroidism and age-related macular degeneration. Curr. Eye Res. 2012, 37, 549–552. [Google Scholar] [CrossRef]
- Gopinath, B.; Liew, G.; Kifley, A.; Mitchell, P. Thyroid Dysfunction and Ten-Year Incidence of Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5273–5277. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.H.; Xirasagar, S.; Kuang, T.T.; Chang, W.W.; Cheng, Y.F.; Kuo, N.W.; Lin, H.C. Association of Age-Related Macular Degeneration with Prior Hyperthyroidism and Hypothyroidism: A Case-Control Study. J. Pers. Med. 2022, 12, 602. [Google Scholar] [CrossRef] [PubMed]
- Farvardin, M.; Mousavi, S.E.; Zare, K.; Bazdar, S.; Farvardin, Z.; Johari, M. Thyroid Dysfunction as a Modifiable Risk Factor for Wet Type Age-Related Macular Degeneration: A Case-Control Study. J. Curr. Ophthalmol. 2022, 33, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, H.; Cheng, J.; Wang, M.; Zhong, Y.; Shi, G.; Yu, A.Y. Causal Associations of Thyroid Function and Age-Related Macular Degeneration: A Two-Sample Mendelian Randomization Study. Am. J. Ophthalmol. 2022, 239, 108–114. [Google Scholar] [CrossRef] [PubMed]
- ARED Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-Related Eye Disease Study Report Number 3. Ophthalmology 2000, 107, 2224–2232. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, M.; Zhang, Q.; Xu, T.; Tao, L. Thyroid Disease Is Associated with Higher Age-Related Macular Degeneration Risk: Results from a Meta-Analysis of Epidemiologic Studies. Ophthalmic Res. 2021, 64, 696–703. [Google Scholar] [CrossRef]
- Balzan, S.; Del Carratore, R.; Nardulli, C.; Sabatino, L.; Lubrano, V.; Iervasi, G. The stimulative effect of T3 and T4 on human myocardial endothelial cell proliferation, migration and angiogenesis. J. Clin. Exp. Cardiol. 2013, 4, 1–7. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, N.; Shi, Y.N.; Yuan, J.; Li, L. Thyroid hormone induced angiogenesis through the integrin αvβ3/protein kinase D/histone deacetylase 5 signaling pathway. J. Mol. Endocrinol. 2014, 52, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Mrugacz, M.; Bryl, A.; Falkowski, M.; Zorena, K. Integrins: An Important Link between Angiogenesis, Inflammation and Eye Diseases. Cells 2021, 10, 1703. [Google Scholar] [CrossRef]
- Ma, H.; Yang, F.; Ding, X.Q. Inhibition of thyroid hormone signaling protects retinal pigment epithelium and photoreceptors from cell death in a mouse model of age-related macular degeneration. Cell Death Dis. 2020, 11, 24. [Google Scholar] [CrossRef]
- Ma, H.; Yang, F.; Ding, X.Q. Deficiency of thyroid hormone receptor protects retinal pigment epithelium and photoreceptors from cell death in a mouse model of age-related macular degeneration. Cell Death Dis. 2022, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Thapa, A.; Morris, L.; Redmond, T.M.; Baehr, W.; Ding, X.Q. Suppressing thyroid hormone signaling preserves cone photoreceptors in mouse models of retinal degeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 3602–3607. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Ma, H.; Belcher, J.; Butler, M.R.; Redmond, T.M.; Boye, S.L.; Hauswirth, W.W.; Ding, X.Q. Targeting iodothyronine deiodinases locally in the retina is a therapeutic strategy for retinal degeneration. FASEB J. 2016, 30, 4313–4325. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Gong, J.; Xu, Z.; Wei, Y.; Duh, E.J. Inhibition of pathological retinal angiogenesis by the integrin αvβ3 antagonist tetraiodothyroacetic acid (tetrac). Exp. Eye Res. 2012, 94, 41–48. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolini, G.; Casini, G.; Posarelli, C.; Amato, R.; Lulli, M.; Balzan, S.; Forini, F. Thyroid Hormone Signaling in Retinal Development and Function: Implications for Diabetic Retinopathy and Age-Related Macular Degeneration. Int. J. Mol. Sci. 2024, 25, 7364. https://doi.org/10.3390/ijms25137364
Nicolini G, Casini G, Posarelli C, Amato R, Lulli M, Balzan S, Forini F. Thyroid Hormone Signaling in Retinal Development and Function: Implications for Diabetic Retinopathy and Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2024; 25(13):7364. https://doi.org/10.3390/ijms25137364
Chicago/Turabian StyleNicolini, Giuseppina, Giovanni Casini, Chiara Posarelli, Rosario Amato, Matteo Lulli, Silvana Balzan, and Francesca Forini. 2024. "Thyroid Hormone Signaling in Retinal Development and Function: Implications for Diabetic Retinopathy and Age-Related Macular Degeneration" International Journal of Molecular Sciences 25, no. 13: 7364. https://doi.org/10.3390/ijms25137364