
Inference of Essential Genes of the Parasite Haemonchus contortus via Machine Learning
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
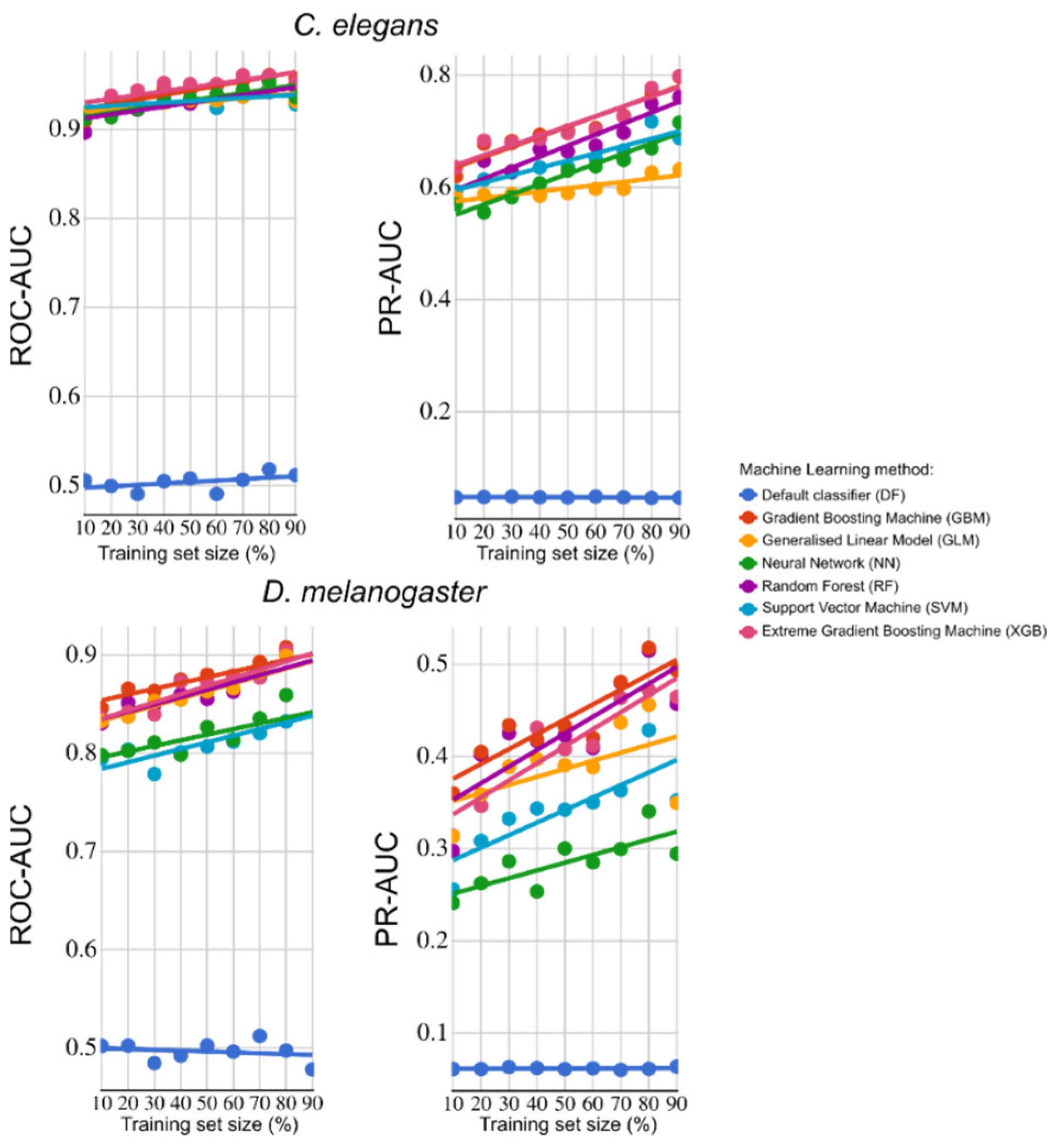
2.1. Identification of Strong Predictors of Essential Genes in H. contortus
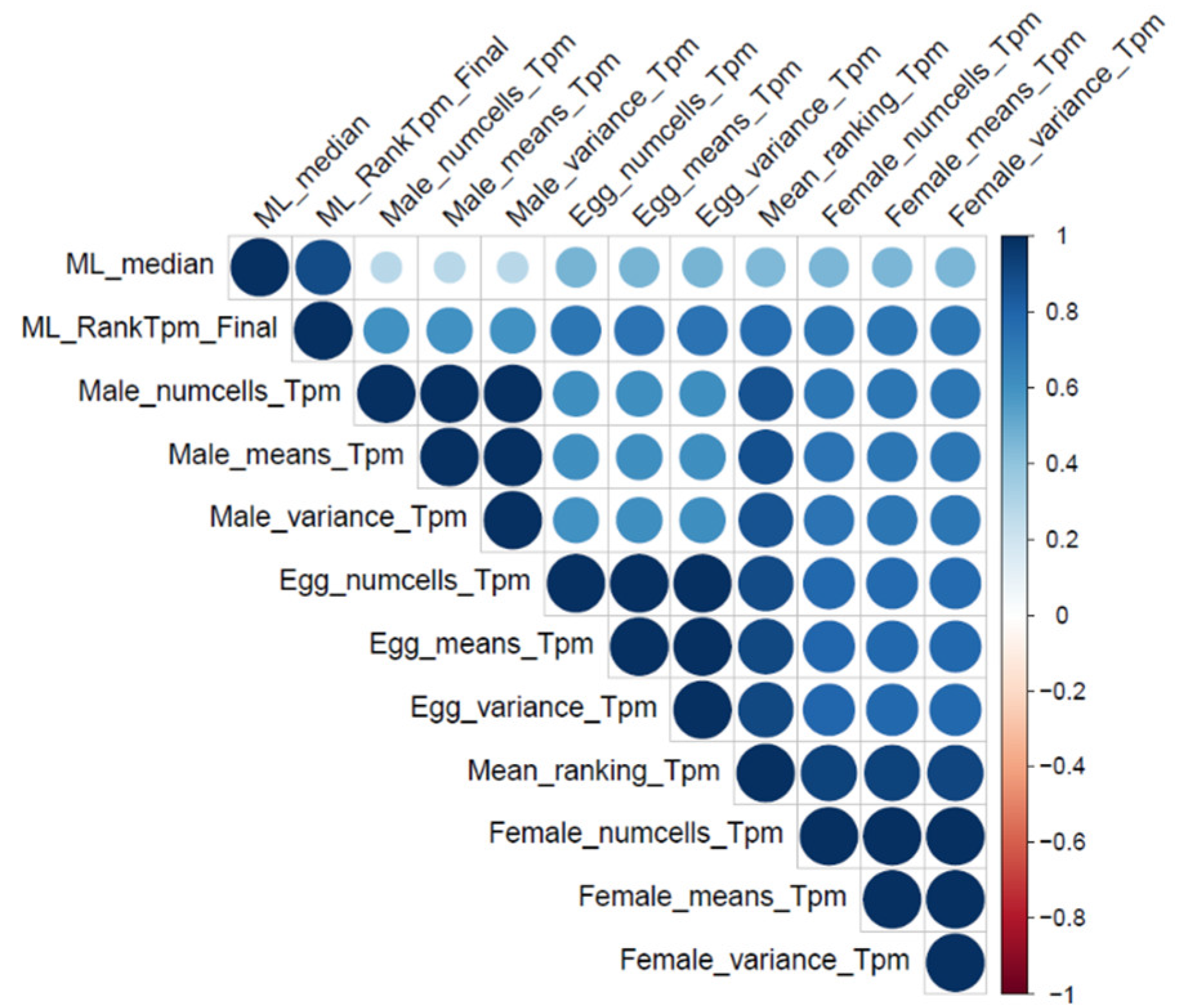
2.2. Clear Association between Essential Genes and Their Transcription Profiles
2.3. Essential Genes of H. contortus Are Inferred to Be Involved Predominantly in Ribosome Biogenesis, Translation, RNA Binding/Processing, and Signalling
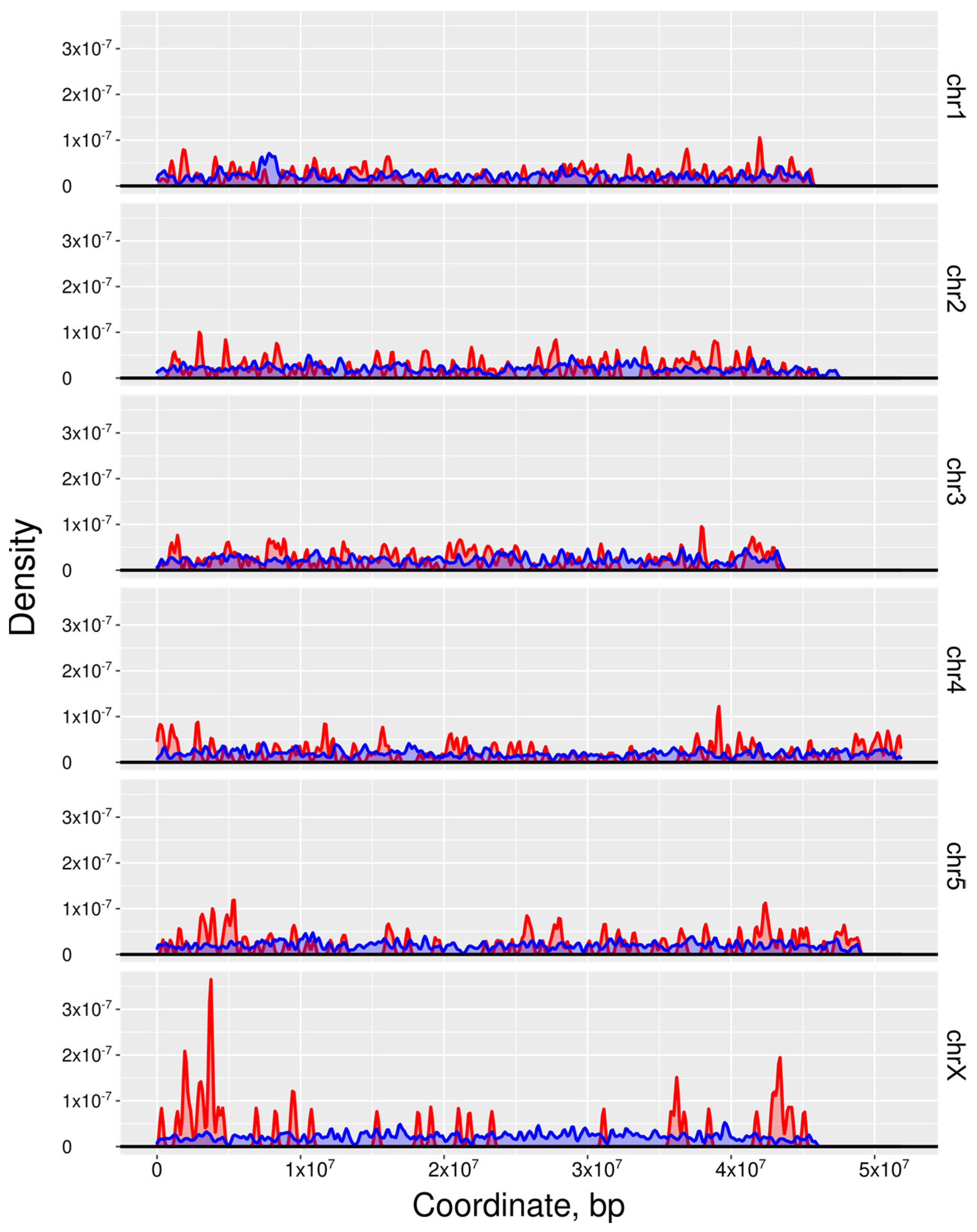
2.4. Linking Essential Genes to Genome Locations and Their Transcription to Cell Type or Tissue
3. Discussion
4. Materials and Methods
4.1. RNA Sequence Data Sets
4.2. Feature Extraction/Engineering for Subsequent ML
4.3. Predicting Gene Essentiality through ML
4.4. Establishing the List of Genes, Ranked According to the Probability of Being Essential
4.5. Gene Clustering
4.6. Methods Used to Infer Genome Locations and Transcription Profiles for Essential Genes in H. contortus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Charlier, J.; van der Voort, M.; Keyon, F.; Skuce, P.; Vercruysse, J. Chasing helminths and their economic impact on farmed ruminants. Trends Parasitol. 2014, 30, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Emery, D.L.; Hunt, P.W.; Jambre, L.F.L. Haemonchus contortus: The then and now, and where to from here? Int. J. Parasitol. 2016, 46, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Selzer, P.; Epe, C. Antiparasitics in animal health—Quoad vadis? Trends Parasitol. 2020, 37, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Preston, S.; Hofmann, A.; Taki, A.; Baell, J.; Chang, B.C.H.; Jabbar, A.; Gasser, R.B. A perspective on the discovery of selected compounds with anthelmintic activity against the barber’s pole worm—Where to from here? Adv. Parasitol. 2020, 108, 1–45. [Google Scholar] [PubMed]
- Kaminsky, R. Drug resistance in nematodes: A paper tiger or a real problem? Curr. Opin. Infect. Dis. 2003, 16, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, H.A. Anthelmintics resistance; how to overcome it? Iran J. Parasitol. 2013, 8, 18–32. [Google Scholar] [PubMed]
- Kotze, A.C.; Prichard, R.K. Anthelmintic resistance in Haemonchus contortus: History, mechanisms and diagnosis. Adv. Parasitol. 2016, 93, 397–428. [Google Scholar] [PubMed]
- Geary, T.G.; Sakanari, J.A.; Caffrey, C.R. Anthelmintic drug discovery: Into the future. J. Parasitol. 2015, 101, 125–133. [Google Scholar] [CrossRef]
- Sepúlveda-Crespo, D.; Reguera, R.M.; Rojo-Vásquez, F.; Balaña-Fouce, R.; Martínez-Valladares, M. Drug discovery technologies: Caenorhabditis elegans as a model for anthelmintic therapeutics. Med. Res. Rev. 2020, 40, 1715–1753. [Google Scholar] [CrossRef] [PubMed]
- Campos, T.L.; Korhonen, P.K.; Sternberg, P.W.; Gasser, R.B.; Young, N.D. Predicting gene essentiality in Caenorhabditis elegans by feature engineering and machine-learning. Comput. Struct. Biotechnol. 2020, 15, 1093–1102. [Google Scholar] [CrossRef]
- Campos, T.L.; Korhonen, P.K.; Hofmann, A.; Gasser, R.B.; Young, N.D. Combined use of feature engineering and machine-learning to predict essential genes in Drosophila melanogaster. NAR Genom. Bioinform. 2020, 22, lqaa051. [Google Scholar] [CrossRef] [PubMed]
- Marygold, S.J.; Crosby, M.A.; Goodman, J.L. FlyBase Consortium. Using FlyBase, a database of Drosophila genes & genomes. In Drosophila: Methods in Molecular Biology; Dahmann, C., Ed.; Springer: Berlin/Heidelberg, Germany,, 2016; Volume 1478, pp. 1–31. [Google Scholar]
- Howe, K.L.; Bolt, B.J.; Shafie, M.; Kersey, P.; Berriman, M. WormBase ParaSite—A comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 2017, 215, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.W.; Arnaboldi, V.; Cain, S.; Chan, J.; Chen, W.J.; Cho, J.; Davis, P.; Gao, S.; Grove, C.A.; Kishore, R.; et al. WormBase: A modern Model Organism Information Resource. Nucleic Acids Res. 2020, 48, D762–D767. [Google Scholar] [CrossRef] [PubMed]
- Kimble, J.; Nüsslein-Volhard, C. The great small organisms of developmental genetics: Caenorhabditis elegans and Drosophila melanogaster. Dev. Biol. 2022, 485, 93–122. [Google Scholar] [CrossRef] [PubMed]
- Campos, T.L.; Korhonen, P.K.; Young, N.D. Cross-predicting essential genes between two model eukaryotic species using machine learning. Int. J. Mol. Sci. 2022, 22, 5056. [Google Scholar] [CrossRef] [PubMed]
- Campos, T.L.; Korhonen, P.K.; Hofmann, A.; Gasser, R.B.; Young, N.D. Harnessing model organism genomics to underpin the machine-learning-based prediction of essential genes in eukaryotes—Biotechnological implications. Biotechnol. Adv. 2021, 54, 107822. [Google Scholar] [CrossRef] [PubMed]
- Britton, C.; Roberts, B.; Marks, N.D. Functional genomics tools for Haemonchus contortus and lessons from other helminths. Adv. Parasitol. 2016, 93, 599–623. [Google Scholar]
- Castelletto, M.L.; Gang, S.S.; Hallem, E.A. Recent advances in functional genomics for parasitic nematodes of mammals. J. Exp. Biol. 2020, 223, jeb206482. [Google Scholar] [CrossRef]
- Gasser, R.B.; Schwarz, E.M.; Korhonen, P.K.; Young, N.D. Understanding Haemonchus contortus better through genomics and transcriptomics. Adv. Parasitol. 2016, 93, 16–67. [Google Scholar]
- Doyle, S.R.; Tracey, A.; Laing, R.; Holroyd, N.; Bartley, D.; Bazant, W.; Beasley, H.; Beech, R.; Britton, C.; Brooks, K.; et al. Genomic and transcriptomic variation defines the chromosome-scale assembly of Haemonchus contortus, a model gastrointestinal worm. Commun. Biol. 2020, 9, 656. [Google Scholar] [CrossRef]
- Doyle, S.R. Improving helminth genome resources in the post-genomic era. Trends Parasitol. 2022, 38, 831–840. [Google Scholar] [CrossRef]
- Schwarz, E.M.; Korhonen, P.K.; Campbell, B.E.; Young, N.D.; Jex, A.R.; Jabbar, A.; Hall, R.S.; Mondal, A.; Howe, A.C.; Pell, J.; et al. The genome and developmental transcriptome of the strongylid nematode Haemonchus contortus. Genome Biol. 2013, 14, R89. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gasser, R.B. Prospects of using high-throughput proteomics to underpin the discovery of animal host-nematode interactions. Pathogens 2021, 10, 825. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Waterson, R.H.; Trapnell, C.; Shendure, J. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Aromolaran, O.; Aromolaran, D.; Isewon, I.; Oyelade, J. Machine learning approach to gene essentiality prediction: A review. Brief. Bioinform. 2021, 22, bbab128. [Google Scholar] [CrossRef] [PubMed]
- Aromolaran, O.; Beder, T.; Oswald, M.; Oyelade, J.; Adebiyi, E.; Koening, R. Essential gene prediction in Drosophila melanogaster using machine learning approaches based on sequence and functional features. Comput. Struct. Biotechnol. J. 2020, 10, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Beder, T.; Aromolaran, O.; Dönitz, J.; Tapanelli, S.; Adedeji, E.O.; Adebiyi, E.; Bucher, G.; Koenig, R. Identifying essential genes across eukaryotes by machine learning. NAR Genom. Bioinform. 2021, 3, lqab110. [Google Scholar] [CrossRef] [PubMed]
- Marques de Castro, G.; Hastenreiter, Z.; Silva Monteiro, T.A.; Martins da Silva, T.T.; Pereira Lobo, F. Cross-species prediction of essential genes in insects. Bioinformatics 2022, 6, btac009. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; McManus, M. Choosing the right tool for the job: RNAi, TALEN, CRISPR. Mol. Cell 2015, 58, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Quinzo, M.J.; Perteguer, M.J.; Brindley, P.J.; Loukas, A.; Sotillo, J. Transgenesis in parasitic helminths: A brief history and prospects for the future. Parasit. Vectors 2022, 15, 110. [Google Scholar] [CrossRef] [PubMed]
- Chien, J.; Wolf, F.W.; Grosche, S.; Yosef, N.; Garriga, G.; Mörk, C. The enigmatic Canal-Associated Neurons regulate Caenorhabditis elegans larval development through a cAMP signalling pathway. Genetics 2019, 213, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Carlton, P.M.; Davis, R.E.; Ahmed, S. Nematode chromosomes. Genetics 2022, 221, iyac014. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Hurt, E.; Bassler, J. Driving ribosome assembly. Biochim. Biophys. Acta 2010, 1803, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liao, W.J.; Liao, J.M.; Liao, P.; Lu, H. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Bolt, B.J.; Rodgers, F.H.; Shafie, M.; Kersey, P.J.; Berriman, M.; Howe, K.L. Using WormBase ParaSite: An integrated platform for exploring helminth genomic data. Methods Mol. Biol. 2018, 1757, 471–491. [Google Scholar] [PubMed]
- Korhonen, P.K.; Wang, T.; Young, N.D.; Byrne, J.J.; Campos, T.L.; Taki, A.C.; Gasser, R.B. Analysis of Haemonchus embryos at single cell resolution identifies two eukaryotic elongation factors as intervention target candidates. Comput. Struct. Biotechnol. J. 2024, 23, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Armenteros, J.J.A.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic ortholog inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Description | Source |
|---|---|---|
| OrthoFinder_species | Orthologs in other species | OrthoFinder analysis |
| num_cells_expressed | Number of cells/nuclei where a gene is transcribed | snRNA-seq data |
| exons | Number of exons | BioMart (WomBase ParaSite) |
| exons_total_length | Total length of exons | BioMart (WomBase ParaSite) |
| Cytoplasm | Subcellular localisation | DeepLoc analysis |
| Mitochondrion | Subcellular localisation | DeepLoc analysis |
| Nucleus | Subcellular localisation | DeepLoc analysis |
| AAC_S | Protein sequence feature | Extracted using protR * |
| APAAC_Pc2.Hydrophobicity.2 | Protein sequence feature | Extracted using protR * |
| CTDC_secondarystruct.Group1 | Protein sequence feature | Extracted using protR * |
| CTDD_prop4.G2.residue0 | Protein sequence feature | Extracted using protR * |
| CTDD_prop4.G2.residue25 | Protein sequence feature | Extracted using protR * |
| CTriad_VS153 | Protein sequence feature | Extracted using protR * |
| CTriad_VS431 | Protein sequence feature | Extracted using protR * |
| CTriad_VS613 | Protein sequence feature | Extracted using protR * |
| DC_HA | Protein sequence feature | Extracted using protR * |
| DC_MP | Protein sequence feature | Extracted using protR * |
| DC_MS | Protein sequence feature | Extracted using protR * |
| DC_VF | Protein sequence feature | Extracted using protR * |
| Geary_CHOC760101.lag7 | Protein sequence feature | Extracted using protR * |
| Moran_CHAM820102.lag7 | Protein sequence feature | Extracted using protR * |
| GC | DNA sequence feature | BioMart (WormBase ParaSite) |
| kmer_3_GCT | DNA sequence feature | Extracted using rDNAse * |
| PseKNC_3_Xc1.CCC | DNA sequence feature | Extracted using rDNAse * |
| PseKNC_5_Xc1.CGT | DNA sequence feature | Extracted using rDNAse * |
| PseKNC_5_Xc1.GCT | DNA sequence feature | Extracted using rDNAse * |
| TACC_Nucleosome.lag2 | DNA Sequence feature | Extracted using rDNAse * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, T.L.; Korhonen, P.K.; Young, N.D.; Wang, T.; Song, J.; Marhoefer, R.; Chang, B.C.H.; Selzer, P.M.; Gasser, R.B. Inference of Essential Genes of the Parasite Haemonchus contortus via Machine Learning. Int. J. Mol. Sci. 2024, 25, 7015. https://doi.org/10.3390/ijms25137015
Campos TL, Korhonen PK, Young ND, Wang T, Song J, Marhoefer R, Chang BCH, Selzer PM, Gasser RB. Inference of Essential Genes of the Parasite Haemonchus contortus via Machine Learning. International Journal of Molecular Sciences. 2024; 25(13):7015. https://doi.org/10.3390/ijms25137015
Chicago/Turabian StyleCampos, Túlio L., Pasi K. Korhonen, Neil D. Young, Tao Wang, Jiangning Song, Richard Marhoefer, Bill C. H. Chang, Paul M. Selzer, and Robin B. Gasser. 2024. "Inference of Essential Genes of the Parasite Haemonchus contortus via Machine Learning" International Journal of Molecular Sciences 25, no. 13: 7015. https://doi.org/10.3390/ijms25137015