
Enhanced Genomic and Transcriptomic Resources for Trichinella pseudospiralis and T. spiralis to Underpin the Discovery of Molecular Differences between Stages and Species
 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
2.1. Creating Enhanced Transcriptomic Resources for T. pseudospiralis and T. spiralis
2.1.1. Nucleic Acid Sequence Data Sets
2.1.2. Predicted Genes, Functional Annotation, and Comparison of Inferred Proteomes
2.1.3. Enriched Biological Pathways
2.1.4. Protein Groups Inferred to Be Involved in Parasite–Host Interplay
2.1.5. Cellular Localisations of Hypothetical Proteins
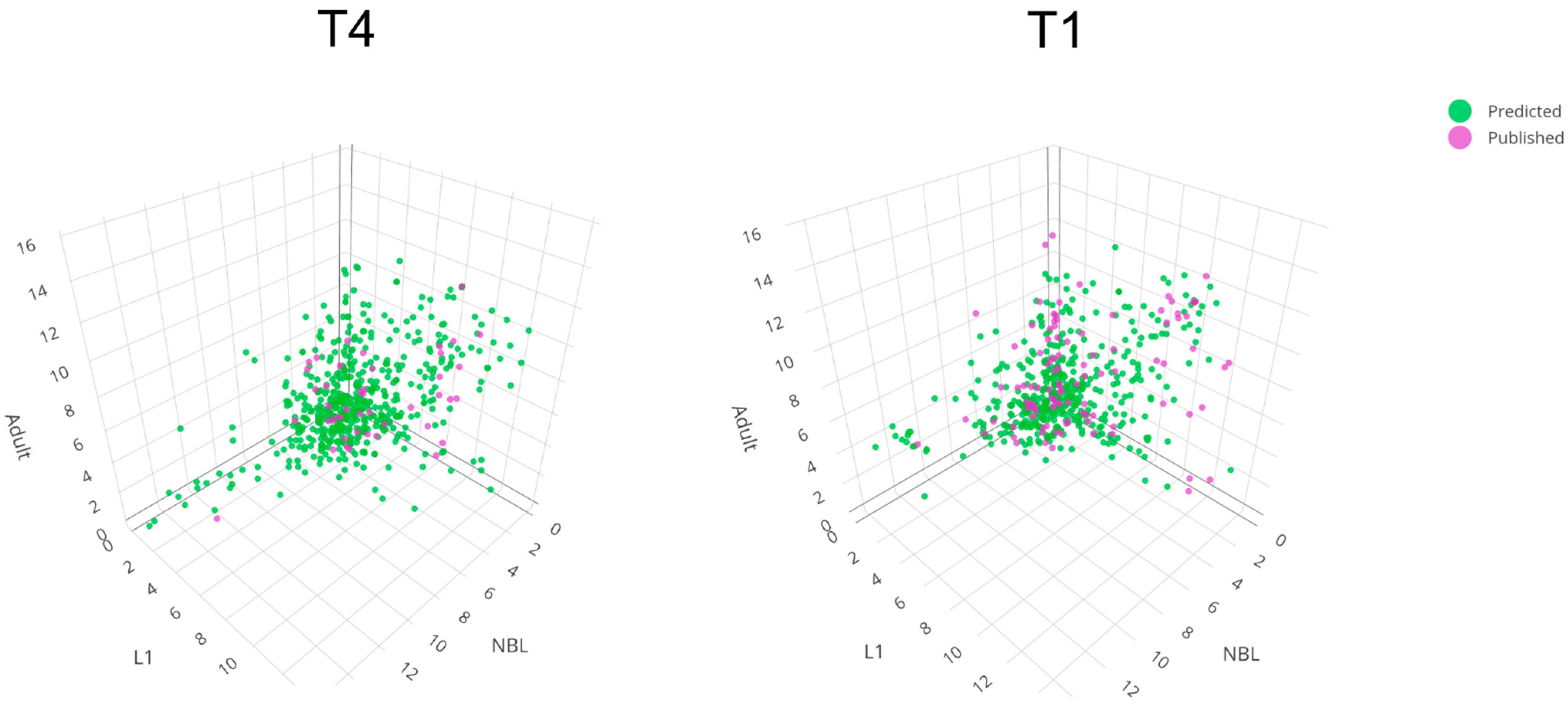
2.2. Linking Transcription within Developmental Stages to Biological Pathways/Processes to Understand Each of the Two Trichinella Species Better at the Molecular Level
3. Discussion
4. Materials and Methods
4.1. Production and Procurement of L1s, Adults, and Newborn Larvae (NBLs) of Trichinella
4.2. Nucleic Acid Isolation and Sequencing
4.3. Nuclear Genomes, Prediction of Repetitive Elements, Protein-Encoding Genes, and Functional Annotation
4.4. Differential Transcription Analysis
4.5. Pathway Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dupouy-Camet, J.; Raffetin, A.; Rosca, E.C.; Yera, H. Chapter 10—Clinical picture and diagnosis of human trichinellosis. In Trichinella and Trichinellosis; Bruschi, F., Ed.; Academic Press: Amsterdam, The Netherlands, 2021; pp. 333–352. [Google Scholar]
- Pozio, E.; Zarlenga, D.S. Chapter 3—Taxonomy of the Trichinella genus. In Trichinella and Trichinellosis; Bruschi, F., Ed.; Academic Press: Amsterdam, The Netherlands, 2021; pp. 185–263. [Google Scholar]
- Zarlenga, D.; Thompson, P.; Pozio, E. Trichinella species and genotypes. Res. Vet. Sci. 2020, 133, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Pozio, E. Scientific achievements of the last 60 years: From a single to a multispecies concept of the genus Trichinella. Vet. Parasitol. 2021, 297, 109042. [Google Scholar] [CrossRef] [PubMed]
- Pozio, E.; Zarlenga, D.S. New pieces of the Trichinella puzzle. Int. J. Parasitol. 2013, 43, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Bilska-Zając, E.; Thompson, P.; Rosenthal, B.; Różycki, M.; Cencek, T. Infection, genetics, and evolution of Trichinella: Historical insights and applications to molecular epidemiology. Infect. Genet. Evol. 2021, 95, 105080. [Google Scholar] [CrossRef] [PubMed]
- Pozio, E. Chapter 6—Epidemiology. In Trichinella and Trichinellosis; Bruschi, F., Ed.; Academic Press: Amsterdam, The Netherlands, 2021; pp. 35–76. [Google Scholar]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Naquin, D.; Gorrichon, K.; Jaszczyszyn, Y.; Ouazahrou, R.; Thermes, C.; Hernandez, C. Genomics in the long-read sequencing era. Trends Genet. 2023, 39, 649–671. [Google Scholar] [CrossRef]
- Luo, J.; Wei, Y.; Lyu, M.; Wu, Z.; Liu, X.; Luo, H.; Yan, C. A comprehensive review of scaffolding methods in genome assembly. Brief. Bioinform. 2021, 22, bbab033. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gasser, R.B. Prospects of using high-throughput proteomics to underpin the discovery of animal host–nematode interactions. Pathogens 2021, 10, 825. [Google Scholar] [CrossRef]
- Korhonen, P.K.; Young, N.D.; Gasser, R.B. Making sense of genomes of parasitic worms: Tackling bioinformatic challenges. Biotechnol. Adv. 2016, 34, 663–686. [Google Scholar] [CrossRef]
- Mitreva, M.; Jasmer, D.P.; Zarlenga, D.S.; Wang, Z.; Abubucker, S.; Martin, J.; Taylor, C.M.; Yin, Y.; Fulton, L.; Minx, P.; et al. The draft genome of the parasitic nematode Trichinella spiralis. Nat. Genet. 2011, 43, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.C.; Zarlenga, D.S.; Liu, M.-Y.; Rosenthal, B.M. Long-read sequencing improves assembly of Trichinella genomes 10-fold, revealing substantial synteny between lineages diverged over 7 million years. Parasitology 2017, 144, 1302–1315. [Google Scholar] [CrossRef]
- Hecht, L.B.; Thompson, P.C.; Rosenthal, B.M. Comparative demography elucidates the longevity of parasitic and symbiotic relationships. Proc. Roy. Soc. B 2018, 285, 20181032. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, P.K.; Pozio, E.; La Rosa, G.; Chang, B.C.; Koehler, A.V.; Hoberg, E.P.; Boag, P.R.; Tan, P.; Jex, A.R.; Hofmann, A.; et al. Phylogenomic and biogeographic reconstruction of the Trichinella complex. Nat. Commun. 2016, 7, 10513. [Google Scholar] [CrossRef]
- Gounaris, K. Nucleotidase cascades are catalyzed by secreted proteins of the parasitic nematode Trichinella spiralis. Infect. Immun. 2002, 70, 4917–4924. [Google Scholar] [CrossRef]
- Song, Y.Y.; Zhang, X.Z.; Wang, B.N.; Weng, M.M.; Zhang, Z.Y.; Guo, X.; Zhang, X.; Wang, Z.Q.; Cui, J. Molecular characterization of a novel serine proteinase from Trichinella spiralis and its participation in larval invasion of gut epithelium. PLoS Negl. Trop. Dis. 2023, 17, e0011629. [Google Scholar] [CrossRef]
- Qi, X.; Yue, X.; Han, Y.; Jiang, P.; Yang, F.; Lei, J.J.; Liu, R.D.; Zhang, X.; Wang, Z.Q.; Cui, J. Characterization of two Trichinella spiralis adult-specific DNase II and their capacity to induce protective immunity. Front. Microbiol. 2018, 9, 2504. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bai, X.; Tang, B.; Zhang, Y.; Zhang, L.; Cai, X.; Lin, J.; Jia, W.; Boireau, P.; Liu, M. Comparative analysis of excretory–secretory products of muscle larvae of three isolates of Trichinella pseudospiralis by the iTRAQ method. Vet. Parasitol. 2021, 297, 109119. [Google Scholar] [CrossRef]
- Robinson, M.W.; Gare, D.C.; Connolly, B. Profiling excretory/secretory proteins of Trichinella spiralis muscle larvae by two-dimensional gel electrophoresis and mass spectrometry. Vet. Parasitol. 2005, 132, 37–41. [Google Scholar] [CrossRef]
- Li, C.; Li, C.; Xu, F.; Wang, H.; Jin, X.; Zhang, Y.; Liu, X.; Wang, R.; You, X.; Liu, M. Identification of antigens in the Trichinella spiralis extracellular vesicles for serological detection of early stage infection in swine. Parasit. Vectors 2023, 16, 387. [Google Scholar] [CrossRef]
- Liu, R.D.; Qi, X.; Sun, G.G.; Jiang, P.; Zhang, X.; Wang, L.A.; Liu, X.L.; Wang, Z.Q.; Cui, J. Proteomic analysis of Trichinella spiralis adult worm excretory-secretory proteins recognized by early infection sera. Vet. Parasitol. 2016, 231, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Guiliano, D.B.; Oksov, Y.; Lustigman, S.; Gounaris, K.; Selkirk, M.E. Characterisation of novel protein families secreted by muscle stage larvae of Trichinella spiralis. Int. J. Parasitol. 2009, 39, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Bien, J.; Cabaj, W.; Moskwa, B. Proteomic analysis of potential immunoreactive proteins from muscle larvae and adult worms of Trichinella spiralis in experimentally infected pigs. Folia Parasitol. 2015, 1, 2022. [Google Scholar] [CrossRef] [PubMed]
- Grzelak, S.; Stachyra, A.; Stefaniak, J.; Mrówka, K.; Moskwa, B.; Bień-Kalinowska, J. Immunoproteomic analysis of Trichinella spiralis and Trichinella britovi excretory-secretory muscle larvae proteins recognized by sera from humans infected with Trichinella. PLoS ONE 2020, 15, e0241918. [Google Scholar] [CrossRef] [PubMed]
- Ilic, N.; Bojic-Trbojevic, Z.; Lundström-Stadelmann, B.; Cujic, D.; Mitic, I.; Gruden-Movsesijan, A. Immunomodulatory components of Trichinella spiralis excretory-secretory products with lactose-binding specificity. EXCLI J. 2022, 21, 793. [Google Scholar]
- Ding, J.; Liu, X.; Bai, X.; Wang, Y.; Li, J.; Wang, C.; Li, S.; Liu, M.; Wang, X. Trichinella spiralis: Inflammation modulator. J. Helminthol. 2020, 94, e193. [Google Scholar] [CrossRef] [PubMed]
- Kobpornchai, P.; Flynn, R.J.; Reamtong, O.; Rittisoonthorn, N.; Kosoltanapiwat, N.; Boonnak, K.; Boonyuen, U.; Ampawong, S.; Jiratanh, M.; Tattiyapong, M. A novel cystatin derived from Trichinella spiralis suppresses macrophage-mediated inflammatory responses. PLoS Negl. Trop. Dis. 2020, 14, e0008192. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, F.; Yang, D.Q.; Jiang, P.; Liu, R.D.; Zhang, X.; Cui, J.; Wang, Z.Q. Molecular characterization of Trichinella spiralis galectin and its participation in larval invasion of host’s intestinal epithelial cells. Vet. Res. 2018, 49, 79. [Google Scholar] [CrossRef] [PubMed]
- Murrell, K.D.; Pozio, E. Trichinellosis: The zoonosis that won’t go quietly. Int. J. Parasitol. 2000, 30, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, L.; Brůna, T.; Hoff, K.J.; Ebel, M.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER3: Fully automated genome annotation using RNA-Seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. Genome Res. 2024, 34, 769–777. [Google Scholar] [CrossRef]
- O’Donnell, S.E.; Newman, R.A.; Witt, T.J.; Hultman, R.; Froehlig, J.R.; Christensen, A.P.; Shea, M.A. Thermodynamics and conformational change governing domain–domain interactions of calmodulin. In Methods Enzymology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 466, pp. 503–526. [Google Scholar]
- Hu, J.; Shi, D.; Ding, M.; Huang, T.; Gu, R.; Xiao, J.; Xian, C.J.; Dong, J.; Wang, L.; Liao, H. Calmodulin-dependent signalling pathways are activated and mediate the acute inflammatory response of injured skeletal muscle. Physiol. J. 2019, 597, 5161–5177. [Google Scholar] [CrossRef]
- Despommier, D. How does Trichinella spiralis make itself at home? Parasitol. Today 1998, 14, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Mayer, A. Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature 1998, 396, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Wanford, J.J.; Odendall, C. Ca2+-calmodulin signalling at the host-pathogen interface. Curr. Opin. Microbiol. 2023, 72, 102267. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.S.; Tomer, K.B. Analysis of the oxidative damage-induced conformational changes of apo-and holocalmodulin by dose-dependent protein oxidative surface mapping. Biophys. J. 2007, 92, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Kawasaki, H.; Suzuki, K.; Yahara, I. The calmodulin-binding domain of the mouse 90-kDa heat shock protein. J. Biol. Chem. 1993, 268, 9604–9610. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Leung-Hagesteijn, C.Y.; Milankov, K.; Michalak, M.; Wilkins, J.; Dedhar, S. Cell attachment to extracellular matrix substrates is inhibited upon downregulation of expression of calreticulin, an intracellular integrin α-subunit-binding protein. J. Cell Sci. 1994, 107, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, D.D.; Duan, Y.; Matsuki, M.; Kunitomo, H.; Hutter, H.; Hedgecock, E.M.; Iino, Y. CASY-1, an ortholog of calsyntenins/alcadeins, is essential for learning in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2008, 105, 5260–5265. [Google Scholar] [CrossRef] [PubMed]
- Florin, A.; Lambert, C.; Sanchez, C.; Zappia, J.; Durieux, N.; Tieppo, A.M.; Mobasheri, A.; Henrotin, Y. The secretome of skeletal muscle cells: A systematic review. Osteoarthr. Cartil. Open 2020, 2, 100019. [Google Scholar] [CrossRef]
- Hintsch, G.; Zurlinden, A.; Meskenaite, V.; Steuble, M.; Fink-Widmer, K.; Kinter, J.; Sonderegger, P. The calsyntenins—A family of postsynaptic membrane proteins with distinct neuronal expression patterns. Mol. Cell. Neurosci. 2002, 21, 393–409. [Google Scholar] [CrossRef]
- Roatta, S.; Farina, D. Sympathetic actions on the skeletal muscle. Exerc. Sport Sci. Rev. 2010, 38, 31–35. [Google Scholar] [CrossRef]
- Ellis, L.A.; McVay, C.S.; Probert, M.A.; Zhang, J.; Bundle, D.R.; Appleton, J.A. Terminal β-linked tyvelose creates unique epitopes in Trichinella spiralis glycan antigens. Glycobiology 1997, 7, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Perteguer, M.; Rodrıguez, E.; Romarıs, F.; Escalante, M.; Bonay, P.; Ubeira, F.; Gárate, M. Minor interspecies variations in the sequence of the gp53 TSL-1 antigen of Trichinella define species-specific immunodominant epitopes. Mol. Immunol. 2004, 41, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Tonanzi, D.; Interisano, M.; Vatta, P.; Galati, F.; La Rosa, G. The International Trichinella Reference Centre database. Report on thirty-three years of activity and future perspectives. Food Waterborne Parasitol. 2022, 27, e00156. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, P.K.; Hall, R.S.; Young, N.D.; Gasser, R.B. Common Workflow Language (CWL)-based software pipeline for de novo genome assembly from long-and short-read data. Gigascience 2019, 8, giz014. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Robert, H.; Kas, A.; Siegel, A.; Gish, W.; Price, A.; Pevzner, P. RepeatModeler, 1.0.5; Institute of Systems Biology: Seattle, WA, USA, 2011; Available online: http://www.repeatmasker.org (accessed on 24 June 2022).
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker; Institute of Systems Biology: Seattle, WA, USA; Available online: http://www.repeatmasker.org (accessed on 24 June 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Sherry, S.T.; Yankie, L.; Karsch-Mizrachi, I. GenBank 2023 update. Nucleic Acids Res. 2023, 51, D141–D144. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2018, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M. UniProt knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI Reference Sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res. 2012, 40, D130–D135. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Thumuluri, V.; Almagro Armenteros, J.J.; Johansen, A.R.; Nielsen, H.; Winther, O. DeepLoc 2.0: Multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 2022, 50, W228–W234. [Google Scholar] [CrossRef] [PubMed]
- Yujian, L.; Bo, L. A normalized Levenshtein distance metric. IEEE Trans. Pattern Anal. Mach. Intell. 2007, 29, 1091–1095. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Features (Parameters) | T. pseudospiralis (ISS13) This Study | T. pseudospiralis (ISS13) Ref. [17] | T. spiralis (ISS534) This Study | T. spiralis (ISS195) Ref. [14] |
|---|---|---|---|---|
| Genome size (bp) | 56,636,606 | 49,162,916 | 63,452,358 | 63,525,422 |
| Number of scaffolds | 320 | 7221 | 568 | 6863 |
| N50 (bp); L50 | 1,024,593; 16 | 235,426; 51 | 438,897; 39 | 6,373,445; 4 |
| N90 (bp); L90 | 66,105; 83 | 60,440; 206 | 31,707; 264 | 2047; 919 |
| Genome GC content (%) | 32.6 | 32.6 | 33.6 | 33.9 |
| Repetitive sequences (%) | 25.3 | 22.3 | 27.7 | 25.6 |
| Exonic proportion; incl. introns (%) | 21.6; 45.9 | 35.7; 76.0 | 19.7; 43.7 | 22.2; 46.7 |
| Number of putative coding genes; isoforms | 9495; 10,773 | 12,659; 17,161 | 10,485; 12,588 | 16,380; 15,840 |
| Mean; median gene length (bp) | 2743; 1792 | 2950; 1241 | 2653; 1700 | 1817; 1078 |
| Mean; median CDS length (bp) | 1361; 951 | 1046; 522 | 1236; 897 | 955; 576 |
| Mean exon number per gene | 7.4 | 6.6 | 7.2 | 5.4 |
| Mean; median exon length (bp) | 175; 126 | 211; 131 | 166; 120 | 178; 129 |
| Mean; median intron length (bp) | 227; 73 | 280; 78 | 234; 72 | 198; 83 |
| Coding GC content (%) | 42.7 | 42.6 | 43.3 | 43.2 |
| BUSCO complete; duplicated; fragmented | 2060; 274; 40 | 2044; 785; 17 | 2051; 644; 44 | 2110; 74; 105 |
| BUSCO completeness: complete; partial (%) | 65.8; 67.1 | 65.3; 65.8 | 65.5; 66.9 | 67.4; 70.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korhonen, P.K.; La Rosa, G.; Sumanam, S.B.; Gomez Morales, M.A.; Ludovisi, A.; Pozio, E.; Tonanzi, D.; Chang, B.C.H.; Young, N.D.; Gasser, R.B. Enhanced Genomic and Transcriptomic Resources for Trichinella pseudospiralis and T. spiralis to Underpin the Discovery of Molecular Differences between Stages and Species. Int. J. Mol. Sci. 2024, 25, 7366. https://doi.org/10.3390/ijms25137366
Korhonen PK, La Rosa G, Sumanam SB, Gomez Morales MA, Ludovisi A, Pozio E, Tonanzi D, Chang BCH, Young ND, Gasser RB. Enhanced Genomic and Transcriptomic Resources for Trichinella pseudospiralis and T. spiralis to Underpin the Discovery of Molecular Differences between Stages and Species. International Journal of Molecular Sciences. 2024; 25(13):7366. https://doi.org/10.3390/ijms25137366
Chicago/Turabian StyleKorhonen, Pasi K., Giuseppe La Rosa, Sunita B. Sumanam, Maria Angeles Gomez Morales, Alessandra Ludovisi, Edoardo Pozio, Daniele Tonanzi, Bill C. H. Chang, Neil D. Young, and Robin B. Gasser. 2024. "Enhanced Genomic and Transcriptomic Resources for Trichinella pseudospiralis and T. spiralis to Underpin the Discovery of Molecular Differences between Stages and Species" International Journal of Molecular Sciences 25, no. 13: 7366. https://doi.org/10.3390/ijms25137366